
Abstract
Self-emulsifying drug delivery system (SEDDS) is the isotropic and thermodynamically stable mixture of oil, surfactant, co‐solvent/surfactant, and drug. It emulsifies spontaneously when introduced into an aqueous phase under a mild agitation. The current study was aimed to prepare SNEDDS to augment solubility, release rate, and oral bioavailability of BCS class II drug, efavirenz (EFV). A series of oil, surfactant, and co-surfactant was screened out by a ternary phase diagram to locate a better homogenous mixture. The prepared SNEDDS was evaluated regarding its appearance, mean droplet size, phase separation, in vitro drug release, and oral bioavailability. Among the screened oil, surfactant, and co-surfactant, Labrafil M 2125 CS, Tween 80, and Transcutol®P mixture exhibited superior solubilizing capacity, respectively. Optimized SNEDDS exhibits 98.39% drug release. SNEDDS dissolution behavior was attributed to oil/surfactant ratios and properties of the surfactant phase. It also demonstrates threefold increments in the area under curve (AUC) in comparison to neat EFV. Furthermore, the optimized SNEDDS does not show any vitrification during its 3-month storage. In the present study, better performance of SNEDDS is explained by various factors like (i) improved surface area of droplets, (ii) superior solubilization potential for hydrophobic drugs due to Labrafil M 2125 CS, and (iii) result of surfactant on mucosal permeability. This study demonstrated that SNEDDS may be an alternative approach for the poorly soluble drugs to improve their solubility and oral bioavailability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Poor aqueous solubility of drug has constantly presented foremost obstacle towards the development of drug delivery systems, and hence, in today’s pharmaceutical industry, solubility enhancement is the most challenging job. Almost 70% of newly discovered drug candidates show poor water solubility (1). To overcome these problems, numerous approaches are exploited including the addition of surfactant, amorphization, complexation, milling, pro-drugs, salt formation, etc. In addition to this variety of particles, engineering methods are available to augment the physicochemical properties of drug molecule (2). Most of these techniques have their own limitations due to the requirement of complicated manufacturing method, specialized equipment, and regulatory complications. Self-emulsifying drug delivery system (SEDDS) is well known for potential of delivering hydrophobic drugs (3). SEDDS is an alternative approach for the delivery which is coupled with low aqueous solubility and poor oral bioavailability (4).
SEDDS is the isotropic and thermodynamically stable mixture of oil, surfactant, co‐solvent/surfactant, and drug. It emulsifies spontaneously when introduced into an aqueous phase under mild agitation (5). In vivo motility of the stomach and intestine provides the agitation required for self-emulsification (6). Selection of a suitable self-emulsifying mixture is based upon the evaluation of the solubility of the drug in various components and the droplet size distribution of the formulated emulsion subsequent to self-emulsification (7). Self-nano-emulsifying drug delivery system (SNEDDS) has the capacity to improve the therapeutic application of various poorly aqueous soluble drug molecules. It helps to improve solubility, bioavailability, and therapeutic efficacy of the drug molecules (8). SNEDDS proves its potential by improving biopharmaceutical properties of indomethacin (9), irbesartan (10), and hydrochlorothiazide (11).
Efavirenz (EFV) is (S)-6-chloro-4(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one; it is a non-nucleoside reverse transcriptase inhibitor (NNRTI). It is an anti-retrovirus agent approved by the USFDA in 1998 for the treatment of HIV type 1 (12). EFV mediates its action by directly inhibiting HIV reverse transcriptase without the need of intracellular phosphorylation (13). EFV is a crystalline powder and insoluble in water. It is strongly basic (pKa 10.2) in nature and has an experimental log P value of 4.90. Major hurdles in poor therapeutic applications of this BCS class II drug are poor aqueous solubility (4 μg/mL), low intrinsic dissolution rate (0.037 mg/cm2/min), poor oral bioavailability (40–50%), and high protein-binding value (>99%) (14,15). Few attempts including bi-continuous nano-structured liquid crystalline particles (16), solid dispersions (17), spray drying (18), hot melt extrusion (19), co-micronization (20), and self-microemulsifying drug delivery systems (SMEDDS) (21,22) have been reported for improvement of solubility, bioavailability, dissolution profile, and physical stability EFV. Mean particle size of prepared liquid EFV-SMEDDS was approximately around 80 nm, and it does not exhibit satisfactory drug release at the end of 1 h. Still, no SNEDDS approach for the improvement of solubility and dissolution profile of EFV has been reported.
The aim of the present study was to formulate EFV-containing SNEDDS which may enhance the bioavailability of EFV. The Labrafil M 2125 CS, Tween 80, and Transcutol®P isotropic mixture was utilized, and its effects on various physicochemical properties were evaluated. The in vitro and in vivo drug release kinetic study was performed to determine the improvement in dissolution rate and bioavailability of SNEDDS. Moreover, the SNEDDS was investigated for vitrification if any during its 3-month storage.
MATERIAL AND METHOD
Material
EFV API was kindly donated by Lupin Research Park, Pvt. Ltd., Pune. Labrafil M 2125 CS, Labrafil 1944, Labrafac, Peceol, Labrasol, Transcutol®P, Lauroglycol, Caproyl 90, and Caproyl P were kindly gifted from Gattefosse (Gennevilliers, France). Capmul, Captex, and Caproyl PGE were kindly gifted from Abitec, USA. All other chemicals and reagents were of analytical grade.
Animals
A total of 12 male Wistar rats weighing 200–250 g were purchased from Yash Farm, Pune, India. All animals were kept in individual plastic cages at a steady temperature and 12-h light-dark cycles. The entire experiment was performed in accordance with the guidelines of Committee for the Purpose of Control and Supervision on Experimental Animals (CPCSEA). The experimental protocol was approved by the Institutional Animal Ethics Committee (IAEC) of Poona College of Pharmacy, Pune (CPCSEA/48/10).
Method
Solubility Studies
The solubility of EFV in various oils, surfactants, and co-surfactants was accomplished by adding an excess amount of EFV in a 2-mL microtube (Tarson-500020) containing 1 mL vehicle. Mixture was vortexed and kept for 24 h at 25°C in a shaking water bath to facilitate the solubilization. The samples were centrifuged (Allegra 64 R, Beckman Coulter, USA) at 4000 rpm for 15 min, and the supernatant was removed. The amount of dissolved drug was determined at 248 nm using a UV-visible spectrophotometer (Jasco, V-530, Japan).
Construction of Ternary Phase Diagram
To determine the concentration range of components for the existing boundary of SNEDDS and to identify the better self-emulsifying region, pseudo-ternary phase diagrams were constructed (23). Ternary phase diagram was plotted using PCP-Disso software (V3; Poona College of Pharmacy, Pune, India). The oil, surfactant, and co-surfactant concentration was varied from 10 to 80% (v/v). These mixtures were diluted dropwise with double-distilled water, to formulate emulsion under moderate agitation.
Preparation of SNEDDS
EFV (150 mg) was dissolved in 1-mL mixture of oil, surfactant, and co-surfactant. All components were mixed on a magnetic stirrer at 50°C and then subjected to vortex. These batches were further screened on the basis of solubility of EFV, particle size of droplets, and phase separation (Table I).
Visual Assessment
For visual assessment of batches, 1 mL of each batch was subjected to 250 mL of distilled water, occasionally shaken, and analyzed visually. The formulations were classified as clear, non-clear, stable, or unstable (7).
Droplet Size Analysis
After dilution of batches (1 mL) in 250 mL of distilled water, the particle size of all SNEDDS batches was measured using a particle size analyzer (Mastersizer 2000 SM, Malvern Instruments, UK) based on laser light scattering phenomenon.
Phase Separation Study
Phase separation study was performed for all SNEDDS batches. Phase separation study was performed in three different media, i.e., distilled water, 0.1 N HCl, and 1% sodium lauryl sulphate (SLS) to check the compatibility of oils, surfactant, and co-surfactant. SNEDDS was placed in individual 5-mL media, and phase separation was observed for up to 24 h.
In Vitro Dissolution Study
The in vitro dissolution studies were done to determine the rate of dissolution of SNEDDS and neat EFV. SNEDDS and EFV (dose equivalent to 150 mg) were filled manually in hard gelatin capsules size 2 and subjected to dissolution study. The dissolution test was executed in USP Type II (Paddle) apparatus (Electrolab, Mumbai, India) with a speed of 50 rpm using 900 mL 1% SLS solution. The temperature of the medium was maintained at 37 ± 0.5°C throughout the experiment. A 5-mL aliquot was withdrawn at predetermined time intervals of 5, 10, 20, 30, 45, 60, 90, 120, 180, and 240 min and filtered through 0.45-μm filters. Then 5 mL of fresh dissolution medium was replaced to maintain the constant volume of dissolution medium. The concentration of EFV was determined spectrophotometrically at 248 nm. Data were analyzed by PCP-Disso software (V3; Poona College of Pharmacy, Pune, India).
In Vivo Oral Pharmacokinetic Study
The rats were divided into two groups. One group was administered SNEDDS orally, dose equivalent to 30 mg/kg body weight of EFV, while the other group was administered EFV dispersed in water, at a constant dose. This dose level was chosen because it was based on body weights when administered orally. Blood sample was collected through retro-orbito puncture under mild anesthesia into heparinized tubes at time intervals 0.5, 1, 2, 3, 4, 5, 6, 12, 24, and 48 h time points after dosing. Plasma was separated by centrifugation (Allegra 64 R, Beckman Coulter, USA) for 15 min at 4°C and 7000 rpm and stored at −20°C prior to analysis.
Analysis of Plasma Samples
Plasma samples were analyzed for the content of EFV by the HPLC system composed of UV/VIS detector (Jasco UV 975). Chromatographic separation was achieved on a Hypersil ODS (250 × 4.6 mm i.d., 5 μm) column and protected by guard column at ambient temperature. The isocratic mobile phase composed of acetonitrile and 50 mM potassium phosphate (55:45 v/v) and pH was adjusted to 4.3 with orthophosphoric acid. Atorvastatin was used as internal standard. The flow rate and detection wavelength were 1 mL/min and 250 nm, respectively. The software used was Jasco BORWIN version 1.5 LC-netll/ADC system (24). The various pharmacokinetic parameters like maximum peak plasma (C max), maximum time to reach peak concentration (t max), and area under curve (AUC) were analyzed.
Stability Studies
The optimized formulation was subjected to chemical and physical stability studies. The stability studies were followed for a 3-month period in refrigeration (5 ± 3°C) and at room temperature (25 ± 3°C). At the end of every month, samples are evaluated for appearance, color, and EFV content also.
RESULT AND DISCUSSION
Solubility Study
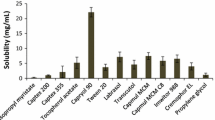
Solubility studies were performed to recognize suitable oil, surfactant and co-surfactant that have the superior solubilizing capacity for EFV. Solubility of the EFV in various oils, surfactants, and co-surfactants is given in Figs. 1 and 2. Among the used oils, the Labrafil M 2125 CS showed maximum solubility for EFV. Moreover, Tween 80 and Transcutol®P were used respectively as surfactant and co-surfactant. It was observed that droplet size decreases as amount of surfactant increases, but after reaching a certain limit, surfactant causes decrease in droplet size and increase in emulsification time; thus, an optimized amount of surfactant was selected with less droplet size and emulsification time.

Solubility of EFV in various oils

Solubility of EFV in various surfactants and co-surfactants
Construction of Ternary Phase Diagram
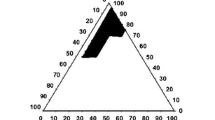
Criteria for selection of oil, surfactant, and co-surfactant were based on their drug solubility potential, hydrophilic-lipophilic balance (HLB), and ability of emulsion formation (25). Figure 3 represents the blue color area of stable nano-emulsion in the presence of EFV. The compositions of optimized SNEDDS with consideration of drug solubilization and lowest mean droplet size are given in Table II. The systems containing around 30–50% (v/v) oil, 30–50% (v/v) surfactant, and 20–40% (v/v) co-surfactant demonstrate clear, transparent, no precipitation, phase separation, cracking, and coalescence of oil droplets. The efficiency of emulsification was better when the oil concentration was more than 50% v/v of SNEDDS formulation. On the basis of ternary phase diagram, specific regions of emulsification was selected, and within that range, the ratio of Labrafil M 2125 CS (30–35%), Tween 80 (40–50%), and Transcutol®P (20–30%) was varied to yield different batches for further study.

Phase diagram of Labrafil M 2125 CS, Tween 80, and Transcutol®P
Visual Assessment
In visual assessment, batch L3 was found to be excellent clear with a faster emulsification rate (Table III).
Droplet Size Analysis
Droplet size of the emulsion is the key variable which determines the rate and extent of drug release as well as drug absorption. Smaller size emulsion droplets may lead to more rapid absorption of drug (26). Mean particle size and uniformity of SNEDDS formulation are shown in Table III and Fig. 4. Batch L3 demonstrates satisfactory particle size and emulsification time (Fig. 5).

Droplet size distribution of all batches

Mean droplet size of batch 3
Phase Separation Studies
Phase separation study was performed in three different media, i.e., distilled water, 0.1 N HCl, and 1% SLS (Table IV). Phase separation study revealed that all batches were stable and no phase separation was occurring between the excipient and drug. Out of all the prepared batches, batch L3 was excellently clear without phase separation and thus was selected for further studies.
In Vitro Dissolution Study
The Q 0 to Q 240 values of capsules containing optimized SNEDDS formulation and EFV are represented in Table V. The 1% sodium lauryl sulphate (SLS) solution was used as a dissolution medium. Figure 6 indicates dissolution profile of optimized SNEDDS formulation and EFV. Optimized SNEDDS exhibits a constantly superior drug dissolution rate compared to that of EFV. Within the period of 30 min, only 35% EFV was dissolved form EFV sample. Conversely, a capsule containing optimized SNEDDS exhibits more than 50% of drug release. The total drug released from EFV was only 53.79% in 3 h, while percentage drug release (Q 120) from optimized SNEDDS was 98.39%. In the present study, SNEDDS exhibits a faster and almost complete dissolution profile compared to the pure drug in 2 h. SNEDDS dissolution behavior was attributed to oil/surfactant ratios and the properties of the surfactant phase. Emulsification time of formulations was increased as oil content was increased. But this may lead to poor self-nano-emulsifying systems due to larger oil droplets (27). Labrafil M 2125 CS is a moderate chain length linoleoyl macrogol glyceride. In comparison to long-chain triglycerides oils, moderate chain length oils are easy to nano-emulsify. Transcutol®P, i.e., amphiphilic solubilizers, is often used in the SNEDDS formulation to improve drug loading and time required for self-nano-emulsification (28). In addition, the presence of Tween 80 as co-surfactants helps build a flexible interracial film, reduces the interfacial tension, and improves the fluidity of the interface (29).

Dissolution profiles of optimized SNEDDS and efavirenz
In Vivo Oral Pharmacokinetic Study
The mean EFV plasma concentration-time profile after oral administration of EFV suspended in water and optimized SNEDDS batch are shown in Fig. 7. The summary of pharmacokinetics of neat efavirenz and SNEDDS is given in Table II. The total plasma concentrations of optimized SNEDDS were found to be superior to EFV. Initial plasma concentrations of animals receiving optimized SNEDDS are significantly higher than those animals receiving EFV. The optimized SNEDDS exhibits noticeably higher AUC and C max than EFV. The AUC of EFV from optimized SNEDDS clearly indicates a threefold higher increment than that of EFV. Yet, the t max value of both optimized SNEDDS and EFV did not differ. Present results indicate that it is possible to improve the bioavailability of EFV if given in the form of SNEDDS. These results were steady with the results from the in vitro dissolution study, indicating that the differences in EFV absorption is primarily accredited to the dissolution profile of EFV. Surfactants help to improve permeability of drugs by maculating tight junctions among the cells and distribute the drug across the cell membrane (30). Oils with medium-chain mono- and di-glycerides, i.e., Labrafil M 2125 CS, have superior solubilization potential for hydrophobic drugs and permeation-enhancing properties (28). Improved bioavailability of EFV was due to medium-chain mono- and di-glycerides. In the current study, better performance of SNEDDS was explained by various factors like (i) improved surface area of droplets, (ii) superior solubilization potential for hydrophobic drugs due to Labrafil M 2125 CS, and (iii) result of surfactant on mucosal permeability.

Plasma profile of optimized SNEDDS and efavirenz
Stability Studies
During the entire 3-month stability study, there were no significant differences in chemical and physical properties of SNEDDS. The appearance, color, and EFV content did not show any significant change in both refrigeration and at room temperature conditions. Thus, these studies assured that formulated SNEDDS was stable up to 3 months.
CONCLUSION
A stable SNEDDS formulation of EFV was formulated successfully. SNEDDS containing EFV showed an appreciable improvement in in vitro and in vivo drug release. Chemical nature and concentration of oil, surfactant, and co-surfactant exhibit prominent effect on emulsifying potential, solubility, and dissolution behavior of EFV. The superior physicochemical properties were shown by SNEDDS due to unique mixture of Labrafil M 2125 CS, Tween 80, and Transcutol®P. It helps to enhance the solubility, stability, dissolution behavior, and bioavailability of EFV. Current results demonstrate SNEDDS as an opportunity to formulate a potential drug delivery system for improving solubility, stability, dissolution rate, and oral bioavailability of various BCS class II candidates.
References
Khadka P, Ro J, Kim H, Kim I, Kim J, Kim H, et al. Pharmaceutical particle technologies: an approach to improve drug solubility, dissolution and bioavailability. Asian J Pharm Sci. 2014;9:304–16.
Rajalakshmi S, Pawar AP, Mali AJ, Bothiraja C. Crystal engineering of bioactive plumbagin using antisolvent precipitation, melt solidification and sonocrystallization techniques. Mater Res Express. 2014. doi:10.1088/2053-1591/1/2/025405.
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self emulsifying and self micro-emulsifying drug delivery system. Eur J Pharm Sci. 2000;11:93–8.
Kim HJ, Yoon KA, Hahn M, Park ES, Chi SC. Preparation and in vitro evaluation of self-micro emulsifying drug delivery systems containing idebenone. Drug Dev Ind Pharm. 2000;26:523–29.
Kumar A, Sharma S, Kamble R. Self emulsifying drug delivery system (SEDDS): future aspects. Int J Pharm Pharm Sci. 2010;2:7–13.
Shah NH, Carvajal MT, Patel CI, Infeld MH, Malick AW. Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm. 1994;106:15–23.
Kommuru TR, Gurley B, Khan MA, Reddy IK. Self emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212:233–46.
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–82.
Obitte NC, Ofokansi KC, Nzekwe IT, Esimone CO, Okoye IE. Self-nano emulsifying drug delivery systems based on melon oil and its admixture with a homo lipid from Bos indicus for the delivery of indomethacin. Trop J Pharm Res. 2011;10:299–307.
Patel J, Patel A, Raval M, Sheth N. Formulation and development of a self-nano emulsifying drug delivery system of irbesartan. J Adv Pharm Technol Res. 2011;2:9–16.
Yadav PS, Yadav E, Verma A, Amin S. Development, characterization, and pharmacodynamic evaluation of hydrochlorothiazide loaded self-nano emulsifying drug delivery systems. Sci World J. 2014;2014:1–10.
Young SD, Britcher SF, Tran LO, Payne LS, Lumma WC, Lyle TA, et al. L-743, 726 (DMP-266): a novel, highly potent non nucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 1965;39:2602–05.
Tripathi KD. Essentials of medical pharmacology. 6th ed. Jaypee; 2003.
Boffito M, Back DJ, Blaschke TF, Rowland M, Bertz RJ, Gerber JG, et al. Protein binding in antiretroviral therapies. AIDS Res Hum Retrov. 2003;19:825–35.
Gaur PK, Mishra S, Bajpai M, Mishra A. Enhanced oral bioavailability of efavirenz by solid lipid nanoparticles: in vitro drug release and pharmacokinetics studies. BioMed Res Int. 2014;2014:1–9.
Avachat AM, Parpani SS. Formulation and development of bi continuous nano structured liquid crystalline particles of efavirenz. Colloids Surf B: Biointerfaces. 2015;126:87–7.
Koh PT, Chuah JN, Talekar M, Gorajana A, Garg S. Formulation development and dissolution rate enhancement of efavirenz by solid dispersion systems. Indian J Pharm Sci. 2013;75:291–01.
Tshweu L, Katata L, Kalombo L, Chiappetta DA, Hocht C, Sosnik A, et al. Enhanced oral bioavailability of the antiretroviral efavirenz encapsulated in poly(epsilon-caprolactone) nanoparticles by a spray-drying method. Nanomedicine (Lond). 2014;9:1821–33.
Sathigari SK, Radhakrishnan VK, Davis VA, Parsons DL, Babu RJ. Amorphous-state characterization of efavirenz-polymer hot-melt extrusion systems for dissolution enhancement. J Pharm Sci. 2012;101:3456–64.
Da Costa MA, Seiceira RC, Rodrigues CR, Hoffmeister CRD, Cabral LM, Rocha HVA. Efavirenz dissolution enhancement I: co-micronization. Pharmaceutics. 2013;5:1–22.
Reddy MS, Reddy NS, Reddy SM. Solubility enhancement of poorly water soluble drug efavirenz by solid self emulsifying drug delivery systems. Int J Pharma Res Rev. 2014;3:20–8.
Deshmukh A, Kulakrni S. Novel self micro-emulsifying drug delivery systems (SMEDDS) of efavirenz. J Chem Pharm Res. 2012;4:3914–19.
Craig DQM, Barker SA, Banning D, Booth SW. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int J Pharm. 1995;114:103–10.
Kamble RN, Mehta PP. Development of simple HPLC method to estimate the blood plasma concentration of efavirenz in rat after oral administration. Int J Pharm Pharm Sci. 2015;7:153–56.
Patil P, Joshi P, Paradkar A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS PharmSciTech. 2004;5:1–8.
Liu L, Pang X, Zhang W, Wang S. Silymarin-loaded self microemulsifying drug delivery systems. Asian J Pharm Sci. 2007;2:150–60.
Eid AM, El-Enshasy HA, Aziz R, Elmarzugi NA. The preparation and evaluation of self nano emulsifying systems containing Swietenia oil and an examination of its anti-inflammatory effects. Int J Nanomedicine. 2014;9:4685–95.
Date AA, Desai N, Dixit R, Nagarsenker M. Self-nano emulsifying drug delivery systems: formulation insights, applications and advances. Nanomedicine. 2010;5:1595–16.
Yang S, Shin H. The functional behaviors of co-surfactant in design of self-nano-emulsifying drug delivery systems. J Pharm Invest. 2010;40:263–67.
Balakrishnan P, Lee B, Oh D, Kim J, Hong M, Jee J, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS). Eur J Pharm Biopharm. 2009;72:539–45.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Kamble, R.N., Mehta, P.P. & Kumar, A. Efavirenz Self-Nano-Emulsifying Drug Delivery System: In Vitro and In Vivo Evaluation. AAPS PharmSciTech 17, 1240–1247 (2016). https://doi.org/10.1208/s12249-015-0446-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0446-2