
Abstract
Background
With 9.6 million deaths in 2018, cancer remains the second leading cause of death worldwide. Breast cancer is the most deadly type of cancer among females, with 55.2% of crude incidence rate and 16.6% of crude mortality rate.
Purpose
The present study was aimed to investigate the anti-breast cancer potential of natural dietary flavonoid, apigenin isolated from Clerodendrum viscosum leaves.
Methods
Apigenin was evaluated for in-depth anticancer activity in MCF-7 cells using cell viability assay, cell cycle analysis, Annexin-V-FLUOS staining, ROS induction, morphological analysis, and western blot analysis.
Results
Apigenin showed selective cytotoxicity on MCF-7 cells with an IC50-56.72 ± 2.35 µM, while negligible cytotoxicity was observed on WI-38 cells. Further, the flow cytometer-based analysis showed that apigenin halted MCF-7 cells in the G2/M phase arrest followed by dose-dependent apoptosis. Moreover, the FACS and confocal microscopy results confirmed the elevation of intracellular ROS and nuclear fragmentation in apigenin-treated MCF-7 cells. Western blots showed up-regulation of cell cycle regulatory proteins, increased p53 expression, Bax/Bcl-2 ratio, activation of caspases, and cleavage of PARP. Finally, apigenin treatment in the presence of Pifithrin-µ showed decreased apoptotic population and it was further confirmed through western blotting study. The results revealed the vital role of p53 in apigenin-induced apoptosis in MCF-7 cells.
Conclusions
In the present findings, treatment of apigenin-induced intracellular ROS in MCF-7 cells followed by induction of G2/M phase cell cycle arrest and further apoptosis through the regulation of p53 and caspase-cascade signaling pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is a group of diseases characterized by the growth of abnormal cells beyond their usual boundaries that can then invade adjoining parts and/or spread to other organs of the body. Cancer is the second leading cause of death globally. Nearly 9.6 million deaths were estimated only in 2018 [1]. Breast cancer is the most deadly type of cancer among females. It has 55.2% of crude incidence rate and 16.6% of crude mortality rate globally in 2018 [1]. In India, more than 1.5 lakhs cases of breast cancer were observed during the year of 2018 only. Among the global cancer deaths, 22% of deaths were found only due to tobacco consumption, 15% due to various infections, 10% due to obesity, 5–10% due to inherited genetic defects, and others due to factors like environmental pollutants and ionizing radiation [2]. Chemotherapy is one of the most widely used treatments for cancer. However, this treatment is very expensive and can produce serious side effects on the body. Paclitaxel, a mitotic inhibitor is one of the most used chemotherapeutic drugs for cancer; still, it has several side effects such as hypersensitivity reactions [3], cardiac rhythm disorder [4], neurotoxicity [5], neutropenia [6], and other miscellaneous toxic effects [7]. Because of the increasing risk of various cancers and limited use of chemotherapeutic drugs, the scientific community moves toward the use of phytochemicals from natural resources due to their huge diversity and lesser side effects. Flavonoids are the most important class of plant secondary metabolites and widely found in fruits, leaves, and vegetables. Flavonoids play a variety of biological activities in plants, animals, and bacteria. In plants, flavonoids have been reported for the synthesis of color and aroma of flowers and fruits, which attracts the pollinators and help in the growth and development of the plant [8] The genus Clerodendrum is widely distributed throughout the tropical and subtropical regions of the world. Several Clerodendrum species have been used in traditional medicine and some of them have been studied for antioxidants and anticancer potentials [9, 10]. The Plant Clerodendrum viscosum (Family- Verbenaceae) has been reported for its prominent activities against the treatment of various skin diseases, tumors, intestinal infections, and kidney-related disorders. Previously, we have published the potent antioxidant and free radical scavenging activities of 70% methanolic extract of C. viscosum leaves (CVLME) [11] as well as evaluated the anti-proliferative activities of various fractions isolated from CVLME [12]. In the present study, we have isolated a 4′,5,7-trihydroxyflavone, commonly known as apigenin from ethyl acetate fraction of CVLME and evaluated its in-depth anti-breast cancer activity in MCF-7 cell line.
Materials and methods
Chemicals
Dulbecco’s Modified Eagle’s Medium (DMEM), antibiotics, Amphotericin-B, and sodium bicarbonate were provided by HiMedia Laboratories Pvt. Ltd, Mumbai, India. Fetal bovine serum (FBS) was purchased from HyClone Laboratories, Inc., Utah, USA. Cell Proliferation Reagent (WST-1), Annexin-V-FLUOS staining kit, and polyvinyl difluoride (PVDF) membrane were provided by Roche Diagnostics, Mannheim, Germany. RNAase A, Triton X-100, and 4′,6-diamidino-2-phenylindole (DAPI) were obtained from MP Biomedicals, France. HPLC grade acetonitrile, BCIP/NBT substrate, and Pifithrin-μ were obtained from Merck, Mumbai, India. Non-Fat dry milk was purchased from Mother Dairy, AMUL, India. Anti-Bcl-2 and anti-p53 antibody were purchased from AnaSpec, Inc., USA. Anti-p21 (WAF1, Cip1) antibody was purchased from eBioscience San Diego, USA. Anti-PARP, anti-caspase 3, anti-caspase 9, anti-Bax, and anti-β-actin antibodies were purchased from OriGene Technologies, Inc, Rockville, USA. The alkaline phosphatase-conjugated anti-rabbit secondary antibody was purchased from Cell Signalling Technology Inc, Danvers, Massachusetts, USA. Dichloro-dihydro-fluorescein diacetate (DCFH-DA) was purchased from Alexis Biochemicals, San Diego, USA. Anti-Cdk-1, anti-Cyclin A1, anti-Cyclin B1, and anti-Cdc25C antibodies were provided by Bioss Antibodies Inc., Massachusetts, USA.
Plant material and ethics
The leaves of Clerodendrum viscosum were collected from the near region of Darjeeling and Siliguri in West Bengal, India. According to the International Union for Conservation of Nature (IUCN), the status of C. viscosum has not yet been assessed for the IUCN Red List. The collected sample was authenticated by taxonomist Prof. A. P. Das from the Department of Botany of the University of North Bengal. A sample specimen was stored at the Herbarium of Botany Department, University of North Bengal, India with accession number 9617.
Sample extraction and isolation of compounds
The collected C. viscosum leaves were washed under tap water to eliminate the excess contaminants, further it was shade dried and used for powder preparation. The 5 kg powder was extracted with 70% methanol and prepared the extract of C. viscosum leaves (CVLME- 1.012 g). CVLME was further fractionated according to the previously reported protocol [12]. From the fractionation, we have collected 36.4 g of CVLE; among these, 31 g of CVLE was subjected for compound isolation process through silica gel column chromatography. Increasing hexane: ethyl acetate gradient (100:0 to 0:100) were applied and through this, 11 sub-fractions (EAF1-11) were obtained. Among these, sample EAF7 contains many spots, so the sample was placed for a re-column process for further purification through silica gel (Mesh size 230–400) column chromatography using firstly with hexane: ethyl acetate gradient and then shift to methanol: dichloromethane (DCM) gradient system. At 10% methanol in DCM, two major spots were eluted together. This mixture was further used for the separation of spots through preparative HPLC using a C18 column (diameter: 10 mm, length: 250 mm and particle size: 10 µm) and eluted with a gradient of methanol and 0.01% TFA in water. Two different peaks at retention time 21.6 and 32.8 min respectively were collected using a fraction collector. The major peak of retention time 32.8 min was concentrated separately using a rotary evaporator and stored at 4 °C by naming CE3. We have isolated the 85 mg of CE3 from the CVLE fraction of C. viscosum leaves.
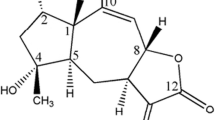
Structure elucidation of the isolated compound
The chemical structure of isolated CE3 was elucidated using liquid chromatography-mass spectrometry (LC–MS), Fourier-transform infrared spectroscopy (FT-IR), and nuclear magnetic resonance (NMR) techniques. Based on these spectroscopic techniques, the compound CE3 was concluded to be apigenin.
In vitro anticancer study of apigenin
Use of cell lines
Both the human breast adenocarcinoma (MCF-7) and normal lung fibroblast (WI-38) cell lines were purchased from National Centre for Cell Sciences (NCCS) Pune, India and supplied with DMEM medium for the proper growth. The media was supplemented with 10% (v/v) Fetal bovine serum (FBS), 100 U/mL of penicillin G, 100 μg/mL of streptomycin, 50 μg/mL of gentamycin sulfate, and 2.5 μg/mL of amphotericin-B. Both the cell lines were maintained in an incubator supplied with 5% CO2 and 37 °C temperature.
Cell cytotoxicity assay
The effect of apigenin on MCF-7 and WI-38 proliferation was evaluated using WST-1 cell proliferation reagent. Briefly, 1 × 104 MCF-7 or WI-38 cells/well were seeded in 96 well cell culture plate and incubated overnight in a CO2 incubator. After incubation, the cells were treated with varying concentrations of apigenin (0–100 μM) and incubated for 24 h. Then, 10 μL of WST-1 reagent was added in each well and again incubated for 2–3 h in a CO2 incubator. The absorbance was recorded at 460 nm on microplate ELISA reader MULTISKAN EX (Thermo Electron Corporation, USA).
Cell cycle analysis of apigenin-treated MCF-7 cells
The cell cycle phase distribution of apigenin-treated MCF-7 cells was performed using a flow cytometer [13]. The MCF-7 cells were seeded in 12 well culture plate and treated with different concentrations of apigenin (0–50 μM) for 24 h. Then the cells were stained with propidium iodide (PI) dye and cell cycle phase distribution was analyzed using a flow cytometer (FACS Verse, Becton Dickinson). The data was analyzed using the FACSuite software Version 1.0.3.2942.
Annexin-V-FLUOS Staining of apigenin-treated MCF-7 cells
As per the manufacturer’s instructions, the cells were seeded and treated with varying concentrations of apigenin (0–50 μM). After incubation, the cells were harvested and labeled with propidium iodide (PI) and FITC stain and analyzed using a flow cytometer (FACS Verse).
Intracellular ROS determination through DCFH-DA staining
Treated and untreated MCF-7 and WI-38 cells were washed with phosphate buffer saline (PBS) and incubated with 20 µM DCFH-DA solution prepared in respective serum-free media. After 30 min of incubation, cells were washed twice with PBS and the intensity of the intracellular ROS was measured using flow cytometer (FACS Verse). Moreover, intracellular ROS intensity in MCF-7 was also observed using a laser scanning confocal microscope and photographed at 400 × magnifications.
DAPI (4′, 6′-diamidino-2-phenylindole) staining
Nuclear fragmentation is one of the key features of apoptosis and it was analyzed using the DAPI staining. The MCF-7 cells were seeded on a coverslip in 6 well culture plate and treated with 0, 30, and 50 µM concentration of apigenin. After the 24 h of treatment, the cells were fixed with 4% paraformaldehyde followed by treatment with 0.5% Triton X-100 for 10 min at room temperature. After proper wash, the cells were stained with DAPI solution (10 μg/mL) and incubated the plates for 30 min. The morphological changes of the apoptotic nuclei were observed at × 63 magnifications under a laser scanning confocal microscope Leica TCS SP8 (Leica, Illinois, United States).
Study of cell cycle arrest and apoptosis-related proteins
The western blotting technique was used to analyze the regulation of cell cycle arrest and apoptosis-related proteins. The MCF-7 cells were treated with 50 µM concentration of apigenin and incubated for varying time intervals (0–24 h). After the treatment, all the cells were harvested and cell lysis was performed using cell lysis buffer (150 mM NaCl, 50 mM Tris–Cl, 0.1% sodium dodecyl sulfate, 0.02% Sodium azide, 1% Triton X-100, 0.5% sodium deoxycholate, 1 µg/mL aprotinin, 100 µg/mL phenylmethylsulfonyl fluoride, pH 8). The cell lysates were centrifuged at 13000 g for 20 min at 4 °C. The protein concentration of the sample was measured by the Folin-Lowry method. 30 μg protein sample was used to resolve on 12% SDS-PAGE and transferred to PVDF membrane using transfer buffer (39 mM glycine, 48 mM Tris base, 0.037% sodium dodecyl sulfate, and 20% methanol). The PVDF membrane was then blocked with TBS solution containing 5% non-fat dry milk and incubated overnight with primary antibodies at 4 °C. The membranes were then washed with TBS-T (0.01% of Tween-20 in TBS) solution and further incubated with alkaline phosphatase-conjugated anti-rabbit IgG secondary antibody for 4 h at room temperature in the dark. Further, the blots were developed using the BCIP/NBT substrate and imaged using EC3 Chemi HR (UVP, USA) imaging system.
Effect of p53 inhibitor on apigenin-induced apoptosis
Pifithrin-µ, a p53 inhibitor, was used to analyze the role of p53 in apigenin-induced apoptosis. The cell proliferation assay using WST-1 reagent and apoptosis study through annexin-V-FLUOS staining was performed with or without Pifithrin-µ treatment (10 µM). Moreover, the protein expression of p53, Bax, cleaved caspase 3, cleaved PARP, and β-actin were analyzed through western blotting upon Pifithrin-µ treatment.
Statistical analysis
The cell cycle and annexin-V-FLUOS staining data were reported as the mean ± SD of 3 measurements. The statistical analysis was determined by KyPlot version 2.0 beta 15 (32 bit). The IC50 values were calculated by the following formula, Y = 100 ∗ A1/(X + A1), where A1 = IC50, Y = response (Y = 100% when X = 0), X = inhibitory concentration. The IC50 values were compared by paired t-test and values with P < 0.05 were considered significant.
Results
Structure elucidation of CE3
LC–MS spectra showed the parent mass (m/z)-270, Melting Point- 345–350 °C. IR spectra were recorded in KBR pellet and the characteristic peaks showed the presence of ester carbonyl group at 1729, aromatic unsaturation at 1652 cm-1. Finally, the 1H 1D NMR, 13C 1D NMR, and DEPT-135 1D NMR technique were used to elucidate the structure of the compound.
NMR data: 1H NMR (500 MHz, DMSO d6)-δ: 6.194 (1H, d, j = 1, Ar-3′H), 6.488 (1H, d, j = 1, Ar-5′H), 6.762 (1H, d, j = 2, Ar-3′H, 5′H), 6.929 (1H, d, C3-H), 7.919 (1H, dd, J = 1.5, J = 1.5, C7-OH, C8), 12.942 (1H, s, C4). 13C NMR (500 MHz, DMSO d6) δ: 94.09 (C-1′), 98.94 (C-3), 102.87 (C-2),103.72 (C-9),116.06 (C-2′, C-6′), 121.22 (C-10), 128.56 (C-3′, C-5′), 157.39 (C-4′), 161.22 (C-6), 161.47 (C-8), 163.86 (C-7), 164.27 (C-5), 181.82 (C-4-CO). Based on these spectroscopic techniques, the compound CE3 was concluded to be a natural trihydroxyflavone, apigenin (Fig. 1).

Structure of apigenin isolated from Clerodendrum viscosum leaves
In vitro anticancer study of apigenin
Cell cytotoxicity assay
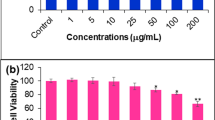
The effect of apigenin on cell proliferation of MCF-7 and WI-38 cells was evaluated through WST-1 assay. The growth of apigenin-treated MCF-7 cells was inhibited dose-dependently. Apigenin inhibited nearly 70% growth of MCF-7 cells in 100 µM concentration (Fig. 2). The IC50 value of apigenin against MCF-7 was 56.72 ± 2.35 µM and more than 500 µM for the WI-38 cell line.

Cytotoxic potential of apigenin on MCF-7 and normal fibroblast cells WI-38. Cell viability was determined with WST-1 cell proliferation reagent. Results were expressed as cell viability (% of control). All data are expressed as mean ± SD (n = 6)
Flow cytometric analysis of cell cycle and apoptosis
The cell cycle distribution of control and apigenin-treated MCF-7 cells was studied using a flow cytometer. The results showed the increase in sub-G1 population (from 0.77% to 15.4%) upon dose-dependent treatment of apigenin. Along with an increase in the sub-G1 population, the G2/M phase population also increased dose-dependently from 19.92% to 32.80% upon 50 µM apigenin treatment (Fig. 3). Also, apigenin-induced apoptosis in MCF-7 cells was analyzed upon annexin-V-FLUOS staining through a flow cytometer and results were depicted in Fig. 4. Annexin-V-FLUOS staining showed 0.03% untreated cells undergoes in apoptosis, while the percentage of apoptotic population increased dose-dependently and 22.72% apoptotic population was observed in 50 µM apigenin treatment.

Flow cytometric cell cycle phase distribution of apigenin-treated MCF-7 cells. Sub G1, G1, S and G2/M phases of MCF-7 cells treated with the indicated doses of apigenin for 24 h. The graphical representation indicates the % cell population in different phases

Flow cytometric plots of Annexin-V-FLUOS and PI staining of apigenin-treated MCF-7 cells with increasing doses. MCF-7 cells were treated for 24 h with different concentrations
Intracellular ROS determination
The intracellular ROS amount of control, apigenin-treated MCF-7 and WI-38 cells were analyzed through flow cytometer upon DCFH-DA staining. As shown in Fig. 5a, the relative intensity of ROS increased with dose-dependent treatment of apigenin in MCF-7 cells while the ROS intensity was constant in control and apigenin-treated WI-38 cells. The results were further confirmed through confocal microscopy, which also showed the absence of ROS amount in control MCF-7 cells while increased ROS amount was observed in apigenin-treated MCF-7 cells (Fig. 5b).

ROS measurement and morphological assessment through flow cytometer and confocal microscopy. a Measurement of ROS amount in apigenin-treated MCF-7 and WI-38 cells through flow cytometer. b Analysis of ROS amount in apigenin-treated MCF-7 cells through a confocal microscope. c Nuclei were stained with DAPI and observed under a confocal microscope in comparison to the untreated cells. The white arrows indicate fragmented nuclei
DAPI (4′, 6′-diamidino-2-phenylindole) staining
Confocal microscopy was used to analyze the nuclear morphology upon DAPI staining. The untreated MCF-7 cells showed intact nuclear morphology, while the fragmented nuclear structure and disturbed morphology were observed in apigenin-treated MCF-7 cells. The result was shown in Fig. 5c.
Study of cell cycle arrest and apoptosis-related proteins
Western blotting was performed to evaluate the expression levels of various cell cycle arrest and apoptosis-related proteins. The results showed the decreased expression levels of cyclin A1, cyclin B1, and cdk-1 upon time-dependent treatment of apigenin in MCF-7 cells. Time-dependent decreased Cdc25C and increased p21 protein expression also suggested the G2/M phase arrest in MCF-7 cells. Furthermore, the up-regulated p53 and Bax while down-regulated Bcl-2 expression was observed upon apigenin treatment. The expression levels of procaspase 9 and 3 were down-regulated and expression levels of cleaved caspase 9 and 3 were up-regulated time-dependently. The cleavage of native PARP also suggested the apoptotic behavior of apigenin in MCF-7 cells while no change in expression of housekeeping gene β-actin upon apigenin treatment (Fig. 6).

Western blot analysis of G2/M phase cell cycle arrest and apoptosis-related proteins of apigenin-treated MCF-7 cells along with graphical representations. The expression of β-actin was used as a protein loading control
Effect of p53 inhibitor on apigenin-induced apoptosis
The cytotoxicity data showed nearly 50% growth inhibition of MCF-7 upon 50 µM apigenin treatment, whereas apigenin treatment along with Pifithrin-µ on MCF-7 cells showed little cytotoxicity (Fig. 7a and b). Further, the annexin-V-FLUOS staining showed 24.18% of apoptotic population in 50 µM apigenin treatment whereas drastically decreased apoptotic population were observed upon 50 µM of apigenin along with 10 µM of Pifithrin-µ treatment (Fig. 7c). Western blot results also confirmed the above results, where the p53 protein is expressed in apigenin and Pifithrin-µ treatment but no change in expression levels of Bax, cleaved caspase 3, and cleaved PARP were observed (Fig. 8).

Effect of p53 inhibitor on apigenin-induced apoptosis. a Cytotoxic potential of apigenin (0–50 µM) on MCF-7 cells. b Cytotoxic potential of apigenin (0–50 µM) along with Pifithrin-µ on MCF-7 cells. Cell viability was determined with WST-1 cell proliferation reagent. c Effects of p53 inhibitor on apigenin-induced apoptosis in MCF-7 cells. The cells were treated with/without Pifithrin-µ in the presence/absence of apigenin (50 μM) for 24 h and analyzed by Annexin-V-FLUOS staining using a flow cytometer. d Graphical representation of Annexin-V-FLUOS staining

Effect of p53 inhibitor on apigenin-induced apoptosis through western blots. The cells were treated with/without Pifithrin-µ in the presence/absence of apigenin (50 μM) for 24 h, proteins were collected and analyzed through western blots
Discussion
Globally, about 1 in 6 deaths is due to cancer, which makes cancer a second leading cause of death worldwide. The most common cancers are Lung (2.09 million cases), Breast (2.09 million cases), Colorectal (1.80 million cases), Prostate (1.28 million cases), Skin cancer (non-melanoma) (1.04 million cases), and Stomach (1.03 million cases) [1]. With 55.2% of crude incidence rate, breast cancer is the most deadly type of cancer in females. Increasing incidences rate and mortality rate of cancer worldwide led the scientist towards natural product research. Medicinal plants remain one of the best reservoirs for new molecules despite the availability of different approaches for the discovery of novel therapeutic agents. Among the natural products, flavonoids are the important class of plant secondary metabolites. Flavonoids have been previously reported for the treatment of various diseases including, cancer, Alzheimer’s disease (AD), and atherosclerosis [14, 15]. Apigenin has been categorized into flavones subgroup of the flavonoids due to the attachment of B ring with the C ring in the structure. The aim of the present report was to evaluate the in-depth anticancer mechanism of apigenin in MCF-7 cells. Although the antioxidant properties of apigenin have been reported previously [16, 17], many reports also showed the potential ROS induction mechanism in various cancer cell lines [18,19,20], which may be due to the dual role of apigenin. Previously, some agents also have been contradictorily regarded as oxidants or antioxidants. For example, emodin has been considered as antioxidant agent before and even now [21], no matter it has been demonstrated to produce a large amount of ROS to kill the cancer cells [22,23,24]. In the present study, we have isolated the apigenin from C. viscosum leaves and studied the anti-breast cancer activity using WST-1 cell cytotoxicity assay, cell cycle phase distribution analysis, annexin-V-FLUOS staining, ROS measurement, DAPI staining and western blot analysis. The selective targeting of cancer cells is the major approach in anticancer drug development. Several studies have reported the cytotoxic nature of apigenin against various cancer cell lines, including ovarian, colon, cervical, hepatoma, as well as breast carcinomas [18,19,20, 25, 26]. Cell viability results of the present study also demonstrated the selective cytotoxic effect of apigenin towards MCF-7 cell line (IC50-56.72 ± 2.35 µM), while it does not hamper the proliferation of normal fibroblast cell line WI-38 (Fig. 2). It is well reported that anticancer drugs target cancer cells through either apoptosis, necrosis, or cell cycle arrest mechanism [27]. The hypodiploid nuclei from apoptotic cells are detected using propidium iodide (PI), a DNA-binding fluorochrome dye that intercalates in between the bases and a flow cytometer technique determines the content of DNA and therefore the number of apoptotic cells in a population [28]. Flow cytometric analysis was performed to verify the effect of apigenin on cell cycle phases of MCF-7 cells and from the results, it is cleared that apigenin firstly induced G2/M phase arrest of cell cycle and followed by induction of apoptosis (sub-G1 phase) dose-dependently (Fig. 3). Another research group also evaluated the effect of apigenin on cell cycle analysis of OVCAR-3 and SKOV-3 cells (ovarian cancer cells), and found that apigenin induces S phase cell cycle arrest in OVCAR-3 cells and G0/G1 as well as G2/M cell cycle arrest in the SKOV-3 cells [25]. The process of apoptosis is highly regulated and characterized by certain changes in cell morphology, such as loss of plasma membrane integrity and nuclear condensation. Phosphatidylserine (PS) is a membrane phospholipid, generally present at the inner side of the plasma membrane, but during the process of apoptosis, PS is translocated from the inner leaflet to the outer leaflet of the plasma membrane [29]. In this study, we have used the annexin-V-FLUOS staining kit to analyze the apoptotic effect of apigenin on MCF-7 cells. As shown in Fig. 4, the apoptotic population increased upon dose-dependent treatment of apigenin and the highest dose of apigenin (50 µM) showed 22.72% of apoptotic population. Reactive oxygen species (ROS) are the by-products of aerobic metabolism. A balanced amount of ROS in a biological system is essential for regulating normal physiological functions, including cell cycle progression and immune response [30]. Although ROS-elevating anticancer therapies are the contradictory matter of cancer treatment, however, many well-known anticancer drugs induce oxidative stress and target the various cancer cells. For example, doxorubicin, a well-known anticancer drug can induce the accumulation of hydroxyl radicals and ultimately target the cancer cells [31]. Moreover, cisplatin, another well-known anticancer agent, directly causes damage to mitochondrial DNA (mtDNA) and interferes with the DNA replication and consequently induce oxidative stress to target especially ovarian cancer cells [32,33,34]. However, the controversial question is, can ROS-elevating therapy in patients put normal cells at risk? Many research groups have explained the selective pro-oxidant approach to target the cancer cells only [20, 22, 35,36,37]. Generally, normal cells have low amounts of ROS as well as antioxidants relative to cancer cells. Loss or gain of a small amount of ROS may not cause much change on ROS homeostasis in normal cells but may cause oxidative burst in cancer cells since cancer cells already contains high level of basal ROS. We have also evaluated the effect of apigenin on ROS induction in MCF-7 cells through a flow cytometer and confocal microscopy upon DCFH-DA staining. The flow cytometer analysis showed that apigenin treatment-induced ROS in MCF-7 cells (6.96 fold as compare to untreated MCF-7 cells), while no ROS induction was observed in apigenin-treated WI-38 cells. The results were further confirmed through confocal microscopy, where green fluorescence of DCF clearly showed apigenin treatment-induced ROS in MCF-7 cells (Fig. 5a and b). This may be due to the selective pro-oxidant approach of apigenin towards the MCF-7 cells only. The ROS induction also leads to fragmentation of the nuclear DNA and it is the key feature of programmed cell death. The mechanism of apoptosis is mainly characterized by the activation of endogenous endonucleases, particularly the caspase 3-activated DNase (CAD), with subsequent cleavage of nuclear DNA [38]. Change in the nuclear morphology was observed through confocal microscopy upon stained with fluorescent dye DAPI. Figure 5c showed the intact nuclear morphology in untreated MCF-7 cells, while nuclear condensation and fragmentation were observed in apigenin-treated MCF-7 cells. The G2/M cell cycle phase/checkpoint is a potential target for anticancer drug development since it prevents the entry of DNA damaged cells to the mitosis phase [39]. The mechanism of cell cycle is tightly regulated through a complex network of positive and negative cell cycle regulatory molecules such as CDKs and cyclins. It has been reported that cyclin A1/Cdk-1 and cyclin B1/Cdk-1 complexes are crucial for the transition of cell cycle from G2 phase to M phase [40]. Therefore, we examined the effect of apigenin on the expression levels of various cell cycle regulatory proteins in MCF-7 cells. Western blotting results revealed that the expression levels of cyclin A1, cyclin B1, and Cdk-1 was decreased dramatically with the increase in time upon apigenin treatment. Cyclin/Cdk complex is activated by Cdc25C phosphatase during the transition of G2 phase to M phase. The expression of Cdc25C was also down-regulated time-dependently upon apigenin treatment, which indicated the G2/M phase cell cycle arrest. A control over cell cycle progression/arrest depends on many crucial factors among those, p21 is a cyclin-dependent kinase (CDK) inhibitor, is one of those factors that promote cell cycle arrest in response to a variety of stimuli [41]. Increased ROS amount in apigenin-treated MCF-7 cells (Fig. 5a and b) may induce DNA damage (Fig. 5c), which may ultimately activate p21. The increased expression of p21 acts as an inhibitor for cdk and blocks the transition of cell cycle from G2 phase to M phase. The increased protein expression of p21 and decreased protein expression of Cyclin/Cdk also confirmed the dose-dependent G2 to M phase cell cycle arrest in MCF-7 cells upon apigenin treatment. Choi et al. [42] reported the effect of apigenin on G2/M phase cell cycle arrest in SK-BR-3 breast cancer cells, where, 50 μM treatment of apigenin induces a 39.9% increase in G2/M phase cell population upon 72 h of treatment. However, our results revealed that 50 μM apigenin treatment on MCF-7 cells induces a 32.8% increase in G2/M phase cell population along with 15.4% increase in sub-G1 (apoptotic) population upon 24 h of treatment. The tumor suppressor protein, p53, plays a very important role in the mechanism of apoptosis. It has been reported that approximately 50% of human cancers bear a mutation in the p53 gene [43]. Also, it is well established that the p53 interacts with members of the Bcl-2 protein family, including Bax and regulates the process of apoptosis. Bax is the most crucial pro-apoptotic member of the Bcl-2 protein family, mainly present in cytosol in healthy mammalian cells and upon initiation of apoptosis, it undergoes in a conformational shift [44]. In response to external apoptosis stimuli, Bax interacts with the mitochondrial outer membrane and activates the mitochondrial voltage-dependent anion channel (VDAC), which releases cytochrome c and other pro-apoptotic factors from the mitochondria [45]. Bcl‑2 is an anti-apoptotic protein, plays an important role in the regulation of apoptosis. The balance between expression levels of Bax (pro‑apoptotic) and Bcl‑2 (anti‑apoptotic), is very crucial for cell survival and death [46]. As shown in Fig. 6, apigenin treatment showed increased p53 and Bax/Bcl‑2 ratio in MCF-7 cells, which may lead to the release of cytochrome c from the mitochondria. The cytochrome c interacts with Apaf-1 (apoptotic protease activating factor-1) protein and initiator caspase, procaspase 9 and forms apoptosome protein complex. As a result, procaspase 9 converted into cleaved caspase 9, which was analyzed through increased expression of cleaved caspase 9 in apigenin-treated MCF-7 cells. Cleaved caspase 9 further activates effector caspase, caspase 3. The effector molecule then after carry out the controlled cell death by activating PARP. The apigenin-treated MCF-7 cells showed down-regulated expression of procaspase 3 and PARP while up-regulation of cleaved caspase 3 and cleaved PARP. The protein expression of housekeeping gene β-actin was also analyzed and it was constant in all conditions. Moreover, some other groups also tried to investigate the involvement of apoptosis-related proteins in apigenin-treated breast cancer cell lines. Madunic et al. showed the cleavage of PARP protein in MCF-7 and MDA MB-231 cells after apigenin treatment [26]. Another group revealed the involvement of Bax, IκBα, and PARP proteins in apigenin mediated apoptosis in MDA MB-231 cells [47]. Nevertheless, our present report revealed the in-depth apoptosis mechanism of apigenin in MCF-7 cells. As mentioned earlier, apigenin treatment activates p53 in MCF-7 cells. The p53 further interacts with pro-apoptotic proteins and continues the mechanism of apoptosis. To address the involvement of p53 in apigenin mediated apoptosis, MCF-7 cells were pre-treated with a Pifithrin-µ (p53 inhibitor). Apigenin showed an anti-proliferative effect against MCF-7 cells, which was confirmed through cytotoxicity study (Fig. 7a). However, apigenin lost the anti-proliferative ability upon combined treatment of apigenin and Pifithrin-µ (Fig. 7b). Further, the annexin-V-FLUOS staining results also showed the decreased apoptotic population of MCF-7 cells upon combined treatment with apigenin and pifithrin-µ (Fig. 7c). Finally, western blot results depict a clear idea about the role of p53 in apigenin-induced apoptosis where combined apigenin and pifithrin-µ treatment showed increased protein expression of p53 but no activation of further apoptosis-related proteins such as Bax, cleaved caspase 3 and cleaved PARP (Fig. 8). The combined results revealed that the apigenin induces apoptosis in MCF-7 cells through the activation of p53 and the caspase-cascade pathway. The proposed anticancer mechanism of apigenin against the MCF-7 cell line was shown in Fig. 9.

Proposed mechanisms of apigenin-induced G2/M phase cell cycle arrest and apoptosis in MCF-7 cells
Conclusion
In summary, herein, we have reported the anti-breast cancer activity of apigenin isolated from C. viscosum leaves. Our findings indicated the inhibitory effect of apigenin against MCF-7 cells without hampering the normal fibroblast cell line WI-38. In the present report, apigenin treatment induces intracellular ROS in MCF-7 cells, which may be responsible for causing DNA damage and activation of p21. Further, p21 may induce the G2/M phase cell cycle arrest by targeting Cdc25C and cdk/cyclin complex. Moreover, ROS induction is also responsible for the activation of p53 protein followed by activation of the caspase-cascade pathway. The collective results revealed the involvement of ROS, p53, and the caspase-cascade signaling pathway in the apoptosis mechanism of apigenin-treated MCF-7 cells.
References
World Health Organization. Global Cancer Observatory. International agency for research on cancer. 2019. https://gco.iarc.fr. Accessed 05 Nov 2019.
Anand P, Kunnumakkara AB, Sundaram C, Harikumar KB, Tharakan ST, Lai OS, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res. 2008;25:2097–116.
Itoh Y, Sendo T, Hirakawa T, Goromaru T, TakasakibS YH, et al. Role of sensory nerve peptides rather than mast cell histamine in paclitaxel hypersensitivity. Am J Respir Crit Care Med. 2004;169:113–9.
Wilson CR, Sauer JM, Hooser SB. Taxines: a review of the mechanism and toxicity of yew (Taxus spp.) alkaloids. Toxicon. 2001;39:175–85.
Scripture CD, Figg WD, Sparreboom A. Peripheral neuropathy induced by Paclitaxel: recent insights and future perspectives. Curr neuropharmacol. 2006;4:165–72.
Safra T, Menczer J, Bernstein RM, Shpigel S, Matcejevsky D, Inbar MJ, et al. Combined weekly carboplatin and paclitaxel as primary treatment of advanced epithelial ovarian carcinoma. Gynecol Oncol. 2009;114:215–8.
Ajani JA, Dodd LG, Daugherty K, Warkentin D, Ilson DH. Taxol induced soft-tissue injury secondary to extravasation: characterization by histopathology and clinical course. J Natl Cancer Inst. 1994;86:51–3.
Griesbach R. Biochemistry and genetics of flower color. Plant Breed Rev. 2005;25:89–114.
Das AD, Chaudhuri D, Ghate NB, Chatterjee A, Mandal N. Comparative assessment of phytochemicals and antioxidant potential of methanolic and aqueous extracts of Clerodendrum colebrookianum walp. leaf from north-east India. Int J Pharm Pharm Sci. 2013;5:420–7.
Srivasatava N, Patel T. Clerodendrum and health care: an overview. Med Aromat Plant Sci Biotechnol. 2007;1:142–50.
Dey P, Chaudhuri D, Tamang S, Chaudhuri TK, Mandal N. In vitro antioxidant and free radical scavenging potential of Clerodendrum viscosum. Int J Pharm Bio Sci. 2012;3:454–71.
Shendge AK, Basu T, Chaudhuri D, Panja S, Mandal N. In Vitro antioxidant and antiproliferative activities of various solvent fractions from Clerodendrum viscosum leaves. Pharmacogn Mag. 2017;13:344–53.
Ghate NB, Chaudhuri D, Sarkar R, Sajem AL, Panja S, Rout J, et al. An antioxidant extract of tropical lichen, Parmotrema reticulatum, induces cell cycle arrest and apoptosis in breast carcinoma cell line MCF-7. PLoS ONE. 2013. https://doi.org/10.1371/journal.pone.0082293.
Ovando C, Hernandez D, Hernandez E, et al. Chemical studies of anthocyanins: a review. Food Chem. 2009;113:859–71.
Lee Y, Yuk D, Lee J, et al. Epigallocatechin-3-gallate prevents lipopolysaccharide-induced elevation of β-amyloid generation and memory deficiency. Brain Res. 2009;1250:164–74.
Romanova D, Vachalkova A, Cipak L, Ovesna Z, Rauko P. Study of antioxidant effect of apigenin, luteolin and quercetin by DNA protective method. Neoplasma. 2001;48:104–7.
Salehi B, Venditti A, Sharifi-Rad M, Kręgiel D, Sharifi-Rad J, Durazzo A. The Therapeutic Potential of Apigenin. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20061305.
Wang B, Zhao XH. Apigenin induces both intrinsic and extrinsic pathways of apoptosis in human colon carcinoma HCT-116 cells. Oncol Rep. 2017;37:1132–40.
Souza RP, Bonfim-Mendonca PS, Gimenes F, et al. Oxidative stress triggered by apigenin induces apoptosis in a comprehensive panel of human cervical cancer-derived cell lines. Oxid Med Cell Longev. 2017. https://doi.org/10.1155/2017/1512745.
Choi SI, Jeong CS, Cho SY, Lee YS. Mechanism of apoptosis induced by apigenin in HepG2 human hepatoma cells: involvement of reactive oxygen species generated by NADPH oxidase. Arch Pharm Res. 2007;30:1328–35.
Jung HA, Chung HY, Yokozawa T, Kim YC, Hyun SK, Choi JS. Alaternin and emodin with hydroxyl radical inhibitory and/or scavenging activities and hepatoprotective activity on tacrine-induced cytotoxicity in HepG2 cells. Arch Pharm Res. 2004;27:947–53.
Jing X, Ueki N, Cheng J, Imanishi H, Hada T. Induction of apoptosis in hepatocellular carcinoma cell lines by emodin. Jpn J Cancer Res. 2002;93:874–82.
Yi J, Yang J, He R, et al. Emodin enhances arsenic trioxide-induced apoptosis via generation of reactive oxygen species and inhibition of survival signaling. Cancer Res. 2004;64:108–16.
Yang J, Li H, Chen YY, et al. Anthraquinones sensitize tumor cells to arsenic cytotoxicity in vitro and in vivo via reactive oxygen species-mediated dual regulation of apoptosis. Free Radic Biol Med. 2004;37:2027–41.
Tavsan Z, Kayali HA. Flavonoids showed anticancer effects on the ovarian cancer cells: Involvement of reactive oxygen species, apoptosis, cell cycle and invasion. Biomed Pharmacother. 2019. https://doi.org/10.1016/j.biopha.2019.109004.
Vrhovac Madunic I, Madunic J, Antunovic M, et al. Apigenin, a dietary flavonoid, induces apoptosis, DNA damage, and oxidative stress in human breast cancer MCF-7 and MDA MB-231 cells. Naunyn Schmiedebergs Arch Pharmacol. 2018;391:537–50.
Chan KT, Meng FY, Li Q, Ho CY, Lam TS, To Y. Cucurbitacin B induces apoptosis and S phase cell cycle arrest in BEL-7402 human hepatocellular carcinoma cells and is effective via oral administration. Cancer Lett. 2010;294:118–24.
Darzynkiewicz Z, Huang X. Analysis of cellular DNA content by flow cytometry. Curr Protoc Immunol. 2004. https://doi.org/10.1002/0471142735.im0507s60.
Vermes I, Haanen C, Steffens-Nakken H. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51.
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. 2016. https://doi.org/10.1155/2016/4350965.
Kotamraju S, Chitambar CR, Kalivendi SV, Joseph J, Kalyanaraman B. Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endothelial cells: role of oxidant-induced iron signaling in apoptosis. J Biolog Chem. 2002;277:17179–877.
Marullo R, Werner E, Degtyareva N, et al. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetics functions. PLoS ONE. 2013. https://doi.org/10.1371/journal.pone.0081162.
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
Helm CW, States JC. Enhancing the efficacy of cisplatin in ovarian cancer treatment - could arsenic have a role. J Ovar Res. 2009. https://doi.org/10.1186/1757-2215-2-2.
Jing Y, Yang J, Wang Y, et al. Alteration of subcellular redox equilibrium and the consequent oxidative modification of nuclear factor kappaB are critical for anticancer cytotoxicity by emodin, a reactive oxygen species-producing agent. Free Radic Biol Med. 2006;40:2183–97.
Trachootham D, Zhou Y, Zhang H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–52.
Jing YW, Yi J, Chen YY, et al. Dicoumarol alters cellular redox state and inhibits nuclear factor kappaB to enhance arsenic trioxide-induced apoptosis. Acta Biochim Biophys Sin (Shanghai). 2004;36:235–42.
Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–9.
Wang Y, Ji P, Liu J, Broaddus RR, Xue F, Zhang W. Centrosome-associated regulators of the G2/M checkpoint as targets for cancer therapy. Mol Cancer. 2009. https://doi.org/10.1186/1476-4598-8-8.
Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221–34.
Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst). 2016;42:63–71.
Choi EJ, Kim GH. Apigenin causes G(2)/M arrest associated with the modulation of p21(Cip1) and Cdc2 and activates p53-dependent apoptosis pathway in human breast cancer SK-BR-3 cells. J Nutr Biochem. 2009;20:285–90.
Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10.
Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17:3878–85.
Weng C, Li Y, Xu D, Shi Y, Tang H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J Biol Chem. 2005;280:10491–500.
Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–33.
Chen D, Landis-Piwowar KR, Chen MS, Dou QP. Inhibition of proteasome activity by the dietary flavonoid apigenin is associated with growth inhibition in cultured breast cancer cells and xenografts. Breast Cancer Res. 2007. https://doi.org/10.1186/bcr1797.
Acknowledgements
AKS and DC are grateful to the Council of Scientific and Industrial Research (CSIR) Govt. of India and Tapasree Basu is grateful to the University Grants Commission (UGC), Govt. of India for providing the fellowships. The authors are thankful to Dr. Nikhil Baban Ghate for his constant guidance during the experiments and Mr. Ranjit Kumar Das for his technical assistance.
Funding
This research did not receive any specific grant from funding agencies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AKS, DC, TB, and NM declared no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals.
Informed consent
The manuscript does not contain clinical studies or patient data.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shendge, A.K., Chaudhuri, D., Basu, T. et al. A natural flavonoid, apigenin isolated from Clerodendrum viscosum leaves, induces G2/M phase cell cycle arrest and apoptosis in MCF-7 cells through the regulation of p53 and caspase-cascade pathway. Clin Transl Oncol 23, 718–730 (2021). https://doi.org/10.1007/s12094-020-02461-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-020-02461-0