
Abstract
Thalassemia is a congenital hemolytic disease which is treated by repeated blood transfusion. Chronic iron overload is currently considered to be the primary cause of mortality in β-thalassemia, mainly due to the induction of left-sided cardiac failure. Iron overload results from a number of mechanisms associated with the disease itself. In addition to chronic iron overload thalassemic patients are more prone for procoagulant status which in turn lead to clinical thrombotic events. The hypercoagulable state in thalassemia is due to multiple elements, a combination of which is often the drive behind a clinical thromboembolic events. PAI-1 study was done in thalassemia major patients receiving multiple blood transfusion as a marker for procoagulant status. Total of 30 thalassemic patients on repeated blood transfusion was included in the study and total of 30 healthy age and sex matched controls were included in the study. It was also found that there was significant differences between cases and controls. The mean level of PAI 1 in controls was 3047 ± 414 pg/ml, the value in cases was 3683 ± 358 pg/ml. The level was significantly increased (p < 0.05) in the cases compared to controls. PAI-1 levels were also compared with the total number of blood transfusion which correlates well.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thalassemia is a congenital hemolytic disease which is treated by repeated blood transfusion. Chronic iron overload is currently considered to be the primary cause of mortality in β-thalassemia, mainly due to the induction of left-sided cardiac failure. Iron overload results from a number of mechanisms associated with the disease itself. In addition to chronic iron overload thalassemic patients are more prone for procoagulant status which in turn lead to clinical thrombotic events. The hypercoagulable state in thalassemia is due to multiple elements, a combination of which is often the drive behind a clinical thromboembolic events (TEE) [1]. Several factors have been implicated in the pathogenesis of this hypercoagulable state, such as changes in the lipid membrane composition of the abnormal RBCs with increased expression of negatively charged phosphatidylserine (PS) at the outer surface, platelet activation, deficiency of natural anticoagulants and others [2].
Materials and Methods
Source of Data
The present study comprises of 30 clinically diagnosed cases of β-thalassemia major who were registered with haematology centre for regular blood transfusion. The study was conducted at tertiary health care centre. All the patients were in the age group of 12–20 years. Age and sex matched 30 healthy individuals were taken as controls.
Criteria for the Selection of Cases
-
(1)
Inclusion criteria Clinically diagnosed and proven cases of β-thalassemia major on regular blood transfusion, children to young adults, both males and females were included in this study.
-
(2)
Exclusion criteria Sickle cell anaemia or any other hemoglobinopathy.
Informed consent was taken from all the cases and the controls and the study was approved by the ethical and research committee.
Collection and Storage of Blood Samples
Twenty ml of blood was collected from cases and controls in clot activator tube and allowed to clot for 30 min. Serum was separated, aliquoted and kept at −70 °C. Timing of collection was just before starting of blood transfusion. Haemolyzed, icteric and lipemic samples were discarded. All samples were brought to room temperature just before the analytical procedure.
Methods of Assay
PAI-1 were analyzed by ELISA.
Test were performed on serum. All patients were on iron chelator. Data was collected by detailed history, clinical examination, questionnaires and medical records of the patients.
The clinical details of patients were recorded in a proforma, which included age, sex, residence, frequency of transfusions, blood grouping, status of iron chelation therapy, prior history of any complications or admission due to the disease activity.
All samples and controls were run in duplicates. ELISA based kit for PAI-1 was used from Invitrogen with intra-assay CV 3.68 % and inter-assay CV 9.05 %. Test is based on the principle of solid phase sandwich enzyme linked immunosorbent assay.
Study results were analyzed by SPSS version 17. Unpaired t test and Mann–Whitney U test were used to analyze the data. Correlation was used using Pearsons coefficient correlation and Spearmanns Rho coefficient.
Results
The present study comprises of 30 proven cases of β-thalassemia major registered to the Haematology dept, receiving multiple blood transfusion and 30 age and sex matched healthy controls. The age group ranges from 12 to 20 years. Study results were analyzed by SPSS version 17. Differences were considered significant for p < 0.05. Result was expressed as mean ± standard deviation (S.D.). The variable PAI-1 were analyzed using unpaired t test and it was found that there was significant differences between study group (cases and control). PAI-1 were also analyzed using non parametric test because of non normality. it was also found that there was significant differences between cases and controls.
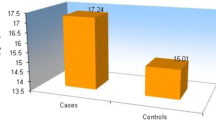
The mean level of PAI 1 in controls was 3047 ± 414 pg/ml, the value in cases was 3683 ± 358 pg/ml (Table 1). The level was significantly increased (p < 0.05) in the cases (Fig. 1) compared to controls.

PAI-1 levels (pg/ml) in cases and in controls
The level of PAI-1 were compared with number of blood transfusion (Table 2). It shows that mean levels of PAI-1 is well correlated with number of blood transfusion in increasing order (Table 2).
Discussion
In the present study, PAI-1 were studied in 30 diagnosed cases of β-thalassemia major along with 30 healthy controls. Aim of the study is to assess prothrombotic state in thalassemic patients receiving multiple blood transfusions. State of hypercoagulability has been measured by estimating serum PAI-1.
The mean levels of serum PAI-1 in control was 3047.1 ± 413.8 pg/ml, the value in cases was 3683.7 ± 357.9 pg/ml (Table 1), which was statistically significant (p < 0.05). The findings of the present study are in accordance with the results of Angchaisuksiri et al. [3]. Hypercoagulability is a well-established characteristic of β-thalassemia. Increased levels of PAI-1 in these patients is responsible for increased risk of thrombosis. According to Angchaisuksiri et al. there is evidence of chronic low grade coagulation and platelet activation, chronic low-grade inflammation, endothelial cell injury, impaired fibrinolysis and decreased naturally occurring anticoagulants in β-thalassemia patients. These changes may account for the increased risk of thrombosis in these patients. Increased level of PAI-1 in these subjects can be explained as follows. Thrombosis is typically an episodic complication associated with a temporary activation of hemostasis. In contrast, markers of platelet and coagulation activation are persistently and consistently elevated in most thalassemic patients (adults and children alike), even in the absence of overt thromboembolic events. The presence of a persistent hypercoagulable state combined with the infrequent occurrence of significant thrombotic events suggests that thrombosis is largely a subclinical process in thalassemia and has been associated with autopsy findings of platelet and fibrin thrombi in the microvasculature in the lungs and the brain [4, 5].
However, in our study none of the cases gave previous history of thromboembolic events like seizures, hemiparesis, pulmonary hypertension, severe calf pain, etc. But mean value of PAI-1 in cases (3682.7 ± 357.9 pg/ml) is raised compared to controls (3047.1 ± 413.8 pg/ml) (p < 0.05). It clearly depicts that raised serum PAI-1 level in cases may lead to procoagulant status and in future they may develop any of thrombotic complications. Several studies done by Sonakul et al. [6], Sumiyoshi et al. [7], Grisaru et al. [8], Luyt et al. [9], Grant et al. [10], Koren et al. [11], Du et al. [12], Chotivittayatarakorn et al. [13] shows that thalassemic patients receiving multiple blood transfusion are more prone for thromboembolic events.
Thalassemic patients are also prone for cardiac disease due to iron overload and also due to polymorphism in PAI-1 gene. PAI-1 is a significant, independent risk factor for coronary artery disease and ischemic stroke. The 4G allele of a recently described common 4/5 guanine tract (4G/5G) polymorphism in the PAI-1 gene promoter region is associated with higher plasma PAI-1 activity [14]. In vitro experiments have shown that both allele of the 4G/5G polymorphism contain a binding site for a transcription activator, and that 5G allele also contains a binding site for a transcription repressor that partially overlaps with the activator binding site [15]. Therefore, 4G allele of the PAI-1 gene is associated with increased basal gene transcription. The 4G/4G genotype of the PAI-1 has been shown to be associated with ischemic heart disease and myocardial infarction [16].
Study done by Patricia Jane Giardina et al. [17] mentioned that brain MRI studes needs to be performed to address the prevalence, pathogenesis, risk factors, neurological and cognitive outcomes of brain infarcts in at risk adolescent and adult thalassemia subjects for the prevention and development of therapeutic strategies.
Conclusion
To conclude, thalassemic patients receiving multiple blood transfusions definitely suffer from increased tendency for procoagulant status. Increased tendency for procoagulant status in these patients may be responsible for thrombotic events in future.
Abbreviations
- MDA:
-
Malondialdehyde
- ROS:
-
Reactive oxygen species
- HIV:
-
Human immunodeficiency virus
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- ELISA:
-
Enzyme linked immunosorbent assay
- CV:
-
Coefficient of variation
- S.D.:
-
Standard deviation
- Gr:
-
Group
References
Hershko C, Graham G, Bates GW, Rachmilewitz EA. Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity. Br J Haematol. 1978;40:255–63.
Elder A, Durst R, Hy-Am E, Goldfarb A, Gillis S, Rachmilewitz EA. A chronic hypercoagulable state in patients with β-thalassemia major is already present in childhood. Br J Haematol. 1999;107(4):739–46.
Angchaisuksiri P, Atichartakaran V, Aryurachai K, Archararit N, Chuncharunee S, Tiraganjana A. Haemostatic and thrombotic markers in patients of hemoglobin E/β-thalassemia disease. Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok. Am J Hematol. 2007; 82(11):1001–4.
Sonakul D, Pachaaree P, Laohaphand T, Fucharoen S, Wasi P. Pulmonary artery obstruction in thalassemia. Southeast Asian J Trop Med Public Health. 1980;11:516–23.
Sumiyoshi A, Thakerngpol K, Sonakul D. Pulmonary microthromboemboli in thalassemic cases. Southeast Asian J Trop Med Public Health. 1992;23:29–31.
Sonakul D, Pachaaree P, Laohaphand T, Fucharoen S, Wasi P. Pulmonary artery obstruction in thalassemia. Southeast Asian J Trop Med Public Health. 1980;11:516–23.
Sumiyoshi A, Thakerngpol K, Sonakul D. Pulminary microthromboemboli in thalassemic cases. Southeast Asian J Trop Med Public Health. 1992;23:29–31.
Grisaru D, Rachmilewitz EA, Mosseri M, Gotsman M, Lafair JS, Okon E. Cardiopulmonary assessment in β-thalassemia major. Chest. 1990;98:1138–42.
Luyt DK, Richards GA, Roode H, Dowdeswell RJ, van Rensburg AJ, Reinach SG. Thalassemia: lung function with reference to iron studies and reactive oxidant status. Pediatr Hematol Oncol. 1993;10:13–23.
Grant GP, Mansell AL, Graziano JH, Mellins RB. The effect of transfusion on lung capacity and material oxygen saturation in patients with β-thalassemia major. Pediatr Res. 1986;20:20–9.
Koren A, Garty I, Antonelli D, Katzuni E. Right ventricular cardiac dysfunction in β-thalassemia major. Am J Dis Child. 1987;141:93–6.
Du ZD, Roguin N, Milgram E, Saab K, Koren A. Pulmonary hypertension in patients with thalassemia major. Am Heart J. 1997;134:532–7.
Chotivittayatarakorn P, Seksarn P, Pathmanand C, Thisyakorn C, Sueblinvong V. Cardiac dysfunction in β-thalassemic children. J Med Assoc Thai. 1993;76:591–6.
Eriksson P, Kallin B, Van’t Hoofi FM. Allele specific increase in basal transcription of the plasminogen activator inhibitor-1 gene is associated with myocardial infarction. Proc Natl Acad Sci USA. 1995;92(6):1851–5.
Kim HS, Hwang KY, Chung IK. Tissue plasminogen activator inhibitor type 1 gene polymorphism in patients with gastric ulcer complicated with bleeding. J Korean Med Sci. 2003;18:58–64.
Boerma M, Forsberg L, Van Zeisl L. A genetic polymorphism in connexin 37 as a prognostic marker for atherosclerotic plaque development. J Intern Med. 1999;246:211–8.
Giardina PJ, Kleinert DA, Salamon OS. Magnetic resonance brain imaging in regularly transfused β-thalassemia major and intermedia subjects and prevalence of thrombophilia mutations. Blood. 2012;120:5175.
Acknowledgments
Statistician, Departmentt of Community Medicine, Armed Forces Medical College, Pune-40.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Kumar, A., Batra, H.S., Banerjee, M. et al. PAI-1 Study in Thalassemia Major Patients Receiving Multiple Blood Transfusion. Ind J Clin Biochem 32, 343–346 (2017). https://doi.org/10.1007/s12291-016-0620-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12291-016-0620-7