
Abstract
Recently, we have published an analytical model describing the resolution and signal-to-noise-ratio of peaks present in an ion mobility spectrum acquired by a drift tube instrument. Based on this model, we designed a combination of a compact 10 cm long drift tube and a fast transimpedance amplifier achieving a resolution above 180. Here, we present results using an improved drift tube with slightly longer drift length of 15 cm and higher drift voltages, resulting in a resolution of R = 250 for the positive reactant ion peak and R = 230 for the negative reactant ion peak. By applying a deconvolution algorithm based on the Jansson method, a two- to three-fold separation improvement is possible in theory. However, as a larger resolution improvement than the initial resolution loss by diffusion may lead to erroneous results, we limited the resolution enhancement to 70 %, reaching a resolution of R = 425 for deconvolved spectra.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Drift tube ion mobility spectrometers separate different ion species depending on the time they require to pass a certain distance through a neutral buffer gas under the influence of an electric field [1]. The performance of this separation can be quantified by different metrics, such as the resolution and signal-to-noise-ratio of the measured peaks or the accuracy of the measured drift times. Being a measure for how well closely spaced peaks can still be separated, the resolution is especially important when a large number of similar ions species are present at the same time during measurements. Key examples are the characterization of proteins, which can possess a variety of different folding, and different isomers of one molecule. Both possess the same mass across all ions species and exhibit only minor differences in collision cross section, resulting in extremely similar ion mobilities.
The resolution, although sometimes the term resolving power is used for the same quantity, is defined as the ratio of drift time t D to full width at half maximum (FWHM) w 0,5 as given by Eq. (1):
Typical values achieved by high resolution ion mobility spectrometers vary depending on the nature of ions analyzed. For small single-charged ions, our setup reached a resolution of R = 183 [2]. To our knowledge, no higher resolution for single-charged ions has been reported yet, although a resolution of R = 172 has been shown in [3]. When measuring multiple-charged ions, better resolution values are typically obtained. For example, the drift tube reported in [3] reached a resolution of R = 240 when analyzing a four times negatively charged ion [4]. Other ion mobility approaches, such as trapped ion mobility spectrometry (TIMS) [5] and overtone cyclotron ion mobility spectrometry [6] have also shown good performance when applied to large, multiple-charged ions, reaching resolutions of up to R = 250 and R = 1000 respectively using extended analysis times.
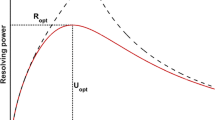
As a guideline for system optimization, it is useful to employ an analytical model which describes the basic relationship between the experimental parameters. This allows identifying the underlying trends and finding a concept for optimization. Here, we use the model presented in [2], which is a refined version of the well-known model for a drift tube ion mobility spectrometer with a certain initial ion packet width and diffusive broadening. The main additions of the refined model are the inclusion of the performance of the transimpedance amplifier used to amplify the ion current and an estimation for the signal-to-noise-ratio of the output signal. It can be shown that an optimum drift voltage U opt exists, at which the maximum resolution R opt is achieved.
The two equations both depend on the same parameters, being the length of the drift tube L, the initial width of the ion packet w Inj , the width added by the amplifier w Amp , the mobility K and charge state z of the ions being analyzed, the absolute temperature T as well as the Boltzmann constant kB and the elementary charge e.
Considering that parameters concerning the measurement, such as the ion mobility, charge state and temperature cannot be varied, the parameters available for optimizing the resolution are the drift length L and the widths created by the injection w In and amplification w Amp . It has been shown that when both widths are chosen the same and the ion mobility spectrometer is operated at its optimum drift voltage, the signal-to-noise-ratio becomes a constant [2]. Thus, both the widths and the drift length can be adjusted in order to increase the resolution without sacrificing signal-to-noise-ratio and we will therefore only consider the optimization of the resolution in this work. However, another important relationship between the optimum drift voltage U opt and optimum resolution R opt must be kept in mind, which is given by Eq. (4). It can be derived by combining Eqs. (2) and (3).
It is obvious that the left factor is constant and therefore the only way to change the relationship between the desired optimum resolution R opt and the required optimum drift voltage U opt is either decreasing the drift cell temperature or increasing the charge state of the ions. This relationship can be intuitively understood from Eq. (5), the Nernst-Einstein-Townsend-equation.
As the ratio T/z controls the ratio between the diffusion coefficient D and the ion mobility K, it controls the ratio between random ion motion and ion motion generated by a certain electric field strength. The lower this ratio, the easier ions can be separated, resulting in a higher resolution at the same drift voltage. This correlates well with the observation that ion mobility spectrometers typically perform significantly better when separating multiple-charged ions as mentioned above.
As shown by Eq. (4), higher resolution for the same ion species will always require significantly higher drift voltage, regardless of the ion shutter performance or the drift tube length. Due to this fundamental limitation, advanced electronics are also a necessity in the design of ultra-high resolution drift tubes.
Improved high resolution drift tube
As the ion mobility spectrometer presented in [2] is already operated using a field switching shutter [7] providing initial ion packet widths of only 5 μs, a further reduction of the widths does not appear to be a feasible optimization path. The drift length however, is just 98 mm, resulting in a compact drift tube setup with an overall length including the amplifier of 185 mm. The new design features a drift length of 154 mm, while the overall length only increased to 223 mm. Therefore, the new drift cell provides a 57 % longer drift length but possesses only 21 % larger overall dimensions. Furthermore, the inner diameter has also been increased to 21 mm to provide better field homogeneity over a larger diameter.
Additionally, the electronics have been refined to provide the increased drift voltage required for increased resolution, see Eq. (4). The drift voltage of up to 35 kV is supplied by a FUG HCP-35-35000. All other voltages are provided by custom-built power supplies. This includes the high voltage power supply for the injection voltage as well as a power supply able to insulate the drift voltage against ground, which is used to power the electronics of the field switching shutter.
For easier comparison, the main operational parameters of the two drift tube setups are summarized in Table 1.
In order to demonstrate the improved resolution, the positive and negative reactant ion spectra are shown in Fig. 1. For the negative reactant ion spectrum, all voltage polarities are reversed, but all further parameters remain the same otherwise.

Positive (left) and negative (right) reactant ion peak of the ultra-high resolution drift tube. The measured peaks are shown by a solid black line, Gaussian fits by a dashed grey line
The positive reactant ion peak as shown on the left of Fig. 1 possesses a resolution of R = 250 and a perfectly Gaussian shape, indicating that diffusion is most likely responsible for most of the observed peak broadening. The negative reactant ion peak as shown on the right of Fig. 1 possesses a resolution of only R = 230 and exhibits slight deviations from a Gaussian shape. The slight visible fronting, which is not present in the positive mode and therefore unlikely to be caused by the instrument, may indicate the presence of different negative reactant ions, clustering reactions or chemical reactions with contaminants occurring inside the drift tube.
Figure 2 shows the spectrum of 10 ppbv DMMP (Dimethyl methylphosphonate, Sigma-Aldrich) which has been averaged for one second. It is clearly shown that the ultra-high resolution is reproducible for substance peaks. Furthermore, exhibiting an excellent signal-to-noise-ratio at a concentration of only 10 ppbv, the ultra-high resolution drift tube is not achieving this resolution improvement at the cost of detection capabilities. In addition to the reactant ion peak at 4.465 ms (K0 = 1.94 cm2 V−1 s−1), the monomer peak at 5.092 ms (K0 = 1.70 cm2 V−1 s−1) and the dimer peak at 6.423 ms (K0 = 1.35 cm2 V−1 s−1) further peaks with extremely low intensities are visible at 4.205, 4.717, 5.449 and 5.710 ms (K0 = 2.06 cm2 V−1 s−1, K0 = 1.84 cm2 V−1 s−1, K0 = 1.59 cm2 V−1 s−1 and K0 = 1.52 cm2 V−1 s−1). These are most likely caused by trace impurities in the gas mixing system and only visible due to the low noise.

Spectrum of 10 ppbv DMMP (averaging time of 1 s). The additional peaks are impurities from the gas mixing system visible due to the low noise
Deconvolution
Fundamental difficulties in improving drift tube ion mobility spectrometers are the scaling rules, which have been outlined by the Eqs. (3) and (4). Every parameter that has to be changed in order to improve the resolution scales superlinearily with the desired resolution improvement. For example, in order to double the resolution, Eq. (3) demands that the drift voltage is increased by a factor of 4 and Eq. (4) demands that either the drift length is increased by \( 2\sqrt{2} \) or the initial width and amplifier width are both reduced by a factor of 8. Thus, improving the resolution of an instrument will become more and more challenging the higher the desired resolution values are. While improved instrumentation and newly available manufacturing technologies and materials will still be able to push the resolution of drift tube ion mobility spectrometers further, it is sensible to also consider additional possibilities for resolution enhancement.
The broadening of peaks through various effects is a phenomenon common to virtually every kind of spectrometer. In the case of ion mobility spectrometers, the main contributor to peak width is usually diffusion. Even at the drift voltage at which the maximum resolution is reached according to Eq. (2) and the initial width and amplifier width become the determining factor, the diffusion still broadens the peak by additional 70 % [2]. As peak broadening by a known spread function can theoretically be reversed by deconvolution, deconvolution algorithms have become an important tool in spectrometry. However, the simple linear analytical deconvolution typically performs abysmally when applied to real-life data, as it contains noise, distorted peaks and various other non-idealities. Therefore, special algorithms have been developed to perform this task more efficiently. One of them is the Jansson method, which was first used for the deconvolution of infrared spectra [8], but has been applied successfully to measurement data from gas chromatographs [9] and also ion mobility spectrometers [10]. It is an iterative, non-linear and constrained method with the basic algorithm given by Eq. (6):
Here, the current estimate ô (k) for the unbroadened spectrum is convolved with the known spread function s, which would be a Gaussian peak for diffusion. Then the difference between this estimate for the observed spectrum and real observed spectrum i is calculated. This correction is then multiplied with a relaxation function r(ô (k)) and added to the current estimate ô (k) in order to obtain the new estimate ô (k + 1). The relaxation function depends on ô (k) and can therefore be used to shape the result by, for example, enforcing boundaries on the solution.
Usually, a modified version of the original algorithm is employed, as Jansson himself already proposed several modifications to improve the convergence speed and quality of the deconvolved spectra [11]. In this work, we employed the following modifications:
-
The relaxation function r(ô (k)) = r 0 ô (k) is used, enforcing positive results, but placing no limit on their maximum value.
-
Point-sequential calculation is employed in order to maximize the convergence speed.
-
The calculation order of the point sequential algorithm is reversed with every iteration in order to avoid skewing the solution in one direction.
-
A time-varying spread function is used in order to account for the fact that the peak broadening across an ion mobility spectrum varies, even at constant resolution. To achieve consistent results across the whole spectrum, the integral of the spread function is kept constant.
-
A zero-phase low pass filter is used to remove noise created by the deconvolution between iterations. Using a zero-phase filter avoids shifting of peak positions.
-
Before the first iteration, the spectrum is filtered using a more aggressive zero-phase low-pass filter to minimize the noise originally introduced into the algorithm. The spread function is also filtered using the same filter in order to remove its broadening effect on the spectrum.
When testing a deconvolution algorithm, it is important to keep in mind that usually every kind of algorithm, even direct analytical inverse filtering of the convolution, will perform well when applied to near ideal data. Furthermore, single peaks can be usually slimmed down to a near-delta peak without problem. The challenge for a deconvolution algorithm is separating overlapping peaks in a noisy measurement. Only under these conditions, the real performance can be evaluated. Therefore, we used the following test procedure on known spectra before applying the algorithm to measurement data. As shown in Fig. 1, the peaks measured by our IMS are nearly perfectly Gaussian. Thus, we numerically generated overlapping Gaussian peaks and added the noise present in our IMS signals in order to create a highly realistic test signal. In order to test whether a certain resolution R target was attainable using the algorithm, the peaks were placed at relative drift times to each other that result in a valley of 12,5 % between each peak at the resolution R target . These spectra were generated at both the resolution R target and a resolution of R = 250, which corresponds to the resolution achieved by our ultra-high resolution drift tube. The spectrum with a resolution of R = 250 was then deconvolved trying to achieve R target and the result compared to the real spectrum with R target .
Two different tests were performed: First, the “peak-in-the-middle” problem shown in Fig. 3, contains three peaks, where the task is to detect the middle peak. The height of these peaks was chosen to be one fifth of the height of the positive reactant ion peak, as they obviously will be smaller due to the near constant charge expected in a spectrum. Second, the “shoulder” problem shown in Fig. 4 contains only to peaks, but one is five times smaller than the other, rendering it more difficult to detect. Again, the larger peak has a height that is one fifth of the height of the reactant ion peak, hence the smaller peak is only one twenty-fifth as high. It should be noted that due to the constant area under a peak, the deconvolved peaks are significantly higher and therefore, all figures show normalized peaks heights for easier comparison of the peak shapes.

Deconvolution of “peak-in-the-middle” test spectra. The dashed grey line represents the original resolution 250 spectrum, the empty black circles the true high resolution spectrum and the solid black line the high resolution spectrum predicted by deconvolution. The higher resolutions are from left to right 400, 600 and 800

Deconvolution of “shoulder” test spectra. The dashed grey line represents the original resolution 250 spectrum, the empty black circles the true high resolution spectrum and the solid black line the high resolution spectrum predicted by deconvolution. The higher resolutions are from left to right 400, 600 and 800
It is obvious that even for a target resolution of R = 800, the higher resolution spectrum can be predicted reasonably well from the spectrum with a resolution of R = 250 despite the present noise. This is a resolution enhancement by more than a factor of three without any instrumental modifications and would therefore be a tremendous improvement. However, the possibility of such post-processing leads to one fundamental question: How much resolution improvement is sensible using deconvolution? We propose about 70 % when the ion mobility spectrometer is operated at its optimum drift voltage, since this is the amount of peak broadening compared to the initial ion packet width and amplifier width expected to stem from diffusion. Thus, further resolution enhancement by deconvolution of Gaussian peak broadening would mean removing diffusion that has never taken place, which may or may not result in correct spectra. For example, other broadening mechanisms showing a similar shape would still be removed correctly, but also non-ideal peak shapes would be misinterpreted as the presence of multiple peaks.
This problem is illustrated in Fig. 5. Both sides of the figure show a non-ideal ion distribution, which may result from chemical reactions with contaminants inside the drift tube or an ion distribution across a mobility range instead of having a single value. This ion distribution has then been broadened by the amount of diffusion expected in our drift tube at a resolution of R = 250 and deconvolved using the presented algorithm. The left figure shows the result for a target resolution of R = 425, which is a 70 % improvement over R = 250, while the right figure shows the result for a target resolution of R = 800, which would seem achievable based on Figs. 3 and 4. It is obvious that the result on the right is completely off, as the actual ion distribution is known in this simulation. Thus, it is extremely important to employ deconvolution with care, always estimating how much removable broadening is actually present in the spectrum to avoid erroneous results.

Deconvolution of a non-ideal ion distribution broadened by diffusion. The open black circles represent the ion distribution before diffusive broadening, the dashed grey line after diffusion. The solid black lines represent the deconvolution results for a correctly chosen target resolution (left) and an exaggerated target resolution (right), which erroneously results in multiple peaks
Having ascertained that a resolution improvement to R = 425 is feasible and sensible, we deconvolved the reactant ion spectra shown in Fig. 1. The results from this calculation are shown in Fig. 6, with the positive reactant ion peak (left) and the negative reactant ion peak (right). While the positive reactant ion peak only becomes slimmer as expected for a single peak, the negative reactant ion peak divides into a symmetrical peak and a plateau. However, even at a resolution of R = 425, completely separated peaks causing the fronting cannot be revealed yet.

Deconvolution of the positive reactant ion peak (left) and negative reactant ion peak (right) from Fig. 1. The dashed grey line depicts the original peak shapes at a resolution of 250, while the solid black lines show the peaks deconvolved to a resolution of 425
Conclusion
By combining an improved drift tube with a slightly longer drift length of 15 cm, improved electronics allowing for higher drift voltage and a deconvolution algorithm based on the Jansson method, a significant improvement in ion mobility spectrometer resolution has been reached. The current resolution achieved with our drift tube is R = 250 measured for the positive reactant ion peak. In addition to this, the deconvolution algorithm can be used to enhance the resolution to about R = 425. Such an increased resolution may allow differentiating even more complex samples in future experiments.
References
Eiceman GA, Hill HH Jr, Karpas Z (2013) Ion mobility spectrometry, 3rd edn. CRC Press, Boca Raton
Kirk AT, Allers M, Cochems P, Langejuergen J, Zimmermann S (2013) Analyst 138:5200–5207
Dugourd P, Hudgins RR, Clemmer DE, Jarrold MF (1997) Rev Sci Instrum 68:1122–1129
Srebalus CA, Li J, Marshall WA, Clemmer DE (1999) Anal Chem 71:3918–3927
Silveira JA, Ridgeway ME, Park MA (2014) Anal Chem 86:5624–5627
Glaskin RA, Ewing MA, Clemmer DE (2013) Anal Chem 85:7003–7008
Kirk AT, Zimmermann S (2014) Int J Ion Mobil Spectrom 17:131–137
Jansson PA, Hunt RH, Plyler EK (1970) J Opt Soc Am 60:596–599
Crilly PB (1987) J Chemometrics 1:79–90
Bell SE, Wang YF, Walsh MK, Du Q, Ewing RG, Eiceman GA (1995) Anal Chim Acta 303:163–174
PA Jansson (1996) Deconvolution of images and spectra, 2nd edn. Academic Press, San Diego
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kirk, A.T., Zimmermann, S. Pushing a compact 15 cm long ultra-high resolution drift tube ion mobility spectrometer with R = 250 to R = 425 using peak deconvolution. Int. J. Ion Mobil. Spec. 18, 17–22 (2015). https://doi.org/10.1007/s12127-015-0166-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12127-015-0166-z