
Abstract
Objectives
To study plasma levels of Thrombin activable fibrinolysis inhibitor (TAFI) in children with β-thalassemia major.
Methods
Fifty β-thalassemia major patients, 1.4 to 17 y of age, with number of transfusions received varying from 21 to 162 were selected at random and complete blood count (CBC), coagulation parameters [Prothrombin time (PT), Activated partial thromboplastin time (aPTT), fibrinogen, D-dimer, protein C, protein S, antithrombin, Tissue plasminogen activator (t-PA), Plasminogen activator inhibitor (PAI-1)] and TAFI were performed.
Results
PT and aPTT were prolonged in 18 % and 30 % of cases respectively. Reduced activity of Protein C (PC) was observed in 50 % of cases and Protein S (PS) was reduced in 54 % of cases. t-PA levels were significantly higher in cases. TAFI levels were 17.24 ± 4.05 ng/ml which were significantly higher than the control group (15.01 ± 3.28; p = 0.003) No significant correlation of TAFI was observed with Hb, platelet counts, liver enzymes, serum ferritin, PC, PS, D-dimer, t-PA or PAI-1.
Conclusions
There is an ongoing subclinical activation of coagulation cascade and fibrinolytic system in thalassemia major (TM) patients. Higher levels of TAFI in the present study with no significant correlation with other parameters were noted, thus pointing out to its independent role in contribution to hypercoagulable state in thalassemia. TAFI serves as a link between two limbs of hemostasis, with its higher levels promoting inhibition of fibrinolytic system and thus promoting a hypercoagulable state. Performing TAFI levels in thalassemic patients could help to detect the early coagulopathy in these patients and hence these patients can be closely monitored for any evidence of thrombosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thalassemia is a congenital hemolytic disorder caused due to partial/ complete deficiency of α or β globin chain synthesis. A chronic subclinical hypercoagulable state already exists in thalassemia. The various factors involved in its etiopathogenesis include red blood cell membrane abnormality [1], deficiency of various proteins of coagulation cascade and fibrinolytic system [1–3], oxidative damage due to free hemoglobin, increased platelet activation and aggregation [4], and endothelial and leukocyte activation [5, 6]. The sites of predilection of these microthrombi are central nervous system, lungs and pulmonary vasculatures [1, 3]. Also a few studies have described bleeding tendencies in these patients in the form of epistaxis [7, 8]. In the Indian subcontinent, only a few studies are available which document bleeding manifestations in these patients. However, a few case reports document thrombosis in them. Thus these patients can have thrombosis/bleeding depending on the balance between coagulation cascade and fibrinolytic system.
TAFI (Thrombin Activable Fibrinolysis Inhibitor) [ 9–12] is a recently discovered zymogen. It is synthesized in the liver and megakaryocytes [13, 14] and circulates in blood in complex with plasminogen [11]. It has two isoforms based on substitution of a single base pair at amino acid position 147, which do not have functional differences [15].
TAFI is primarily activated by thrombin-thrombomodulin complex [16]. It cleaves lysine residue from fibrin, which is the binding site for plasminogen, thereby reducing the rate of plasmin generation with consequent prolongation of fibrin dissolution. Also, as a result, there is reduced positive feedback by fibrin degradation products and hence, down regulation of clot lysis.
TAFI is an unstable enzyme at physiological conditions and is primarily inactivated by a spontaneous conformational change [17] with a t1/2 of 10 min at 37°C [18], secondary route of inactivation being proteolytic cleavage at Arg302 by thrombin or plasmin [19].
Increased TAFI levels have been correlated with increased clot lysis time in normal individuals [20]. Role of TAFI has been studied in various conditions like stroke [21], deep vein thrombosis [22] and gastric carcinomas [23]. Increased TAFI levels were positively correlated in patients with deep vein thrombosis [22]. A two-fold increased risk of thrombosis in cases who had TAFI levels above 90th percentile of controls (>122 U/dl) as compared to cases having levels below it (odd ratio 1.7, confidence interval 1.1–2.5) was observed.
TAFI, thus serves as a link between the coagulation cascade and the fibrinolytic system.
With this background, the present study was conducted to know the role of TAFI as a contributing factor in causing hypercoagulable state in children with β-thalassemia major.
Material and Methods
Ethical clearance for the study was obtained from institutional ethics committee, Lady Hardinge Medical College. Out of the total of 379 cases registered in Thalassemia Day Care centre at Kalawati Saran Children’s Hospital, 50 randomly selected cases upto 18 y of age were enrolled in the study along with equal number of age and sex matched controls. Informed consents were taken. All the blood samples were drawn before transfusion. Those who received <20 blood transfusions or having any clinical evidence of acute infection/ serum C-reactive protein (CRP) levels >8 mg/L or seropositive (HIV, HBsAg, HCV) were excluded.
The cases were subjected to complete blood count with peripheral smear, serum C-reactive protein, liver function tests, serum ferritin, coagulation tests [prothrombin time (PT), activated partial thromboplastin time (aPTT), fibrinogen, D-dimer; coagulation inhibitors – protein C, protein S, antithrombin; fibrinolytic system-tissue Plasminogen Activator (tPA), Plasminogen Activator Inhibitor (PAI-1) and Thrombin Activable Fibrinolysis Inhibitor (TAFI)].
CBC was performed using Sysmex KX-21 haematology analyzer. Liver function tests [alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP)] were done on Beckman Coulter Synchron CX-9 autoanalyzer.
Semi quantitative test for CRP was done using BIOLATEX CRP_EKO kit.
The quantitative measurement of serum ferritin was done using Cal biotech, Inc. (CBI) ferritin ELISA kit.
All clotting assays of PT, aPTT, PC, PS, fibrinogen were performed on fully automated STA compact analyser using STA diagnostic kits. Calorimetric assay of antithrombin III, using STA- STACHROM® AT III and immuno-turbidimetric assay of D-dimer, using STA-LIATEST® D-DI kits were also performed on fully automated STA compact analyser.
t-PA and PAI-1 were measured using t-PA (human) ELISA DRG diagnostics kit and PAI-1 (human) ELISA BIOVENDOR research and diagnostics kits respectively.
Quantitative determination of TAFI was done by enzyme immunoassay using ASSERCHROM® TAFIa (active form)/ TAFIai (inactive form) ELISA kit. All ELISA based tests were done using Bio-rad micro plate ELISA reader and Bio-rad washer.
The TAFIa / TAFIai to be measured was captured by specific mouse monoclonal anti-human TAFIa/ TAFIai antibody coated on the internal walls of a plastic micro plate well. Next, the second monoclonal anti TAFIa/ TAFIai antibody coupled with peroxidise were bound to the remaining free antigenic determinants of the bound TAFIa/ TAFIai. The bound enzyme peroxidase was revealed by its reaction with a strong acid, the intensity of the color was directly proportional to the concentration of TAFIa/ TAFIai initially present in the plasma sample.
The statistical analysis was done by using Statistical Package for Social Science (SPSS) version 20.0. The data was expressed as mean ± standard deviation. Besides descriptive statistics, the comparison of 10 parameters (PT, aPTT, fibrinogen, D-dimer, protein C, protein S, antithrombin, t-PA, PAI-1, and TAFI) between cases and controls was done using Mann Whitney U test for non parametric data and independent sample t test for parametric data.
The correlation between the various parameters among cases was done using pearson and spearman coefficients. The “p value” of <0.05 was considered to be significant.
Results
The age group of cases ranged from 1.4 y to 17 y with mean age of 9.17 ± 4.43 y. The male to female ratio was 2.57:1. The mean age of controls was 7.78 ± 4.24 y with a range of 1.5 to 17 y and male to female ratio of 2.12 : 1. No significant difference was noted in age and sex of the two groups. The number of blood transfusions received by patients varied from 21 to 162 units. All the cases were non-splenectomized. They were on iron chelation therapy and regular transfusions. No history of clinical evidence of thrombosis was present in any of cases in last 6 mo. The complete blood count (CBC) findings of the two groups are shown in Table 1 and the results of coagulation tests are depicted in Table 2. Table 3 shows the liver function tests in cases.
Patients showed significantly higher values of PT and aPTT as compared to controls. Abnormally prolonged PT and aPTT were observed in 18 % (9/50) and 30 % (15/50) of the patients respectively. The fibrinogen levels of cases ranged from 123 to 408 mg/dl, and were significantly higher (p = 0.034) than controls.
Although the mean D-dimer levels were raised in cases [0.449 ± 0.914 μg/ml (FEU)] as compared to controls, the difference was not statistically significant (p = 0.082).
PC and PS were significantly lower in cases as compared to controls (p < 0.001 for both). AT III levels were comparable in the two groups (p = 0.38).
The tPA levels were significantly higher in cases than controls (p = 0.047). PAI-1 levels were comparable in two groups (p = 0.380).
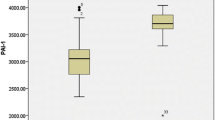
The levels of TAFI in cases ranged from 10.1 to 25.0 ng/ml with mean of 17.24 ± 4.05 ng/ml. It was statistically significantly higher than controls (15.01 ± 3.28 ng/ml; p = 0.003) as shown in Fig. 1.

Comparison of TAFI in cases and controls
TAFI levels in males and females in the case group were 17.6 ng/ml and 16.1 ng/ml respectively. The difference was not statistically significant (p > 0.1). Plasma TAFI levels showed positive correlation with age (r = 0.24, p = 0.09) but the values were not statistically significant. A positive correlation was also noted between plasma TAFI levels and number of blood transfusions (r = 0.28, p = 0.05). TAFI correlated poorly with hemoglobin and platelets (r = 0.18, 0.08 respectively). No significant correlation of TAFI was noted with serum bilirubin or liver enzymes (p > 0.1). No significant correlation of TAFI was noted with PT, aPTT and fibrinogen (p > 0.1). TAFI also correlated poorly with protein C, protein S and antithrombin (p > 0.1).
In control group no significant correlation of TAFI with age or any other parameters was observed (r = 0.16, p = 0.2).
D-dimer levels were high in patients with raised TAFI levels but the correlation was not statistically significant (r = 0.194, p > 0.1). No significant correlation of TAFI was noted with t-PA (p > 0.1) and PAI-1(p > 0.1).
Discussion
Hypercoagulable state in thalassemia is a well recognised phenomenon. The various etiologies implicated, include the altered levels of anticoagulant and procoagulant proteins [1–3], alteration of RBC membrane [1], increased platelet activation and aggregation [4], endothelial, monocyte and granulocyte activation [5, 6].
Studies in literature have documented both hypercoagulability and bleeding tendencies in β-thalassemia major patients. The incidence of thromboembolic events in beta thalassemia (β-TM) varied from 0.9–4.0 % in various studies [1, 3]. However a few studies also reported bleeding tendencies in these patients7.8. None of the patient in present series had any clinical evidence of thrombosis. Although two patients had thrombocytopenia, none had bleeding manifestations.
The mean PT and aPTT in patients were significantly higher than controls (p = 0.001 and <0.001 respectively). Similar results were also obtained by several other authors [7, 8, 24, 25].
Fibrinogen levels in the cases, though were significantly higher than controls, were within normal range. This is in contrast to other studies in which low levels of fibrinogen were observed in the cases [25, 26].
D-dimer, breakdown product of fibrin, is a marker of thrombin activation and secondary fibrinolysis. Although the levels of D-dimer were high in cases as compared to controls, the difference was not statistically significant. However, Tripatara et al. noticed a significant higher values of D-dimer in cases as compared to controls (p < 0.05) [27].
In the present series the mean PC and PS activity were significantly lower than the control group. A similar observation was made by several authors [7, 25, 27, 28]. A few authors had reported significantly reduced AT activity in β thalassemia major patients [3, 7, 26]. However, in the present study AT activity was comparable to controls.
In the current series reduced activity of Protein C and Protein S was observed in 50 % and 54 % of cases respectively. Antithrombin was reduced in only 4 % of the cases. These findings are consistent with other authors who observed a similar decrease in PC, PS and AT [29, 30]. However Naithani et al. observed reduced PC and PS activity in 26.2 % and 28.6 % of cases [7]. They observed reduced AT in a higher number of cases (46.8 %).
Reduced levels of the anticoagulants might be because of deranged liver functions in these patients due to iron overload resulting from repeated blood transfusions. Also, the ongoing subclinical coagulation activation might have contributed to the consumption of these anticoagulant proteins. In the present study as no correlation between deranged liver enzymes and anticoagulant proteins was observed, the latter mechanism seems to be the major contributing factor.
Only a very few studies on levels of t-PA and PAI-1 in patients with β- thalassemia major are available in literature [26]. Angchaisuksiri et al. noticed a significantly higher level of t-PA and PAI-1 in splenectomised HbE/β-thalassemia patients as compared to non-splenectomised HbE/β-thalassemia patients and controls [26].
In the present study the t-PA levels were significantly higher in cases as compared to controls (p = 0.047). Though the PAI-1 levels were also high in cases, the difference was not statistically significant (p > 0.1). Increased t-PA and PAI-1 suggest an activation of fibrinolytic system which is also supported by increased D-dimer levels.
TAFI is a recently discovered protein that attenuates fibrinolysis. Thus, increased levels of TAFI favour hypercoagulability. Only a few studies on role of TAFI in thalassemia are available in literature. Tripatara et al. conducted study on HbE/β-thalassemia children and observed a reduced level of TAFI in both severe non-splenectomised and splenectomised group (p < 0.05) [ 27]. Mokhtar et al. observed a reduced TAFI levels in β-thalassemia patients [8]. The TAFI % activity observed by them was lower (47.33 ± 11.43 %) in TM splenectomised and TM non-splenectomised patients (69.33 ± 12.13 %) as compared to controls (116.22 ± 10.47 %), (p < 0.001). Nine out of fifty one of their patients had bleeding manifestations. The cases with bleeding had significantly lower TAFI activity as compared to the patients without bleeding (p < 0.0001).
TAFI levels in the present study were 17.24 ± 4.05 ng/ml which were significantly higher than control group (15.01 ± 3.28, p = 0.003). No significant correlation of TAFI was observed with Hb, platelet count, liver enzymes, serum ferrittin, PC, PS, and D-dimer, t-PA or PAI-1. However, Mokhtar et al. noticed a significant negative correlation of TAFI% with serum ferritin, liver enzymes, TLC and platelet count whereas no correlation was observed between TAFI% and PT or aPTT [8].
In the present study none of the patients was splenectomised and no clinical evidence of thrombosis was present in last 6 mo. Hypercoagulable state in thalassemia is generally subclinical. Platelets activation and aggregation is a known contributing factor to this hypercoagulable state. Previous two studies showed significant reduction in TAFI levels in cases as compared to controls which is attributed to liver derangements due to iron overload. Also significant reduction of TAFI levels in splenectomised thalassemia major patients was observed as compared to non-splenectomised patients, indicating its utilization in inhibiting fibrinolysis.
The higher levels of TAFI in the present study might be due to release of TAFI from α granules of platelets. Though the liver function tests were marginally deranged in current study due to iron overload attributed to regular blood transfusions and poor compliance to chelation therapy, it did not affect the TAFI levels significantly. All the patients were non-splenectomised, so the abnormal red cells were significantly removed by spleen, thus less number of abnormal red cells were available in circulation and so there was lower activation of the coagulation cascade and fibrinolytic system. This partially explains the high levels of TAFI due to its lower consumption in inhibiting fibrinolysis. The other reason for its higher levels being its release from platelets. Also due to lower number of circulating abnormal red cells because of regular transfusion, no clinically evident thrombotic events were noted in the present study.
In the present study, significantly reduced levels of anticoagulant proteins and increased levels of TAFI favoured increased tendency of hypercoagulability in these patients. The proteins of fibrinolytic system (i.e., t-PA and PAI-1) were elevated along with increased D-dimer in these patients, thus suggesting ongoing fibrinolysis.
Conclusions
TAFI’s role in regulation of fibrinolysis suggests that high levels of TAFI would contribute to thrombophilia, while low levels of TAFI would cause hemorrhage. Performing TAFI levels in thalassemic patients could help to detect the early coagulopathy in these patients and hence these patients can be closely monitored for any evidence of thrombosis/bleeding.
The coagulopathy in thalassemic patients is multifactorial. Along with the well known factors responsible for it, TAFI could also be a potential parameter to determine the hemostatic alteration in them. It has a short half life, so how early it can detect an impending thrombotic event remains yet to be studied. Once this could be determined, timely interventions in form of anticoagulants could be performed and thus the patient could be saved of devastating thromboembolic complications.
As this parameter is not well studied yet, a larger study is required to identify its use for detection of subclinical hemostatic alteration in thalassemic patients. This would in turn help to predict the increased risk of thromboembolic events/bleeding in them and thus, a way to improve their survival.
References
Taher A. Iama’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis. 2006;37:12–20.
Winichagoon P, Fucharoen S, Wasi P. Increased circulating platelet aggregates in thalassemia. Southeast Asian J Trop Med Public Health. 1981;12:556–60.
Cappellini MD. Coagulation in the pathophysiology of hemolytic anemias. Hematology Am Soc Hematol Educ Program. 2007;1:74–8.
Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99:36–43.
Butthep P, Bunyaratvej A, Funahara Y, et al. Possible evidence of endothelial cell activation and disturbance in thalassemia: an in vitro study. Southeast Asian J Trop Med Public Health. 1997;28:141–8A.
Hovav T, Goldfarb A, Artmann G, Yedgar S, Barshtein G. Enhanced adherence of beta thalassemic erythrocytes to endothelial cells. Br J Haematol. 1999;106:178–81.
Naithani R, Chandra J, Narayan S, Sharma S, Singh V. Thalassemia major- on verge of bleeding or thrombosis? Haematology. 2006;11:57–61.
Mokhtar GM, Matter RM, Shawki H, Abdel Aziz MM. Thrombin activable fibrinolysis inhibitor (TAFI): relationship to haemostatic alteration in patients with beta thalassemia. Pediatr Hematol Oncol. 2010;27:363–73.
Nesheim M, Bajzar L. The discovery of TAFI. J Thromb Haemost. 2005;3:2139–46.
Mosneir LO, Bouma BN. Regulation of fibrinolysis by thrombin activable fibrinolysis inhibitor, an unstable carboxypeptidase B that unites the pathway of coagulation and fibrinolysis. Arterioscler Thromb Vasc Biol. 2006;26:2445–53.
Boffa MB, Koschinsky ML. Curiouser and curiouser: recent advances in measurements of thrombin activable fibrinolysis inhibitor (TAFI) and in understanding its molecular genetics, gene regulation and biological roles. Clin Biochem. 2007;40:431–42.
Bertina RM, Tilburg LH, Haverkate F, et al. Discovery of thrombin activable fibrinolysis inhibitor (TAFI). J Thromb Haemost. 2006;4:256–7.
Mosnier LO, Buijtenhuijs P, Marx PF, Meijers JCM, Bouma BN. Identification of thrombin activable fibrinolysis inhibitor (TAFI) in human platelets. Blood. 2003;10:4844–6.
Schadinger SL, Linn JHH, Garand M, Boffa MB. Secretion and antifibrinolytic function of thrombin- activable fibrinolysis inhibitor from human platelets. J Thromb Haemost. 2010;8:2523–9.
Zhao L, Morser J, Bajzar L, Nesheim M, Naqashima M. Identification and characterization of two thrombin activable fibrinolysis inhibitor isoforms. Thromb Hemost. 1998;80:949–55.
Bajzar L, Morser J, Neshim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271:16603–8.
Marx PF, Hackeng TM, Dawson PE, Griffin JH, Meijers JCM, Bouma BN. Inactivation of thrombin activable fibrinolysis inhibitor takes place by process that involves conformational instability rather than proteolytic cleavage. J Biol Chem. 2000;275:12410–5.
Boffa MB, Wang W, Bajzar L, Nesheim ME. Plasma and recombinant thrombin activable fibrinolysis inhibitor (TAFI) and activated TAFI compared with respect to glycosylation, thrombin/thrombomodulin dependent activation, thermal stability and enzymatic properties. J Biol Chem. 1998;273:2127–35.
Boffa MB, Bell R, Stevens WK, Nesheim ME. Roles of thermal instability and proteolytic cleavage in regulation of activated thrombin activable fibrinolysis inhibitor. J Biol Chem. 2000;275:12868–78.
Mosnier LO, von den Borne PA, Meijers JC, Bouma BN. Plasma TAFI levels influences the clot lysis time in healthy individuals in the presence of an intact intrinsic pathway of coagulation. Thromb Haemost. 1998;80:829–35.
Biswas A, Tiwari AK, Ranjan R, et al. Thrombin activable fibrinolysis inhibitor gene polymorphisms are associated with antigenic levels in the Asian-Indian population but may not be a risk for stroke. Br J Hematol. 2008;143:581–8.
Tilburg N, Rosendaal FR, Bertina RM. Thrombin activatable fibrinolysis inhibitor and risk of deep vein thrombosis. Blood. 2000;95:2855–9.
Eser M, Kement M, Balin S. Is there any role of thrombin activable fibrinolysis inhibitor in the development of a hypercoagulable state in gastric cancer? World J Surg Oncol 2012;10:180.
Caocci L, Alberti M, Burrai P, Corda R. Screening coagulation tests and clotting factors in homozygous β- thalassemia. Acta Haematol. 1978;60:358–64.
Musumeci S, Leonardi S, Dio R, Fischer A, Costa G, Protein C. And antithrombin III in polytransfused thalassemic patients. Acta Haematol. 1987;77:30–3.
Angchaisuksiri P, Atichartakarn V, Aryurachai K, et al. Hemostatic and thrombotic markers in patients with hemoglobin E/ beta- thalassemia disease. Am J Hematol. 2007;82:1001–4.
Tripatara A, Jetsrisuparb A, Teeratakulpisarn J, Kuaha K. Haemostatic alterations in splenectomized and non-splenectomized patients with β-thalassemia/ hemoglobin E disease. Thromb Res. 2007;120:805–10.
Shirahata A, Funahara Y, Opartkiattikul N, Fucharoen S, Laosombat V, Yamada K. Protein C and protein S deficiency in thalassemic patients. Southeast Asian J Trop Med Public Health. 1992;23:65–73.
Teli A, Economou M, Tzovaras F, et al. Subclinical central nervous system involvement and thrombophilic status in young thalassemia intermedia patients of Greek origin. Blood Coag. Fibrinolysis. 2012;23:195–202.
Kemhali S, Gurman C, Egin Y, et al. Hypercoagulability in children with thalassemia major. Clin Appl Thromb Haemostat. 1997;3:129–32.
Contributions
AC: Preparation of manuscript, critical analysis and statistics analysis; SS and AN: Overall supervision of the study; JC: Provided with clinical details. SS will act as guarantor for the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
None.
Source of Funding
None.
Rights and permissions
About this article

Cite this article
Chhikara, A., Sharma, S., Chandra, J. et al. Thrombin Activable Fibrinolysis Inhibitor in Beta Thalassemia. Indian J Pediatr 84, 25–30 (2017). https://doi.org/10.1007/s12098-016-2208-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-016-2208-x