
Abstract
Objective
To investigate if expanded newborn screening using tandem mass spectroscopy (TMS) is adequate to detect low excretor phenotype in Indian Glutaric aciduria type I (GA-I) patients.
Methods
Ten GA-I patients were investigated for blood glutaryl carnitine (C5DC) levels on dried blood spot (DBS) by tandem mass spectroscopy and urine glutaric acid (GA) and 3-hydroxyglutaric acid (3-OH-GA) by gas chromatography-mass spectroscopy. The student’s T test and Pearson’s correlation were applied to draw a relationship between various biochemical parameters. Further confirmation of low excretors by DNA mutation analysis in the glutaryl CoA dehydrogenase (GCDH) gene was performed by polymerase chain reaction and Sangers sequencing.
Results
Among 10 GA-I patients, 7 patients were found to have high excretor, and 3 were found to have low excretor phenotype. The low excretors were found to have GCDH gene mutations. The mean C5DC levels in high and low excretors were 2.61 ± 2.02 μmol/L and 2.31 ± 1.00 μmol/L, respectively. In high excretors, C5DC levels correlated with GA (r = 0.95). In low excretors, C5DC levels correlated with 3-OH-GA (r = 0.99). No significant difference was found between C5DC levels of high and low excretors (p = 0.82).
Conclusions
The MS/MS, C5DC screening is a sensitive technique and detected 10 GA-I patients. Irrespective of the urine organic acid levels, Indian GA-I patients including low excretors seem to have a significantly elevated C5DC level and well above the stipulated cut-off values and therefore, expanded newborn screening is probably adequate to diagnose them.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glutaric aciduria type I (GA-I) (OMIM # 231670) is one of the major inherited causes of acute metabolic brain damage in early childhood [1]. This neurodegenerative inborn error of metabolism has been reported worldwide with a rate of 1 in 30, 000 to 100,000 newborns [2, 3]. In India, GA-I is one of the significantly reported organic acidurias with an incidence of 14 in 17,100 [4, 5]. Clinically, GA-I is presented with acute encephalopathy crises in infancy or early childhood between 6 and 18 mo with a high risk of developing an irreversible dystonia or movement disorder and many patients develop macrocephaly [6]. Other clinical outcomes associated with this disease are developmental delays, seizures, dystonia, hypotonia, ataxia, etc. In some patients, this disorder may also occur insidiously without any clinically deceptive crises [7, 8].
Glutaric aciduria type I is caused due to mutations in the glutarylCoA dehydrogenase (GCDH) gene, causing translational defects resulting in a malfunctioned GCDH enzyme (E.C 1.3.99.7). The GCDH enzyme is a mitochondrial matrix protein, which catalyses the oxidative decarboxylation of glutaryl-CoA, an intermediate in the degradation of the amino acids lysine and tryptophan. Defective GCDH enzyme blocks the catabolism of lysine and tryptophan leading to the increase in intermediary by-products i.e., glutaric acid (GA), 3-hydroxyglutaric acid (3-OH-GA), glutaconic acid (GC) and glutaryl carnitine (C5DC) in body fluids [blood, cerebrospinal fluid (CSF), urine etc.] and tissues [9,10,11]. The C5DC is the diagnostically relevant metabolite in dried blood spot (DBS) to detect GA-I [10]. These metabolites accumulate in body fluids (plasma, urine, and CSF) and tissues and can be detected by electrospray ionization tandem mass spectrometry (TMS; MS/MS) or gas chromatography/mass spectrometry (GC/MS). Based on the urinary GA concentration, GA-I is further divided into two patient biochemical subgroups, low excretors (GA in urine <100 mmol/mol Cr) and high excretors (GA in urine >100 mmol/mol Cr) [7, 11]. Both the subgroups are thought to have the same risk of striatal necrosis and the same severity of outcome when a crisis occurs [3, 10, 11].
Glutaric aciduria type I is included in the panel of diseases that are recognized by expanded newborn screening in some countries [5, 12, 13]. The newborn screening for GA-I is performed using TMS by detecting the levels of C5DC in dried blood spots of affected individuals [11, 14]. This has provided promising results in some population-wide newborn screening programs. However, a major diagnostic pitfall is that some GA-I patients, especially patients with no intermittent increase in C5DC level or with mild biochemical phenotype (i.e., low excretor) can be missed [11, 15, 16]. To circumvent this problem, some laboratories are now using additional tests to follow-up investigations of initial positive screening results such as analysis of urinary GA and 3-OH-GA by GC/MS. However, some patients may again show no biochemical abnormalities in the urine [7, 17]. The missing of positive cases may also be either due to mistakes in technical (cut-off) or medical (false interpretation) validation [16, 18]. In certain genetically isolated populations like Oji-Cree, an alternative strategy of DNA-based neonatal screening has been followed with no reported false-positive or false-negative screening results for GCDH deficiency. In this population, MS/MS for mass neonatal screening or DNA-based neonatal screening are considered reliable approaches to detect GCDH-deficient patients before the onset of neurological deterioration [17].
In India, such newborn screening programmes are not routinely done in mainstream health care and still are confined to a few centers and projects [19]. The second-tier tests, especially GCMS, is laborious and adds to the costs of screening, hence economically not feasible by many low-income families. There are very few studies on the biochemistry of GA-I patients in India with no reported studies on the expediency of newborn screening to detect Indian low excretor GA-I patients. There is a lack of understanding of the biochemical and molecular profiles of the low excretor phenotypes of GA-I and/or if additional tests such as GC-MS and DNA based screening are essential to identify Indian GA-I patients. Glutaryl carnitine was found elevated in present cohort with a high urinary excretion of GA as well as with intermittently normal urinary organic acids or isolated elevation of urinary 3-OH-GA. This prompted the authors to perform this preliminary study, where they correlated the blood C5DC levels and urine GA and 3-OH-GA levels of GA-I patients to establish if expanded newborn screening to detect C5DC levels is efficient and adequate in detecting the Indian GA-I patients irrespective of the high and low excretor biochemical phenotype.
Material and Methods
This study was approved by the institutional ethics committee, and informed consent forms were obtained from all study participants. The clinical history, biochemical, and demographic data was collected from the patient records. Dried blood spots and 5 ml of midstream urine samples of screened GA-I patients from August 2016 through August 2018 were received from K.L.E.S. Dr. Prabhakar Kore Hospital & Medical Research Centre, Belgaum, and KIMS Hubli. The values of blood C5DC calculated at the onset of disease were obtained retrospectively and if not available were estimated by TMS using DBS (Neogen labs Pvt. Ltd., Bangalore, India). The diagnosis was further confirmed by GCMS analysis using the collected urine samples to analyse the GA and 3-OH-GA levels as follows. The urine sample of the patient was collected, and derivatization was done by silylation of organic compounds by BSTFA+TMCS [N, O-bis(trimethylsilyl) trifluoroacetamide with trimethylchlorosilane]. The residue was derivatized at 80 °C and heated for 30 min. The GCMS analysis was performed using gas chromatography instrument Shimadzu GC-2010 Plus and a gas chromatography capillary column Zebron ZB-5 (30 m × 0.32 mm × 0.25 μm, Phenomenex, India). The column temperature was initially maintained at 100 °C for 2 min and then increased from 100 °C to 280 °C by 4 °C/min, where it was held for 3 min. Run time was 56 min. The results were calculated in μmol/mmol creatinine using a calibration curve of the organic acid of interest, which was processed under the same conditions.
Ten patients were selected based on the following inclusion criteria i.e., i) clinical presentations such as macrocephaly, acute encephalitis-like crisis, spasticity, dystonia, choreoathetosis, ataxia, dyskinesia and seizures, ii) neuroimaging studies [Computerized tomography (CT)/Magnetic resonance imaging (MRI)] showing generalized cerebral atrophy, subdural hematomas, changes in the basal ganglia or caudate and putamen and iii) elevated C5DC in plasma and/or elevated GA or 3-OH-GA in urine. The cases which could not be followed up to obtain a confirmation of the diagnosis and children older than 14 y were excluded from the study.
The recruited GA-I patients were categorised as two biochemical phenotypes, high excretors, with urine GA levels >100 mmol/mol creatinine and low excretors of GA with urine GA levels <100 mmol/mol creatinine. Further, the whole gene sequencing by polymerase chain reaction using exon specific primers and direct DNA Sangers sequencing was performed (Chromous Biotech Pvt. Ltd., Bangalore, India) for all the low excretor GA-I patients.
The statistical analysis was done by SPSS software version 17. The data were expressed as median, mean, and standard deviation. The correlation coefficient was determined by Pearson’s correlation analysis and for significance analysis, “students T test” was performed. The level of significance was fixed at p < 0.05 for evaluation.
Results
Ten GA-I patients from ten unrelated families were recruited for the study. The median age at diagnosis (Interquartile range) of these patients was 0.7 y (0.6 to 2.3 y). The study group comprised of 3 males and 7 females. Parental consanguinity was seen in 70% of the families. Macrocephaly was the major clinical sign present in 70% GA-I patients. Other clinical symptoms presented were developmental delay (40%), regression in motor activities (30%), dystonia (20%), hypotonia (10%), and seizures (10%). The neuroimaging data revealed cerebral atrophy (5%), widened Sylvian fissure (20%), frontotemporal atrophy (20%), and basal ganglia hypersensitivity (20%) patients. None of the patients were under treatment at the time of diagnosis. Details including the age of onset, clinical and neurological signs, and biochemical values of all the patients are presented in Table 1.
All GA-I patients showed elevated C5DC levels with an average of 2.5 ± 1.64 μmol/L (ref range: 0.00–0.56 μmol/L), ranging from 0.98 to 6.5 μmol/L with a minimum increase of 1.75% to a maximum of 12%. The graph showing the cut-off of C5DC range-based distribution of GA-I cases is shown in Fig. 1. There was a weak, however, negative correlation (r = −0.2) between the age of the GA-I patients and their C5DC levels (Fig. 2).

The graph showing the cut-off based distribution of GA-I positive cases. Comparison of the C5DC values in micromols/L (μmol/L) in the presumptive positive patients. C5DC Glutaryl carnitine

Correlation of the blood C5DC levels and the age of the GA-I patients. C5DC Glutaryl carnitine
In the ten GA-I patients, the results of Urine organic acid analysis (UOA) by GC-MS showed average GA elevation of 652 ± 888 mmol/mol creatinine (range 0–2805 mmol/mol creatinine) (Ref range: 0.05–13 mmol/mol creatinine), and average 3-OH-GA elevation of 58 ± 60 mmol/mol creatinine (range 4–221 mmol/mol creatinine) (Ref range: 0–4.7 mmol/mol creatinine). Interestingly, authors found a positive and significant correlation (r = 0.85) of C5DC with elevations of GA. However, a weak correlation (r = 0.22) was found between the C5DC and 3-OH-GA levels.
Among the ten patients, 7 patients showed high GA levels (>100 mmol/mol creatinine) ranging from 200 to 2805 mmol/mol creatinine. Hence, these seven patients were categorized as the high excretor patient group. In these patients, the increase in 3-OH-GA was comparatively lesser, however, ranged from 20.03 to 63 mmol/mol creatinine. The mean C5DC levels in these high excretor patients was found to be 2.61 ± 2.02 μmol/L ranging from 0.98 to 6.5 μmol/L. In these patients, a positive and strong correlation (r = 0.95) of C5DC with GA level was found (Fig. 3a). However, a very weak correlation (r = 0.17) was found between the C5DC and 3-OH-GA level (Fig. 3b).

(a) The urine GA elevation and (b) the Urine 3-OH-GA elevation in the high excretor patients respectively and correlation of the C5DC levels with (c) the Urine GA elevation and (d) the Urine 3-OH-GA elevation in the low excretor phenotype patients respectively. 3-OH-GA 3-hydroxyglutaric acid; C5DC Glutaryl carnitine; GA Glutaric acid; GC-MS Gas chromatography-mass spectrometry; TMS Tandem mass spectroscopy
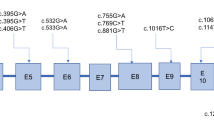
On the other hand, rest of the three patients were categorized as low excretors as these patients showed nil to mildly elevated GA (<100 mmol/mol creatinine) and elevated 3-OH-GA levels. Genetic analysis of GCDH gene in these patients revealed the presence of R94Q (g.5966 G > A); P286S (g.10754 C > T), I152M (g. 7445C > G) and L221P (g.10072 T > C) mutations. The P286S (g.10754 C > T) and I152M (g.7445C > G) mutations were present in combined heterozygous form in a single patient. While the other two were in the homozygous state in one patient each. Among these three low excretors, one patient showed only very slight elevation of 3-OH-GA level with no elevation in GA levels. The other two patients showed a mild elevation in both GA and 3-OH-GA, however, the levels of 3-OH-GA were much higher than the GA. The mean C5DC levels in these low excretor patients was found to be 2.31 ± 1.00 μmol/L ranging from 1.5 to 3.43 μmol/L. In these patients, a positive and very strong correlation (r = 0.99) of the blood C5DC with elevations of 3-OH-GA in urine was found (Fig. 3d). Conversely, a weak correlation was found between the C5DC levels and GA (Fig. 3c). Further, there was no statistically significant difference (P = 0.8231) between the C5DC levels of high and low excretor GA-I patients.
Discussion
The newborn screening aims for timely detection and intervention of treatable inborn errors of metabolism (IEM) to prevent disability and death [20]. The routine newborn screening is done using DBS of patients by TMS [21]. It has emerged as a sensitive technique to detect IEMs utilizing microliter quantities of whole blood from newborns [22]. Among many organic acidurias, GA-I can be detected using TMS by screening elevated levels of C5DC, the primary analyte in the affected individuals [10, 11]. Expanded newborn screening has been deliberated beneficial, disease-changing intervention for GA-I [8]. The early diagnosis of GA-I is essential for timely treatment before irreversible neurological damage occurs, which would result in a better outcome than intervention after the onset of neurological disease [5]. The second-tier tests are generally recommended to confirm the diagnosis of GA-I, which includes urine analysis for elevated organic acids such as GA, 3-OH-GA, and GC by GCMS [5, 11, 23]. In India, IEMs are diagnosed in high-risk babies, thus, making diagnosis in symptomatic phase when maximum insult and damage to vital organs such as brain has already been done.
In this study, 10 GA-I patients based on elevated C5DC levels were detected, which were confirmed by GC-MS urine analysis results and further among them 7 high and 3 low excretors of GA were identified. Thus, this study, suggests that expanded newborn screening using TMS is a sensitive technique to screen GA-I patients based on only elevated C5DC levels irrespective of the biochemical phenotypes. In other studies, from India as well, low excretors have been reported and were found to have a detectable blood C5DC levels even when GA levels were low or not evident. [24, 25]. Further, DNA-based mutation analysis has been recommended and is considered more reliable for high-risk screening and detection of low excretors [11, 12]. Therefore, genetic analysis of GCDH gene was conducted in low excretor patients, which revealed the presence of R94Q, P286S, I152M, and L221P mutations. These mutations were previously reported and were associated with low excretor phenotype [24, 25].
A weak correlation of C5DC with age of patients not under treatment was observed, which could probably hint that the C5DC levels in blood decrease with age. This might be due to the rapid urinary excretion of C5DC with an increase in age, and hence GA-I cases can be missed by TMS, if not identified at an early stage. This was also suggested in a previous study where the authors reported a deficiency of carnitine and acyl carnitines in the blood of GA-I patients beyond the neonatal period [22]. In the present study, the levels of C5DC in high excretors positively correlated with GA levels and were found to be high if GA levels were elevated. This might be due to an increased accumulation of toxic GA, resulting in increased formation of C5DC in blood and urine, which further might lead to secondary carnitine depletion if untreated [22, 26].
On the contrary, even in the low excretor GA-I patients, C5DC was elevated despite the low to undetectable GA in urine. A study by Tortorelli et al. in 2005 suggested that C5DC may remain elevated in the urine of low excretors as well as in GA-I patients with secondary carnitine depletion [27]. However, all these low excretor patients had detectable levels of urinary 3-OH-GA. Furthermore, in these low excretor patients, the C5DC levels and 3-OH-GA, correlated. On the contrary, in this study no correlation was found between C5DC and GA levels in low excretors. A probable relation between the 3-OH-GA and C5DC levels has been suggested earlier in a study by Keyser et al., in 2008 [28]. The 3-hydroxyglutaric acid is the diagnostic metabolite and has been found in low excretors even if GA levels are low [9]. An alternative method for newborn screening using TMS to detect 3-OH-GA for high- or low-excretor variants has been reported [29]. Further studies are needed to explain these correlations. Hence, a newborn screening method using TMS, where it is possible to detect both C5DC and 3-OH–GA together, would prove useful and there will be no need to take up second tier GCMS analysis in Indian GA-I patients. This might further reduce the chances of missing low-excretors at the initial screening stages. Irrespective of whether the GA-I patient is a high or low excretor, the levels of GA and the C5DC were found to be elevated/above the cut-off, in Indian GA-I patients. A previous Indian study has also shown high sensitivity and specificity of the newborn screening tests establishing it as reliable and sensitive with a sensitivity and specificity of 93.33% and 99.42%, respectively, where the TMS was able to pick up as many as 14 positive cases in a 3 y period, and all the patients had an elevated C5DC level [20]. There was no significant correlation between biochemical and the clinical course of the disorder [10, 11].
Hence, it can be concluded that newborn screening using MS/MS may be sufficient, allowing the opportunity to identify Indian newborns with GA-I prior to catastrophic insult. However, before confirmation, the results should be correlated with the clinical and neuroimaging data. Further confirmation can be done by genetic testing or enzyme analysis depending upon the availability. The GA-I patients in India have a high elevated C5DC range and can thus be identified easily using newborn screening method such as TMS. The high and detectable C5DC levels are probably due to the fact that the mutations found in the Indian gene pool are probably pathogenic, resulting in reduced activity of GCDH enzyme to an extent that results in increased blood C5DC levels in GA-I patients [24, 25]. Detection of GA-I in countries like India, where the C5DC is probably, easily detected using blood/DBS by TMS in newborn screening can prove economical and additional confirmation of disease by second tier tests such as GCMS may not be required. The limitations of this study are the small cohort size and a low number of low excretor phenotype. Although it should be noted that difficulty exists in collecting the data, as GA-I is a rare disease with a low incidence and low excretor phenotypes are arbitrarily only 5% of all GA-I patients [15]. Poor follow-up and lack of awareness in India, further adds up to the poor diagnosis and recruitment of positive GA-I cases. The findings of this pilot study will serve as a requisite initial step, which can be taken up and further confirmed, conceivably by multicentric studies. In addition, the newborn tests must be improved, aiming towards increased sensitivity with the diagnosis being made in pre symptomatic phase resulting in a favourable outcome in GA-I cases as against late diagnosis.
References
Barić I, Baraka K, Maradin M, et al. Glutaric aciduria type 1: an example of the importance of early detection of so-called cerebral organic aciduria. [Article in Croatian]. Lijec Vjesn. 2003;125:312–6.
Strauss KA, Puffenberger EG, Robinson DL, Morton DH. Type I glutaric aciduria, part 1: natural history of 77 patients. Am J Med Genet. 2003;121C:38–52.
Kölker S, Garbade SF, Greenberg CR, et al. Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr Res. 2006;59:840–7.
Vaidyanathan K, Narayanan MP, Vasudevan DM. Organic acidurias: an updated review. Indian J Clin Biochem. 2011;26:319–25.
Babu RP, Bishnupriya G, Thushara PK, et al. Detection of glutaric acidemia type 1 in infants through tandem mass spectrometry. Mol Genet Metabol Rep. 2015;3:75–9.
Fraidakis MJ, Liadinioti C, Stefanis L, et al. Rare late-onset presentation of glutaric aciduria type I in a 16-year-old woman with a novel GCDH mutation. JIMD Rep. 2015;18:85–92.
Busquets C, Merinero B, Christensen E, et al. Glutaryl-CoA dehydrogenase deficiency in Spain: evidence of two groups of patients, genetically, and biochemically distinct. Pediatr Res. 2000;48:315–22.
Boy N, Mengler K, Thimm E, et al. Newborn screening: a disease-changing intervention for glutaric aciduria type 1. Ann Neurol. 2018;83:970–9.
Hedlund GL, Longo N, Pasquali M. Glutaric acidemia type 1. Am J Med Genet P C Semin Med Genet. 2006;142C:86–94.
Kolker S, Christensen E, Leonard JV, et al. Diagnosis and management of glutaric aciduria type I-revised recommendations. J Inherit Metab Dis. 2011;34:677–94.
Boy N, Mühlhausen C, Maier EM, et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision. J Inherit Metab Dis. 2017;40:75–101.
Heringer J, Boy N, Burgard P, Okun JG, Kolker S. Newborn screening for glutaric aciduria type I: benefits and limitations. Int J Neonatal Screen. 2015;1:57–68.
Horster F, Kolker S, Loeber JG, Cornel MC, Hoffmann GF, Burgard GF. Newborn screening programmes in europe, arguments and efforts regarding harmonisation: focus on organic acidurias. JIMD Rep. 2017;32:105–15.
Chace DH, Kalas TA, Naylor EW. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin Chem. 2003;49:1797–817.
Heringer J, Boy SP, Ensenauer R, et al. Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol. 2010;68:743–52.
Wilcken B, Wiley V, Hammond J. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348:2304–12.
Lindner M, Kölker S, Schulze A, Christensen E, Greenberg CR, Hoffmann GF. Neonatal screening for glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:851–9.
Smith WE, Millington DS, Koeberl DK, Lesser PS. Glutaric acidemia, type I, missed by newborn screening in an infant with dystonia following promethazine administration. Pediatrics. 2001;107:1184–7.
Kaur G, Thakur K, Kataria S, et al. Current and future perspective of newborn screening: an Indian scenario. J Pediatr Endocrinol Metab. 2016;29:5–13.
Devi ARR, Naushad SM. Newborn screening in India. Indian J Pediatr. 2004;71:157–60.
Banta-Wright SA, Steiner RD. Tandem mass spectrometry in newborn screening: a primer for neonatal and perinatal nurses. J Perinat Neonatal Nurs. 2004;18:41–58.
Rakheja D, Jones VK, Burlina AB, Bennett MJ. Diagnosis of glutaric acidemia type I: a cautionary note. Lab Med. 2005;36:174–7.
Couce ML, López-Suárez O, Bóveda MD, et al. Glutaric aciduria type I: outcome of patients with early- versus late-diagnosis. Eur J Paediatr Neurol. 2013;17:383–9.
Radha Rama Devi A, Ramesh VA, Nagarajaram HA, Satish SP, Jayanthi U, Lingappa L. Spectrum of mutations in Glutaryl-CoA dehydrogenase gene in glutaric aciduria type I--study from South India. Brain Dev. 2016;38:54–60.
Gupta N, Singh PK, Kumar M, et al. Glutaric acidemia type 1-clinico-molecular profile and novel mutations in GCDH gene in Indian patients. JIMD Rep. 2015;21:45–55.
Flanagan JL, Simmons PA, Vehige J, Willcox MD, Garrett Q. Role of carnitine in disease. Nutr Metab. 2010;7:30.
Tortorelli S, Hahn SH, Cowan TM, Brewster TG, Rinaldo P, Matern D. The urinary excretion of glutarylcarnitine is an informative tool in the biochemical diagnosis of glutaric aciduria type I. Mol Genet Metab. 2005;84:137–43.
Keyser B, Glatzel M, Stellmer F, et al. Transport and distribution of 3-hydroxyglutaric acid before and during induced encephalopathic crises in a mouse model of glutaric aciduria type 1. Biochim Biophys Acta Mol Basis Dis. 2008;1782:385–90.
Al-Dirbashi OY, Kölker S, Ng D, et al. Diagnosis of glutaric aciduria type 1 by measuring 3-hydroxyglutaric acid in dried urine spots by liquid chromatography tandem mass spectrometry. J Inherit Metab Dis. 2011;34:173–80.
Author information
Authors and Affiliations
Contributions
All the authors have revised the manuscript and contributed to the drafting of the article. They confirm that the manuscript is an original version and has not been published in any other scientific journal or elsewhere; MS has performed the biochemical analysis and genetic analysis and drafted the manuscript. MK has been the referral physician for neonatal resuscitation and metabolic and nutritional management of the patient and has validated the results. KVTP and VAB have helped in cross checking the genetic reports and framing the script. All authors read and approved the final manuscript. Prof. Shyam Kumar Vootla, Professor and Chairman, Department of Biotechnology and Microbiology, Karnatak University, Dharwad, is the guarantor for this article.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Source of Funding
Maulana Azad National Fellowship F1–17.1/2017–18/MANF-2017-18-KAR-75132.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shaik, M., T. P., KV., Kamate, M. et al. Is Expanded Newborn Screening Adequate to Detect Indian Biochemical Low Excretor Phenotype Patients of Glutaric Aciduria Type I?. Indian J Pediatr 86, 995–1001 (2019). https://doi.org/10.1007/s12098-019-03017-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-019-03017-z