
Abstract
Mutations in NPM1, FLT3 and CEBPA genes are found in 25–35 % of adult acute myeloblastic leukemia (AML) cases and correlate with prognosis. To date, there have been no reports about these mutations in pediatric AML from Argentina. The aims of the present study were to describe the incidence of NPM1, FLT3 and CEBPA mutations and to analyze their prognostic impact in this population. The incidences of these mutations within a population of 216 pediatric AML cases were: NPM1-mutated 4.2 %, CEBPA-mutated 1.9 %, FLT3-ITD 10.2 % and FLT3-TKD 7.9 %. Among 33 patients with normal karyotype, we found significantly higher frequencies for NPM1-mutated 24.2 % and CEBPA-mutated 12.1 %. Overall survival (pOS) for the 163 eligible non-acute promyelocytic leukemia cases was 46.2 ± 4.3 %, while leukemia-free survival probability was 51.0 ± 4.4 % (n = 135). The NPM1-mutated/FLT3-ITD-negative genotype showed better outcome than any other combined NPM1/FLT3 genotype; this difference was statistically significant within the group of high-risk patients (pOS ± SE 83.3 ± 15.2 % versus 33.1 ± 4.7 %; p = 0.0251). This is the first report of the frequencies of these mutations in Argentina. Despite the limited number of patients, a favorable prognosis of AML with genotype NPM1-mutated/FLT3-ITD-negative was confirmed. This is especially relevant within the high-risk group of patients, as it may contribute to the detection of patients with better prognosis, and thus avoid unnecessary treatment intensification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloblastic leukemia (AML) is a genetically heterogeneous disease, with 50–60 % of patients showing recurrent chromosomal alterations which are related to the leukemogenic process and provide relevant prognostic information [1]. In AML patients without recurrent chromosomal abnormalities, a large number of alterations have been involved in the pathogenesis of the disease. It has been proposed that leukemia results from the acquisition of at least two complementary molecular alterations: Class I mutations which confer a proliferative advantage to hematopoietic progenitors, and Class II mutations which cause impaired differentiation of normal cells [1–3]. In recent years, new genetic alterations have been identified among adult AML patients, including mutations in NPM1 (nucleophosmin), FLT3 (fms-related tyrosine kinase 3), and CEBPA (alpha enhancer binding protein/CCAAT) genes that have been reported in 6–12, 18–25 and 6–8 %, of cases, respectively, according to previously published data [4–14]. Mutations in NPM1 and CEBPA are mutually exclusive and are generally associated with normal karyotype-AML [15]. In the absence of other prognostic factors, patients with AML who carry mutations in NPM1 and CEBPA seem to be part of a group of AML with better prognosis [16–20]. Other studies have reported that approximately 40 % of patients with NPM1 mutations show mutations in FLT3 gene, especially internal tandem duplications (ITD) [21]. These patients with NPM1-mutated and FLT3-ITD show a lower overall survival (pOS) and event free survival probabilities [4]. Based on clinical studies, genetics, and their prognostic value, the World Health Organization (WHO) classification of AML has recently incorporated NPM1 and CEBPA mutations as a new group of leukemia within “Acute myeloid leukemia and related neoplasms” [22].
The aims of the present study were to report the incidence of mutations in NPM1, FLT3 and CEBPA genes in pediatric patients diagnosed with AML, to correlate the presence of these mutations with clinical and biological features and to evaluate the role of NPM1, FLT3, and CEBPA mutations as predictors of clinical outcome in pediatric AML patients from Argentina. The analysis of the impact of these mutations on the outcome of our population of AML patients is the first step to achieve better stratification criteria for future AML protocols.
Patients and methods
The present study included 216 patients diagnosed with AML admitted at the Hematology and Oncology Department of Garrahan Hospital, from March 2000 to May 2014.
AML diagnosis was performed according to morphological, cytochemical, immunophenotypic and cytogenetic criteria of the WHO classification [23]. In 92 % of cases, molecular studies for the detection of fusion transcripts PML/RARA, RUNX1/RUNX1T1, CBFB/MYH11 and/or KMT2A/MLLT3 were performed by standard techniques [24, 25].
The analysis of treatment response and survival was performed on a population of 163 eligible patients. Thirty three acute promyelocytic leukemia (APL), 9 patients with treatment-related leukemia, 1 patient who was lost-in-follow up, and 6 patients that had received chemotherapy before admission to the hospital and who did not have complete diagnostic studies were excluded. The group of AML cases with CEBPA mutations were also excluded because of the small sample size (n: 4). The leukemia-free survival probability (pLFS) was analyzed in the population of 135 patients who achieved CR.
Patients were classified into two risk groups according to the cytogenetic–molecular findings and evaluation of early response to chemotherapy in bone marrow aspirates on day 15 of treatment. Patients with t(15;17)(q24;q21) or PML/RARA, t(8;21)(q22;q22) or RUNX1/RUNX1T1, inv(16)(p13;q22) or CBFB/MYH11 and early good response on day 15 of therapy (less than 10 % of blasts in bone marrow aspirate) were defined as standard-risk group. Acute promyelocytic leukemia patients were not evaluated for response on day 15. Those patients in whom the presence of these translocations was ruled-out and any AML case who presented more than 10 % of blasts in bone marrow on day 15 were stratified as high-risk group. Non-APL AML patients were treated according to 2 consecutive AML protocols based on Berlin–Frankfurt–Münster (BFM) study group treatment strategy [26]. AML patients defined as high risk were eligible for receiving allogeneic hematopoietic stem cell transplantation (HSCT) as consolidation of their treatment, if they had an available matched familial donor (MFD).
The characterization of mutations in NPM1, FLT3 and CEBPA genes was retrospectively carried out on mononuclear cells obtained by density gradient centrifugation from bone marrow samples and preserved at −80 °C until use. NPM1 and CEBPA mutations were studied from genomic DNA in all cases. Total DNA was isolated using DNA extraction kit (QIAamp DNA Mini Kit (Qiagen, Germany). FLT3 mutations were studied using copy DNA (cDNA) in all cases. Total RNA was reverse-transcribed according to the BIOMED-1 protocol from 1 μg of RNA using random hexamers in a final volume of 20 μl [24].
The detection FLT3-ITD was performed by polymerase chain reaction (PCR) amplification of exons 14 and 15 of the FLT3 gene, using primers R5 and R6, as described in the literature [27]. The presence of internal duplications was analyzed by 9 % polyacrylamide gel electrophoresis and ultraviolet visualization after ethidium bromide staining. Detection of FLT3-TKD (FLT3-tyrosine kinase domain) was performed by PCR amplification of exon 20 of the FLT3 gene with primers 17F′ and 17R′, similar to those already reported [28], and subsequent digestion with EcoRV restriction enzyme that recognizes the cleavage site corresponding to codons 835–836 (GATATC) of the wild-type sequence. Mutations in CEBPA and NPM1 were studied by GeneScanning analysis of PCR products as previously published [15]. Briefly, fluorescent PCR products were subjected to capillary electrophoresis in an automated DNA sequencer (ABI-Prism Model 3130, Applied Biosystems) and fragment analysis was performed using GeneMapper software v4.0.
All cases positive for NPM1, FLT3 and CEBPA mutations were sequenced to characterize mutations at the molecular level, using the sequencing kit Big Dye Terminator v1.1 (Applied Biosystems) and an automated DNA sequencer (model ABI-Prism 3130, Applied Biosystems).
Statistical analysis
The descriptive statistics of the results were performed using Statistix (Analytical Software, Florida, USA), version 7.0. The correlation of discrete diagnostic features of AML patients with the presence of NPM1, CEBPA and FLT3 mutations was analyzed by bivariate methods. Statistical significance was calculated using Chi-square or Fisher exact test for nominal variables and Student’s test or Wilcoxon test for continuous numerical variables. p values <0.05 were considered as statistically significant.
Outcome events were defined as death during induction, failure to achieve complete remission (CR), death during remission, and relapse. Leukemia-free survival was defined as the time from CR to the occurrence of an adverse event (relapse or death in CR due to any cause); patients alive and relapse-free at last follow-up were censored. Overall survival (OS) was defined as the time from the diagnosis to the date of the death from any cause, with patients last known to be alive without report of relapse censored at the date of last contact. The LFS and OS probabilities were estimated by the Kaplan–Meier method, and standard error was calculated by the method of Peto. The survival probabilities reported were expressed as percentage of estimated 5-year probability ± standard error (SE). Differences between curves were analyzed by the log-rank test [29–31].
Results
Mutations in NPM1, FLT3 and CEBPA: incidence and clinical correlation
The clinical characteristics, French–American–British (FAB) subtypes and genetic alterations of the 216 AML patients are shown in Table 1. The mean age was 7.0 years (range 1 day–17.9 years) and 44 infants (patients younger than 1 year of age) were included in population of patients.
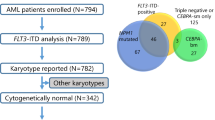
The incidences of the studied mutations were: NPM1-mutated 4.2 %, CEBPA-mutated 1.9 %, FLT3-ITD 10.2 % and FLT3-TKD 7.9 % in the total population of analyzed cases. Normal karyotype was observed in 15 % of cases (n: 33) and the incidences of the studied mutations within this group of cases were: NPM1-mutated 24.2 % (p < 0.00001), CEBPA-mutated 12.1 % (p = 0.0001), FLT3-ITD 18.2 % and FLT3-TKD 6.1 %. The combined data of the genetic aberrations studied in the population of 216 AML cases is shown in Fig. 1.

Scheme showing the combined data of the genetic aberrations observed in the group of 216 AML patients
The mean age was significantly higher in patients with mutations, being NPM1-mutated 13.4 years (p = 0.0001), CEBPA-mutated 11.6 years (p < 0.00001) and FLT3-ITD 13.8 years (p = 0.0368), showing a gradual increase of incidence with older ages. There were no significant differences between NPM1, FLT3 and CEBPA-mutated and wild-type patients in terms of sex and white blood cell (WBC) count at presentation, although the median WBC of those in the FLT3-mutated group was higher than that of those within the non-mutated FLT3 group.
Regarding patients younger than 1 year old, the mean age was 4.5 (range 0–11.0) months and median WBC was 74.4 (range 2.1–500.0) × 109/L. Fifty percent of infants presented alterations in the KMT2A (lysine methyltransferase 2A) gene and FLT3-TKD was the only mutation detected in this group of patients (11.4 %) (Table 2).
The median length of duplicated fragments in FLT3-ITD cases was 57 nucleotides (range 15–111 nucleotides), being 48 nucleotides for duplications (range 12–111) and 6 nucleotides for insertions (range 1–18).
FLT3-TKD mutation was detected in 17 out of 216 patients with AML: 5 of 44 infants (11.4 %) and 12 of 172 children older than 1 year (7.0 %). The mutation most frequently observed was the substitution Asp835Glu.
NPM1 gene mutations were detected in 9 out of 216 patients studied. Four of these nine patients showed type A mutation, while the remaining 5 patients presented mutations not previously reported. The detailed sequences are shown in Table 3.
In 3 of the 5 previously unreported mutations, the resulting newly reported NPM1 variant protein retains the tryptophan at position 288, whereas loss of tryptophan at position 290 is observed in all cases, with generation of the same last five amino acid residues (VSLRK). Eight out of 9 patients who harbored mutations in NPM1 had normal karyotype (p < 0.00001), while the ninth case corresponded to an AML case with no metaphases evaluable for cytogenetic study and no recurrent rearrangements detected by reverse transcription followed by polymerase chain reaction analysis. In 2 cases NPM1 mutations coexisted with FLT3-ITD, one of them with a TGCC insertion in NPM1 and duplication of 48 nucleotides in FLT3 (UPN 256), and the other one with type A mutation in NPM1 and a duplication of 73 nucleotides plus a 2 nucleotides-insertion in FLT3 gene (UPN 843).
Eighteen double signals were detected in the electropherograms of the fragment analysis from the 216 patients studied for the presence of CEBPA mutations. Fourteen of the 18 alterations detected corresponded to mutations within the transactivation domain-2 (TAD2) region where an insertion of 6 nucleotides (GCACCC) was observed in the genomic DNA sequence which results in a histidine–proline duplication. This variation has been reported to be a polymorphism, thus these patients were not considered as being mutated for the present study [15]. The remaining 4 mutated cases showed insertions of 3–12 nucleotides in the basic leucine zipper (b-ZIP) domain (Table 3), preserving the reading frame. These four patients had normal karyotype and the absence of recurrent translocations. One patient with CEBPA mutation (UPN 1238) showed a mutated FLT3-TKD with a substitution of aspartic acid to tyrosine at position 836 of the FLT3 protein.
Response to treatment and outcome
Parameters of response to treatment, the observed events and survival analysis for the population of 163 evaluable non-APL patients are shown in Table 1. The mean follow-up time for the total population was 63 months, ranging from 13 to 158 months and the 5-year pLFS ± SE and pOS ± SE were 51.0 ± 4.4 and 46.2 ± 4.3 %, respectively.
Regarding the response to treatment for the total group, 83 % of patients achieved complete remission, 22 patients (13 %) died during the induction phase, while the remaining 3 % showed no response to treatment.
The pLFS ± SE for FLT3-ITD group was 44.4 ± 16.6 %, and for FLT3-TKD it was 55.6 ± 16.6 % (see Table 1). The pLFS ± SE for NPM1 mutated patients was 75.0 ± 15.3 % while it was 49.4 ± 4.5 % for NPM1 wild-type AML cases (p = 0.2081). All patients with NPM1 mutations were early responders on day 15 and achieved complete remission after induction phase. One patient who presented mutated NPM1 and FLT3-ITD relapsed in bone marrow within 6 months and died due to disease progression, while another patient died in CR due to sepsis. The six remaining patients keep staying in continuous CR with a median follow-up of 66 months. Two of them underwent HSCT as consolidation treatment.
One patient with mutated CEBPA and FLT3-TKD died during the induction phase of treatment. The remaining 3 cases achieved CR and 2 of them were high-risk patients with available MFD who received HSCT. This subgroup of patients was considered too small to draw further conclusions regarding their survival probability.
The pLFS for 135 patients with AML other than acute promyelocytic leukemia according to the combined genotypes of FLT3-ITD and NPM1 is shown in Fig. 2a. The best outcome was observed for the FLT3-ITD-negative and NPM1-mutated group of patients (83.3 ± 15.2 %).

Kaplan–Meier curves for leukemia-free survival for 135 non-APL patients according to a the different combinations of FLT3-ITD and NPM1 status, and b combined genotype FLT3-ITD negative and NPM1 positive versus other genotypes
Regarding the 127 high-risk patients, 21 died on induction, 100 (79 %) of them achieved CR, 13 died in CR, 42 relapsed and 45 remain in CR. Considering the subgroup of 26 AML patients with normal karyotype, 4 died on induction, 21 (81 %) of them achieved complete remission, 2 died in CR, 8 relapsed and 11 remain in CR. The pOS and pLFS for the high-risk group and for the normal karyotype patients according to the combined genotypes are shown in Fig. 3.

Kaplan–Meier curves for overall survival and leukemia-free survival stratified by the status of FLT3-ITD and NPM1 in high risk group (a, b), and in normal karyotype AML patients (c, d)
Discussion
The present study shows the retrospective analysis of the incidence of NPM1, FLT3 and CEBPA mutations, the clinical impact and the outcome of pediatric AML according to their presence in 216 patients, from a single Institution in Argentina.
NPM1 mutations were detected in 4.2 % of patients with AML, showing a slightly lower frequency than previously published data in other series of pediatric patients where incidences ranged between 6 and 8 % [4, 5, 8, 9]. The lower frequency observed might be due to the composition of the study population, since 20 % of our patients were children younger than 1 year of age. This bias is due to the fact that Garrahan Hospital, as national referral center, receives this particular group of patients from many different institutions. When infant patients are excluded from the incidence analysis, the frequency of mutated NPM1 increases to 5.2 %. Mutation A, reported as the most frequent mutation in all series, was detected in 44.4 % of cases and five additional novel NPM1 mutations could be identified. All mutations detected in this study resulted in the loss of tryptophan at position 290, and shared the same last five amino acid residues (VSLRK) comparable to what has already been reported [32].
In our series of patients, a significant increase of the incidence of NPM1 mutations with age was observed and no mutations were detected in children younger than 6 years of age. This is in agreement with studies reported by other authors, detecting a tendency to higher incidence of mutations in NPM1 with increasing age [4, 8, 33]. NPM1 mutations were most often observed in the FAB-M2 subtype and no significant differences were detected in terms of gender and WBC association. Similar to what has been reported in adult AML patients [32, 34], our results show that in children with AML diagnosis, the frequency of NPM1 mutations is significantly higher in the group of patients with normal karyotype, which has been historically included in the group of high-risk AML patients [4, 5, 18, 35].
CEBPA mutations were detected in 1.9 % of patients with AML, being all of them insertions of nucleotides within the b-ZIP domain, maintaining the reading frame. We also found an insertion of 6 nucleotides in the TAD2 domain in 6.5 % of the studied patients, a mutation that has been reported as being a polymorphism [36]. In contrast to what has been described for adult AML publications, no cases of double CEBPA mutations were detected [17, 37]. This can be due to the characteristics of our cohort which included 20 % of patients under 1 year of age, although limitations of the method selected to detect small insertions, deletions or substitutions cannot be excluded. Similar to what we found for patients with NPM1, the frequency of mutations in CEBPA was significantly higher in patients with normal karyotype and in older patients. We also observed a significant association with FAB-M2 subtype.
FLT3 gene mutations were detected in 18.1 % of cases, of which 10.2 % were FLT3-ITD and 7.9 % FLT3-TKD. Patients with FLT3 mutations presented a higher mean white cell count, although this difference was not statistically significant. We also observed an increase of the incidence for FLT3-ITD mutation with age, a fact not observed for FLT3-TKD. Data from series of adult AML patients, show an incidence of internal tandem duplications of 20–30 % [1, 38], whereas in pediatric series the reported incidence is lower [39], which is also consistent with our findings. The only mutation detected in infants was FLT3-TKD, which occurred more frequently in the group of patients with abnormalities in KMT2A gene, although this association was not statistically significant. We observed that FLT3-ITD mutations were significantly associated with FAB-M3 subtype and the presence of fusion transcript PML-RARA.
Concerning the prognostic value of these mutations, there were no statistically significant differences between survival probabilities according to FLT3 status. Our results show that AML patients with mutations in NPM1 have a higher LFS probability than NPM1 wild-type patients. Regarding CEBPA mutations, although most patients showed a favorable outcome, the number of cases is too small to draw definitive conclusions about the prognostic value of this mutation.
However, when we analyzed the results of pLFS for the population of non-APL AML patients and in different groups according to the combined presence of FLT3-ITD and/or NPM1 mutations, we observed a group of patients with a clearly better prognosis, corresponding to patients with absence of FLT3-ITD and presence of NPM1 mutations (pLFS ± SE 83.3 ± 15.2 %, p = 0.5012, see Fig. 2a).
Moreover, survival analysis for high-risk-group patients showed a better prognosis in terms of pOS and pLFS for the combined genotype NPM1 mutated and FLT3-ITD negative (pOS ± SE 83.3 ± 15.2 versus 33.1 ± 4.7 %, p = 0.0251, see Fig. 3a; pLFS ± SE 83.3 ± 15.2 versus 41.5 ± 5.2 %, p = 0.0668, see Fig. 3b).
Considering the high prevalence of mutations in NPM1 in AML with normal karyotype, we analyzed the potential prognostic significance of these alterations within this specific group of high-risk patients. Despite the relatively small number of patients, the mutations were associated with a better prognosis in the group of patients with NPM1 mutation and absence of FLT3-ITD (pLFS ± SE 80.0 ± 17.9 versus 43.8 ± 12.4 %, p = 0.2258, see Fig. 3d).
Taking into account the important prognostic significance of the genotype FLT3-ITD negative with NPM1 mutated in children with AML, we intend to include the presence of these mutations as part of the risk-group definition for all patients at the moment of initial diagnosis in our next protocol for non promyelocytic AML patients. Within the high-risk group, the identification of a subgroup of patients with better prognosis, as has been observed, would allow considering the possibility of reducing the intensification of the chemotherapy to decrease the morbidity and mortality of these patients. It is noteworthy that any decrease in the intensity of treatment should be considered with caution, taking into account that the excellent response of this group of AML patients has indeed been obtained by administering an intensive scheme of treatment.
Finally, this report contributes to the progress in the characterization of NPM1, FLT3 and CEBPA mutations in pediatric AML patients, with the intention of achieving a better understanding of their contribution to the leukemogenic process.
In addition, we are convinced that this study makes a significant contribution in assessing the prognostic value of these mutations, allowing the planning of a different stratification of patients in risk groups to achieve a better tailoring of treatment in pediatric patients with AML.
References
Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18.
Kelly LM, Gilliland DG. Genetics of myeloid leukemia. Annu Rev Genom Hum Genet. 2002;3:179–98.
Suzuki T, Kiyoi H, Ozeki K, Tomita A, Yamaji S, Suzuki R, et al. Clinical characteristics and prognostic implications of NPM1 mutations in acute myeloid leukemia. Blood. 2005;106:2854–61.
Brown P, McIntyre E, Rau R, Meshinchi S, Lacayo N, Dahl G, et al. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood. 2007;110:979–85.
Cazzaniga G, Dell’ Oro MG, Mecucci C, Giarin E, Masetti R, Rossi V, et al. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood. 2005;106:1419–22.
Chou WC, Tang JL, Lin LI, Yao M, Tsay W, Chen CY, et al. Nucleophosmin mutations in de novo acute myeloid leukemia: the age-dependent incidences and the stability during disease evolution. Cancer Res. 2006;66:3310–6.
Thiede C, Creutzig E, Reinhardt D, Ehninger G, Creutzig U. Different types of NPM1 mutations in children and adults: evidence for an effect of patient age on the prevalence of the TCTG-tandem duplication in NPM1-exon 12. Leukemia. 2007;21:366–7.
Braoudaki M, Papathanassiou C, Katsibardi K, Tourkadoni N, Karamolegou K, Tzortzatou-Stathopoulou F. The frequency of NPM1 mutations in childhood acute myeloid leukemia. J Hematol Oncol. 2010;3:41–5.
Balgobind BV, Hollink IHIM, Arentsen-Peters S, Zimmermann M, Harbott J, Beverloo HB, et al. Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica. 2011;96(10):1478–87.
Matsuo H, Kajihara M, Tomizawa D, Watanabe T, Saito AM, Fujimoto J, Horibe K, et al. Prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia: a report from the Japanese Pediatric Leukemia/Lymphoma Study Group. Blood Cancer J. 2014;11(4):e226.
Meshinchi S, Woods WG, Stirewalt DL, Sweetser DA, Buckley JD, Tjoa TK, et al. Prevalence and prognostic significance of FLT3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001;97:89–94.
Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006;108:3654–61.
Kottaridis PD, Gale RE, Linch DC. FLT3 mutations and leukemia. Br J Haematol. 2003;122:523–38.
Greaves M. Childhood leukemia. Br Med J. 2002;324:283–7.
Lin LI, Lin TC, Chou WC, Tang JL, Lin DT, Tien HF. A novel fluorescence-based multiplex PCR assay for rapid simultaneous detection of CEBPA mutations and NPM mutations in patients with acute myeloid leukemias. Leukemia. 2006;20:1899–903.
Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, et al. Nucleophosmin gene mutations are predictors of favourable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106(12):3733–9.
Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CAJ, Wouters BJ, et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 2011;117(8):2469–75.
Martelli MP, Sportoletti P, Tiacci E, Martelli MF, Falini B. Mutational landscape of AML with normal cytogenetics: biological and clinical implications. Blood Rev. 2013;27:13–22.
Schneider F, Hoster E, Unterhalt M, Schneider S, Dufour A, Benthaus T, et al. NPM1 but not FLT3-ITD mutations predict early blast cell clearance and CR rate in patients with normal karyotype AML (NK-AML) or high-risk myelodysplastic syndrome (MDS). Blood. 2009;113:5250–3.
Becker H, Marcucci G, Maharry K, Radmacher MD, Mrózek K, Margeson D, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:596–604.
Falini B, Martelli MP, Bolli N, Sportoletti P, Liso A, Tiacci E, et al. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood. 2011;117(4):1109–20.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Swerdlow SH, Campos E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, et al. Standarized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Leukemia. 1999;13:1901–28.
Alonso CN, Longo PL, Gallego MS, Medina A, Felice MS. A novel AF9 breakpoint in MLL-AF9-positive acute monoblastic leukemia. Pediatr Blood Cancer. 2008;50(4):869–71.
Felice MS, Rossi JG, Alonso CN, Gallego MS, Eberle SE, Alfaro EM, et al. Experience with four consecutive BFM-based protocols for treatment of childhood with non-promyelocytic acute myeloblastic leukemia in Argentina. Leuk Lymphoma. 2016;6:1–10.
Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the FLT3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8.
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3. Blood. 2001;97:2434–9.
Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1995;53:457–81.
Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, et al. Design and analysis of randomized clinical trials requiring prolonged observation on each patient. II. Analysis and examples. Br J Cancer. 1977;35:1–39.
Tarone RE, Wara J. On distribution-free test for equality of survival distribution. Biometrika. 1997;64:156–60.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–66.
Schneider F, Hoster E, Schneider S, Dufour A, Benthaus T, Kakadia PM, et al. Age-dependent frequencies of NPM1 mutations and FLT3-ITD in patients with normal karyotype AML (NK-AML). Ann Hematol. 2012;91:9–18.
Döhner K, Schlenk RF, Habdank M, Scholl C, Frank G, Rücker FG, for the AML Study Group (AMLSG), et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–6.
Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107:4011–20.
Wouters BJ, Louwers I, Valk PJ, Lowenberg B, Delwel R. A recurrent in-frame insertion in a C/EBPA transactivation domain is a polymorphism rather than a mutation that does not affect gene expression profiling-based clustering of AML. Blood. 2007;109:389–90.
Leroy H, Roumier C, Huyghe P, Biggio V, Fenaux P, Preudhomme C. CEBPA point mutations in hematological malignancies. Leukemia. 2005;19:329–34.
Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35.
Mukda E, Pintaraks K, Sawangpanich R, Wiangnon S, Pakakasama S. FLT3 and NPM1 gene mutations in childhood acute myeloblastic leukemia. Asian Pac J Cancer Prev. 2011;12:1827–31.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
About this article

Cite this article
Rubio, P., Campos, B., Digiorge, J.A. et al. NPM1, FLT3 and CEBPA mutations in pediatric patients with AML from Argentina: incidence and prognostic value. Int J Hematol 104, 582–590 (2016). https://doi.org/10.1007/s12185-016-2064-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-2064-5