
Abstract
Organic acidurias are an important class of inherited metabolic disorders arising due to defect in intermediary metabolic pathways of carbohydrate, amino acids and fatty acid oxidation. This review summarizes the current knowledge about the important organic acidurias in the Indian population. Specifically, diagnosis and principles of treatment of organic acidurias are covered. The salient features of common organic acidurias as well as their prevalence in various parts of the world are reviewed in some detail.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Organic acidurias (Organic acid disorders, OADs) are an important class of inherited metabolic disorders (IMD) arising due to defect in intermediary metabolic pathways of carbohydrate, amino acids and fatty acid oxidation. It leads to accumulation of organic acids in tissues and their subsequent excretion in urine [1]. It is now known that along with amino acidurias, organic acidurias form the most important class of IMD in high-risk population and among severely-ill children [2–5]. Although individually rare, the conjunct frequency of OAD in preselected high-risk group may be up to 200 times higher than that identified in the general population [6]. Several studies have pointed out the main clinical and laboratory characteristics that lead to the suspicion of inborn error of metabolism especially OAD in critically ill children [7, 8].
In Western countries, aminoacidurias, especially phenylketonuria, are the most common IMD. PKU is very common in USA, China, Taiwan, Turkey, Ireland, Italy, UK, British Columbia and other parts of Europe, whereas it is rare in India [9–15]. However organic acidurias are comparatively rare in UK, USA and other Western countries [13]. In our previous paper, we have showed that organic acidurias (OAD) are quite common in our part of the world [16]. There are also other studies which report a higher prevalence of OAD in India (Please see details mentioned below). OADs were seen in some Western countries, for example in North Carolina, as reported by Frazier et al. [17].
There is an accelerating demographic switch from communicable diseases to genetic disorders. India is a country with a very high birth-rate and the number of children born with IMD is also very high making it a problem of tremendous importance. In one of the earlier studies in India, biochemical screening of 4400 cases of mental retardation revealed that 5.75% (256 cases) were due to various inherited metabolic disorders [18, 19]. There is no sex difference reported in the incidence of IMD [20]. Age of clinical presentation varies from child to child and the form of presentation is also variable according to age. It also depends upon the degree of accumulation of toxic substances before the metabolic block. Environmental factors may trigger the onset and severity of disease. e.g. diet, intercurrent infections, fasting, drugs etc.
Common Organic Acidurias: Basic Considerations
Important organic acidurias (See Table 1) include propionic acidurias (PA), methyl malonic acidurias (MMA), branched chain organic acidurias (which includes isovaleric aciduria), glutaric aciduria Type I, multiple carboxylase deficiency (which is due to deficiency of 4 biotin dependent enzymes) etc. Rarer OADs are summarized in Table 3. Maple syrup urine disease (MSUD) is due to elevated branched chain amino acids (valine, leucine and isoleucine) and is considered as branched chain aminoaciduria; it is also referred to as organic aciduria. Propionic aciduria, methyl malonic aciduria, MSUD and isovaleric aciduria are sometimes referred to as classical organic acidurias. All organic acidurias are inherited as autosomal recessive conditions.
Diagnosis of Organic Acidurias
Organic acidurias generally present with hyperammonemia and high anion gap metabolic acidosis. Additional biochemical features include hypoglycemia, ketonuria etc. The major clinical features are developmental delay/mental retardation, seizures, lethargy, coma, hypotonia, vomiting, failure to thrive, hepatomegaly, respiratory distress, and cardiac dysfunction. The symptoms worsen in the absence of supporting care and can proceed to coma/death. A diagnosis in the first 24–48 h is vital and can prevent chronic sequel including physical/mental retardation. The prevalence of organic acidurias is high in India, a rapidly developing country with a high birth rate and relatively frequent occurrence of consanguineous marriages.
There is a clinically well demarcated sub-group of OAD known as ‘cerebral’ OADs where neurological manifestations are predominant or sometimes even exclusive [21].
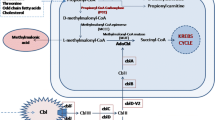
Analysis of organic acids in urine is of paramount importance for the diagnosis of organic acidurias. More than 100 different organic acids are excreted in urine in these conditions (See Table 2). Confirmatory diagnosis of organic acidurias require expensive instruments like HPLC, GC/MS or tandem mass spectrometry and enzyme assays/DNA analysis. High-field proton NMR is a promising technique for the diagnosis of OAD. Screening tests like DNPH test (Di nitro phenyl hydrazine test) and thin layer chromatography are also employed when these are not available, but they do not confirm the diagnosis. A flow chart for the laboratory diagnosis of common organic acidurias is given in Fig. 1.

Flow chart for diagnosis of organic acidurias
Treatment
Most of the common organic acidurias can be effectively treated; therefore diagnosis becomes much more important. Amino acid based formulas provide energy, nitrogen, vitamins and minerals which can promote anabolism and growth. Goal of nutritional therapy is to provide all essential nutrients to promote physical and mental development [23]. Synthetic amino acid based formulas should provide approximately 50% of daily protein requirement. At the same time, the offending dietary precursor amino acid has to be restricted. Fasting also has to be avoided [24]. Secondary carnitine deficiency is common and hence L-Carnitine (as well as biotin) is also given [25]. Metronidazole is given to reduce endogenous gut propionate. Treatment of the acute case includes dialysis, correction of fluid and electrolyte imbalances, correction of acidosis as well as maintenance of cerebral function with adequate perfusion, oxygen and glucose as the case demands. Selective detoxificants like glycine and carnitine are given in some cases.
Sometimes, patients with OAD present in a critical condition, with eventual death. Even when the treatment is not available, the identification of OAD is important for the genetic counseling and for making the pre-natal diagnosis possible in a future gestation [26].
Indian Studies
In the multicentric Indian Council of Medical Research (ICMR) study, 4.9% of the genetic causes of mental retardation were due to metabolic disorders. Screening of mentally retarded children in India revealed that 0.5–2.4% of children had amino acid disorders [27]. In a multi-centric study by ICMR in 1991 on 1314 children, 5% were found to have a metabolic defect. In a hospital based study in India biochemical screening of 4400 cases of mental retardation revealed that 256 (5.75%) cases were due to metabolic disorders [18, 19, 28].
Other studies from South India reported the presence of disorders of amino acid metabolism among 0.66–2.4% of children with mental retardation [27, 29–31]. Kaur et al. [32] screened 2560 cases for IMD and 62 positive cases were reported. The frequency pattern of the various amino acid disorders in North India was found to be remarkably different from that observed in Western countries.
Swarna et al. [33] detected 41 cases of IMD among 4500 children screened from Andhra Pradesh. Uma et al. [34] also reported a new entity, threoninaemia from Andhra Pradesh.
Screening is a basic tool for clinically suspected cases of inborn metabolic disease by a simple economical and effective technique like paper chromatography, TLC and some biochemical tests. However, it is not reasonable to make firm decision on the basis of screening test. HPLC is an acceptable technique to analyze and quantification of amino acids, organic acids and metabolites from biological fluids [35]. It is speculated that late suspicion was an obstacle for early diagnosis of OA [36].
Nagaraja et al. [37] identified 47 cases of organic acidurias and 61 cases of amino acidurias in a total of 113 children with IMD. Study conducted by Mamta et al. [38] on the incidence of organic acidurias in India found out that out of 365 patients with IEM diagnosed over a period of 20 years, organic aciduria accounted for 27% of cases (MMA 18%, PA 9.2%).
In our own study published in the previous issue of IJCB, we had screened 420 high-risk children and identified 45 children (10.7%) with organic acidurias. These included 16 cases of propionic aciduria, 15 cases of methyl malonic aciduria, 13 cases of MSUD and 1 case of isovaleric aciduria. Out of the 13 MSUD patients diagnosed 7 were of classic form [16].
International Studies
Global surveys conservatively estimate the occurrence of inherited metabolic disorders in the range of three to four per thousand live born infants [39]. The World Health Organization (WHO) estimated amino acid disorders to account for approximately 10% of profoundly retarded children.
In one review of cases of IEM diagnosed in British Columbia (a predominantly Caucasian population) between 1969 and 1996, organic acidurias were found at 3.7 per 100,000 [14]. In another review of cases of IEM diagnosed in the West Midlands of the United Kingdom (where approximately 11 percent of the population is from black and ethnic minority groups) during 1999 to 2003 found that the incidence of organic acidemias was 12.6 per 100,000 [40].
A study by Satwani et al. [41] from Pakistan found 10 cases of organic acidemias in a total of 62 children screened. There were a total of 16 cases of IMD in this study which also include lactic acidosis and OTC deficiency.
Survival and quality of life for organic acidemias (OA) is improving due to early diagnosis with newborn screening and vigorous management as reported by Filiano et al. [42], Glass et al. [43] and Dionisi et al. [44].
In contrast, in countries without newborn screening, patients may not be diagnosed, are misdiagnosed or diagnosed late with permanent sequels, as the key factor determining diagnosis and subsequent prognosis is physician’s acumen. Hans et al. [45] studied 3070 children suspected with IMD in China and diagnosed 212 cases with IMD including 92 amino acidurias and 107 organic acidurias.
Study conducted by Tan et al. [46] revealed the incidences of organic acidurias is 32.3%. Among the organic acidurias, 24.4% were glutaric aciduria, 19.6% propionic aciduria, 17.1% methylmalonic aciduria, 12.2% glutaric aciduria Type I and 2.4% were alkaptonuria. The overall detection of organic acidurias revealed by Daisuke Hori et al. [47] was 3.1% comprising mainly MMA and PA i.e, the predominance of MMA over PA was seen in Asian children [47]. MMA is the most common organic acidemias in many studies [47, 48].
A recent study by Moammar et al. (2010) [49] spanning 25 years between 1983 and 2008 in Saudi Arabia, organic acidurias was identified as the most common IMD. They identified 48 OAD cases out of a total of 248 cases of IMD (19%). Niu et al. [12] identified MSUD, MMA and GA-1 as the commonest OADs in Taiwan in a study spanning 2000–2009. Wajner et al. [50] identified 218 cases of OADs between 1994 and 2008 in Brazil.
Maple Syrup Urine Disease
Classic MSUD is the most common form of MSUD with symptoms developing in neonates aged 4–7 days, depending on feeding regimen. In cases of nonclassic MSUD, onset may be later. In the original family with MSUD reported by Menkes, four sibs died in the newborn period. MSUD with a neonatal onset of encephalopathy is now considered as the classic form, and represents the most severe and most common form of the disease. The levels of branched chain amino acids (BCAA), particularly leucine are greatly increased in the blood, cerebrospinal fluid (CSF) and urine. In all patients with the classic form of MSUD, there was more than 10 fold increase in leucine levels and significant elevation in valine and isoleucine. In classic MSUD 50% or more of the branched chain ketoacid (BCKA) are derived from leucine. A reduction in glutamate, glutamine and γ amino butyric acid (GABA) was noted in these patients. Free amino acids in the brain from a 25-day-old infant with untreated MSUD showed marked elevation of BCAA and significant reduction of glutamate, glutamine and GABA [51]. Mutations are known in BCKDHA, BCKDHB, DBT and DLD genes.
Propionic Acidurias
The first documented case of propionic aciduria was described in 1961 by Childs et al. They described a male infant with episodic metabolic ketoacidosis, protein intolerance, and very high levels of plasma glycine. Propionic acidurias are in reality a heterogenous group of disorders and may result from massive propionate accumulation in blood, impaired propionate oxidation, defective carboxylase enzyme in fibroblasts or a combination of these factors. It can also be a feature of multiple carboxylase deficiency. The original patient described by Childs died at age 7, but a sister of the patient lived a relatively normal life till age 15. Propionic acidurias have also been identified in adults. Relapse in precipitated by dietary factors, especially branched chain amino acids, threonine and methionine, and infections. Butanone, a 4-carbon ketone which is a byproduct of isoleucine metabolism, has been reported uniquely in PA [51]. A large number of mutations in pccA and pccB genes have been described.
Methyl Malonic Acidurias
Methyl malonic aciduria as a separate entity was known from 1967 from the separate reports by Oberholzer et al. and Stokke et al. They described critically ill children with profound metabolic acidosis and developmental retardation who accumulated large amounts of methylmalonate in blood and urine. Many reports of methylmalonic acidurias followed. As with PA, methylmalonic acidurias are heterogeneous groups of diseases and distinct genetic mutations in mut, cblA and cblB genes are known. Methylmalonic acidurias are also associated with homocystinuria due to defects in cblC, cblD and cblF genes [51] as well as nutritional deficiency of Vitamin B12.
Isovaleric Acidurias
Initial report of isovaleric acidurias was by Tanaka et al. in 1966. Isovaleric aciduria is the first OAD discovered by GC/MS analysis of metabolites. Two varieties are described, the acute neonatal type and the chronic intermittent type. One of the characteristic features of isovaleric aciduria is the presence of “sweaty foot” odour to urine due to elevated isovaleric acid. Normal development occurs with leucine restriction and administration of carnitine/glycine [51].
Glutaric Aciduria Type I
The condition was first described in 1975. Glutaric acid concentrations are elevated in all tissues, including liver, brain, kidney, eyes, muscles etc. Treatment by restriction of glutarigenic amino acids, lysine, tryptophan and hydroxyl lysine, and supplementation with L- carnitine and riboflavin has been successful. Laboratory features are present only during the acute phase of the disease [51].
Multiple Carboxylase Deficiency
The first case of multiple carboxylase deficiency was described by Gompertz et al. in 1971. There was a dramatic improvement in symptoms by the administration of biotin. The disease could be due to two distinct diseases of biotin metabolism. The first group is due to holocarboxylase deficiency, where there are distinct clinical features including the characteristic skin rash and/or alopecia. The second larger group is due to biotinidase deficiency, but both groups respond dramatically to biotin therapy. Urinary metabolite identification is very important to prevent misdiagnosis as well as therapeutic purposes [51]. “Tomcat urine” smell is characteristic.
Other Organic Acidurias
There are a large number of rarer OAD entities; some of the important ones are summarized in Table 3. Most of them are very rare and in some cases, only a few cases have been reported.
References
Ozand PT, Gascon GG. Organic acidurias: a review. Part I. J Chil Neurol. 1991;6:196–219.
Chalmers RA, Lawson AM. Organic acids in man. Analytical chemistry, biochemistry and diagnosis of the organic acidurias. 1st ed. London: Chapman and Hall; 1982.
Wajner M, Wannmacher CMD, Gaidzinski D, Dutra Filho CS. Detection of inborn errors of metabolism in patients of pediatric intensive care units of Porto Alegre, Brazil. Comparison between the prevalence of such disturbances in a selected and an unselected sample. Braz J Genet. 1986;9:331–40.
Wajner M, Barschak AG, Luft AP, Pires R, Grillo E, Lohr A, et al. Organic aciduria: diagnosis in high risk Brazilian patients. J Pediatr 2001;77(5), 401–6.
Hoffmann GF. Selective screening for inborn errors of metabolism-past present and future. Eur J Pediatr. 1994;153(Suppl 1):S2–8.
Wanmacheer CMD, Wajner M, Giugliani R, Giugliani ERJ, Costa MG, Giugliani MCK. Detection of metabolic disorders among high-risk patients. Rev Brasil Genet. 1982;5:187–94.
Coelho JC, Wajner M, Burin MG, Vargas CR, Giugliani R. Selective screening of 10,000 high- risk Brazilian patients for the detection of inborn errors of metabolism. Eur J Pediatr. 1997;156:650–4.
Green A. Guide to diagnosis of inborn errors of metabolism in district general hospital. J Clin Pathol. 1989;42:84–91.
Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376(9750):1417–27.
van Spronsen FJ. Phenylketonuria: a 21st century perspective. Nat Rev Endocrinol. 2010;6(9):509–14.
Zhan JY, Qin YF, Zhao ZY. Neonatal screening for congenital hypothyroidism and phenylketonuria in China. World J Pediatr. 2009;5(2):136–9.
Niu DM, Chien YH, Chiang CC, Ho HC, Hwu WL, Kao SM, Chiang SH, Kao CH, Liu TT, Chiang H, Hsiao KJ. Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. J Inherit Metab Dis. 2010;33(Suppl 2):S295–305.
Seymour CA, Thomason MJ, Chalmers RA, Addison GM, Bain MD, Cockburn F,Littlejohns P, Lord J, Wilcox AH. Newborn screening for inborn errors of metabolism: a systematic review. Health Technol Assess. 1997;1(11):i–iv, 1–95.
Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics. 2000;105(1):e10.
Loeber JG. Neonatal screening in Europe; the situation in 2004. J Inherit Metab Dis. 2007;30(4):430–8.
Narayanan MP, Vaidyanathan K, Vinayan KP, Vasudevan DM. Diagnosis of major organic acidurias: two years experience at a tertiary care center. Indian J Clin Biochem (Accepted for publication).
Frazier DM, Millington DS, McCandless SE, Koeberl DD, Weavil SD, Chaing SH, Muenzer J. The tandem mass spectrometry newborn screening experience in North Carolina: 1997–2005. J Inherit Metab Dis. 2006;29(1):76–85.
Verma IC. Burden of genetic disorders in India. Indian J Pediatr. 2000;67(12):893–8.
ICMR collaborating centers & central coordinating unit. Multicentric study on genetic causes of mental retardation in India. Indian J Med Res. 1991;94:161–9.
Choudhuri T, Sengupta S. Inborn errors of metabolism—an Indian perspective. Int J Hum Genet. 2006;6(1):89–91.
Hoffmann GF, Gibson KM, Trefz FK, Nyhan WL, Bremer HJ, Rating D. Neurological manifestations of organic acid disorders. Eur J Pediatr. 1994;153(Supplement 1):S94–100.
Kumps A, Duez P, Mardens Y. Metabolic, nutritional, iatrogenic, and artifactual sources of urinary organic acids: a comprehensive table. Clin Chem 2002;48(5):708–17.
Yannicelli S. Nutrition therapy of organic acidaemias with amino acid-based formulas: emphasis on methylmalonic and propionic acidaemia. J Inherit Metab Dis 2006;29:281–7.
Thompson GN, Chalmers RA. Increased urinary metabolite excretion during fasting in disorders of propionate metabolism. Part 1 of 4. Pediatr Res. 1990;27:413–6.
Chalmers RA, Stacey TE, Tracey BM, et al. L-Carnitine: insufficiency in disorders of organic acid metabolism: response to L-carnitine by patients with methylmalonic aciduria and 3-hydroxy-3-methylglutaric aciduria. J Inherit Metab Dis. 1984;7:109–10.
Burlina AB, Bonafe L, Zacheloo F. Clinical and biochemical approach to the neonate with a suspected inborn error of amino acid and organic acid metabolism. Sem Perinatol. 1999;23:162–73.
Ambani LM, Patel ZM, Dhareshwar SS. Clinical biochemical and cytogenetic studies in mental retardation. Indian J Med Res. 1984;79:384–7.
Kumta NB. Inborn errors of metabolism (IEM)—an Indian perspective. Indian J Pediatr. 2005;72:325–32.
Rama Rao BSS, Subhash MN, Narayanan HS. Metabolic anomalies detected during a systematic biochemical screening of mentally retarded cases. Indian J Med Res. 1977;65:241–5.
Reddi OS, Kumar CK, Reddy PP. Screening of aminoacidopathies in India. In Verma IC editors. Medical genetics in India, Vol. 1 Pondichery: Auroma Enterprises 1978;93–8.
Devi ARR, Rao NA, Bittles AH. Inbreeding and the incidence of childhood genetic disorders in Karnataka, South India. J Med Genet. 1987;24:362–5.
Kaur M, Das GP, Verma IC. Inborn errors of amino acid metabolism in North India. J Inherit Met Dis. 1994;17:230–3.
Swarna M, Jyothy A, Usha Rani P, Reddy PP. Amino acid disorders in mental retardation: a two-decade study from Andhra Pradesh. Biochem Genet. 2004;42(3–4):85–98.
Uma SM, Jyothy A, Reddy PP, Reddi O. Aminoacidopathies in Andhra Pradesh; report of a screening programme. J Inherit Metab Dis. 1982;5(4):211–4.
Rao AN, Kavitha J, Koch M, Suresh Kumar V. Inborn errors of metabolism: review and data from a tertiary care center. Indian Journal of Clinical Biochemistry. 2009;24(3):215–22.
Agarwal RL, Muranjan MN. Diagnostic practice for organic acidemias: barriers to early diagnosis. Arch Dis Child 2008;93;1000.
Nagaraja D, Mamatha SN, De T, Christopher R. Screening for inborn errors of metabolism using automated electrospray tandem mass spectrometry: study in high-risk Indian population. Clin Biochem 2009.
Muranjan MN, Kondurkar P. Clinical features of organic acidemias: experience at a tertiary care center in Mumbai. Indian Pediatr. 2001;38:518–24.
Chalmers RA, Watts RWE, Lawson AM. A comprehensive screening method for detecting organic acidurias and other metabolic diseases in acutely sick infants and children. Ann Clin Biochem. 1977;14:149–56.
Sanderson S, Green A, Preece MA, Burton H. The incidence of inherited metabolic disorders in the west midlands UK. Arch Dis Child. 2006;91(11):869–99.
Satwani H, Raza J, Hanai J, Nomachi S. Prevalence of selected disorders of inborn errors of metabolism in suspected cases at a tertiary care hospital in Karachi. J Pak Med Assoc. 2009;59(12):815–9.
Filiano JJ, Bellimer SG, Kunz PL. Tandem mass spectrometry and newborn screening: pilot data and review. Pediatr Neurol. 2002;26:201–4.
Glass HC, Feigenbaum A, Clarke JTR. A study on the nature of genetic metabolic practice at a major pediatric referral centre. J Inherit Metab Dis. 2006;29:175–8.
Dionisi-Vici C, Deodato F, Röschinger W, et al. ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: Longterm outcome and effects of expanded newborn screening using tandem mass spectrometry. J Inherit Metab Dis. 2006;29:383–9.
Han LS, Ye J, Qiu WJ, Gao XL, Wang Y, Gu XF. Selective screening for inborn errors of metabolism on clinical patients using tandem mass spectrometry in china: a four-year report. J Inherit Metab Dis. 2007;30(4):507–14.
Tan I-K, Gajra B, Lim MSF. Study of inherited metabolic disorders in Singapore-13 Years experience. Ann Acad Med Singapore 2006;35:804–13.
Hori D, Hasegawa Y, Kimura M, Yang Y, Verma IC, Yamaguchi S. Clinical onset and prognosis of Asian children with organic acidemias, as detected by analysis of urinary organic acids using GC/MS, instead of mass screening. Brain Dev. 2005;27:39–45.
Song Y-Z, Li B-X, Hao H, Xn R-L, Zhang T, Zhang C-H, et al. Selective screening for inborn errors of metabolism and secondary methylmalonic aciduria in pregnancy at high risk district of neural tube defects: a human metabolome study by GC-MS in China. Clin Chem. 2008;41:616–20.
Moammar H, Cheriyan G, Mathew R, Al-Sannaa N. Incidence and pattern of inborn errors of metabolism in the Eastern province of Saudi Arabia 1983–2008. Ann Saudi Med. 2010;30(4):271–7.
Wajner M, Coelho Dde M, Ingrassia R, de Oliveira AB, Busanello EN, Raymond K et al. Selective screening for organic acidemias by urine organic acid GC-MS analysis in Brazil: fifteen-year experience. Clin Chim Acta. 2009;400(1–2):77–81.
Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease, 7th ed. New York: Mc Graw-Hill 2001:1.
Acknowledgments
NMP is Senior Research Fellow of ICMR, Financial assistance received from Kerala state Council for Science, Technology and Environment (KSCSTE), Government of Kerala, Indian Council of Medical Research (ICMR), Government of India is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vaidyanathan, K., Narayanan, M.P. & Vasudevan, D.M. Organic Acidurias: An Updated Review. Ind J Clin Biochem 26, 319–325 (2011). https://doi.org/10.1007/s12291-011-0134-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12291-011-0134-2