
Abstract
CCN1 is encoded by an extracellular matrix protein-gene that is essential for the proper development of the cardiovascular system and the control of angiogenesis, inflammation, progenitor cell lineage commitment and extracellular matrix protein remodeling during the adult life. High-precision genetic models of tissue-specific gene deletion demonstrated a pivotal role of CCN1 in providing positional information to angiogenic endothelial cells (ECs) during the outgrowth and maturation of nascent blood vessel sprouts, fine-controlling Notch-dependent inter-endothelial cell communications and mediating interaction with inflammatory cells. Some of these pleiotropic activities of CCN1 are unique among proteins of the extracellular matrix. Thus, CCN1 represents a model molecule for investigating and unraveling novel aspects of extracellular protein signaling in vascular development and diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
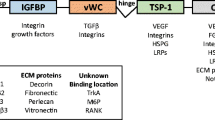
Formation and/or regeneration of blood vessels by sprouting angiogenesis depends on the coordinated actions of both cellular signaling molecules and extracellular growth, guidance and morphogenic signals, which work together to produce highly ordered stable and functional vascular networks (Tung et al. 2012). However, despite the complexity involved in angiomodulation, angiogenesis is centrally driven by a single growth factor, vascular endothelial growth factor (VEGF). VEGF expression and signaling is required at every step of the formation of a new functional vascular branch including the selection of endothelial tip cells and induction of the stalk cell phenotype in adjacent cells, migration and proliferation, mural cell recruitment, fusion of sprouts, lumen formation and pruning of instable vessels (Olsson et al. 2006). VEGF levels and signaling are so important that even VEGF haplo-insufficiency in mice results in embryonic lethality (Haigh et al. 2000). Similarly, slight variations of VEGF signaling results in highly variable cellular responses depending on ligand availability, VEGF receptor regulation at the transcriptional, translational and post-translational levels (e.g., VEGF receptor phosphorylation, internalization, degradation, recycling and dephosphorylation), and the abundance of co-receptors (Bautch 2012; Darland et al. 2011). However, while the VEGF pathway is necessary for angiogenesis, it is far from being sufficient to achieve a functional vasculature. An exquisite network of regulators tightly controls various aspects of VEGF expression and signaling. One such important factor is the matricellular protein CCN1 aka Cyr61.
Lester Lau’s group pioneered “landscape studies” on CCN1 and showed that this matricellular protein exhibits several proangiogenic activities through binding to integrin receptors (e.g., αv subtype) (Chen et al. 2004). These include promoting cell adhesion, migration and survival and potentiating the mitogenic activity of growth factors. Other studies further demonstrated that CCN1 activates angiogenic gene expression such as that of matrix metalloproteinases, αv integrin subunit, and even VEGF (Chen et al. 2001; Zhou et al. 2005). A study by Guillon-Munos et al. demonstrated that CCN1 physically interacts with VEGF and may regulates its bioavailability as well (Guillon-Munos et al. 2011).
Brahim chaqour’s group has recently investigated the crosstalk between CCN1 and VEGF using mouse genetics and the murine retina as a model of postnatal vascular development (Chintala et al. 2015). In rodents, the retina begins to be invested by blood vessels on the day of birth when blood vessels emerge from the optic nerve and subsequently give rise to a system of three vascular plexuses that are interconnected (Chaqour 2013). This angiogenic process occurs without perturbing the retinal architecture in place due to an exquisitely well-orchestrated cross-talk between the neural compartment and invading blood vessels. Subsequently, this coordinated interaction promotes the formation of the neurovascular unit that underlies the blood–retinal barrier and regulates blood flow to match electro-retinal activity.
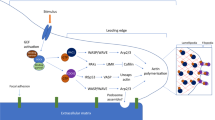
In the retina model, CCN1 is largely expressed by angiogenic EC of the expanding vascular front, precisely in endothelial tip and stalk cells indicating a potential role of CCN1 in the phenotypical plasticity of angiogenic ECs. A precise ratio of EC stalk and tip cells is required for the outgrowth of blood vessels and correct sprouting and branching patterns, a process controlled by VEGF and the Notch signaling pathway (Trindade et al. 2008). The latter involves interaction between adjacent cells, one presenting a Notch ligand, either Jagged or Dll4, and the other exposing a Notch receptor. Interestingly, a study by Koziol et al. has shown that the ability of ECs to acquire a tip cell phenotype is dependent on the matrix-metalloproteinase MMP-14/MT1-MMP and its proteolytic activity vis-à-vis several proteins, including CCN1 (Koziol et al. 2012). Loss of MMP-14 in stimulated ECs impaired tip cell selection blunting capillary sprouting and favoring quiescence of the vasculature. Although CCN1 is, indeed a substrate for MMP-14 as reported by Choi et al. (Choi et al. 2013), the mere involvement of CCN1 in tip cell differentiation and sprout regulation in vivo has not been envisaged. The study by Chintala et al. examined the in vivo function of endothelial CCN1 through generation and characterization of mice with EC-specific deletion of CCN1. Loss of EC CCN1 caused retinal vessels to coalesce into large flat hyperplastic sinuses with subsequent loss of their hierarchical organization. The dysmorphic vasculature was poorly unsheathed with mural cells (e.g., pericytes) and highly permeable which is indicative of a breach of barrier function. The retinal vascular front exhibited a significant increase of the number of endothelial tip cells together with EC hyperplasia at the retinal vascular edge compared with wild-type control retinas. CCN1 deletion caused integrin-dependent downregulation of Dll4 in the numerous tip cells with filipodial extension further demonstrating an important role of CCN1 in Notch-dependent inter-endothelial signaling. Although hypoxia-driven VEGF-VEGF-R2 signaling has been presumed to be the principal inducer of the expression of Dll4 in tip cells (Lobov et al. 2007), recent studies using mouse genetics demonstrated that Dll4 protein expression in retinal tip cells is only weakly modulated by VEGF-R2 signaling suggesting that one or more additional factor(s) is (are) involved (Zarkada et al. 2015). Chintala et al’s study further demonstrated that extracellular effectors such as CCN1 upregulates Dll4 expression and Notch signaling pathway downstream targets such as Hes1 and Hey1. Loss of CCN1 reduced the levels of Dll4 despite a concomitant hyperphosphorylation of specific tyrosine residues on VEGF-R2 owing to disrupted activity of the Src homology 2 domain-containing protein tyrosine phosphatase-1 (SHP-1). The mere absence of CCN1 during the sprouting process altered the expression and activity of downstream VEGF R2 signals suggesting that CCN1 action lies in the organization of a hierarchy in responsiveness and/or receptiveness to VEGF signaling along tip, stalk and phalanx cells.
Additional studies by Yan et al. further demonstrated that ubiquitous deletion of CCN1 altered resident microglia recruitment, distribution, and reactivity in the retina suggesting that non-endothelial effects contribute to the phenotypic differences between the wild-type mice and those with CCN1 deficiency (Yan et al. 2015). Microglial cells are local inflammatory cells that contribute to the initiation and maintenance of inflammatory responses which are of paramount importance in physiological and pathological angiogenesis. Ubiquitous CCN1 deficiency increased the number of microglia in the retina and upregulated the expression of several interleukins including IL-10. The latter is the principal default response of retinal microglia during inflammatory responses. IL-10 is a proangiogenic molecule as IL-10 deficiency in mice blunted neovascular growth in the retina (Dace et al. 2008). Interestingly, IL-10 also controls the activation of other inflammatory cells such as macrophage and their ability to regulate angiogenesis (Apte 2010). Low levels of IL-10 have been associated with “classically activated” macrophages, or M1 macrophages, that display an antiangiogenic phenotype. Thus, CCN1 signaling has far reaching effects on both vascular cells and accessory inflammatory cells. Further investigations of the autocrine and paracrine signaling of CCN1 will elucidate how the integration of multiple signaling networks determines the behavior of ECs, the reactivity of inflammatory cells and the shaping of the vascular network. The valuable information generated thus far from mice with tissue-specific deletion of CCN1 serves as a fertile ground for the basic challenges and therapeutic opportunities that lie ahead (Afzal et al. 2007).
References
Afzal A, Shaw LC, Ljubimov AV, Boulton ME, Segal MS, Grant MB (2007) Retinal and choroidal microangiopathies: therapeutic opportunities. Microvasc Res 74:131–144
Apte RS (2010) Regulation of angiogenesis by macrophages. Adv Exp Med Biol 664:15–19
Bautch VL (2012) VEGF-directed blood vessel patterning: from cells to organism. Cold Spring Harb Perspect Med 2:a006452
Chaqour B (2013) Molecular control of vascular development by the matricellular proteins (CCN1/Cyr61) and (CCN2/CTGF). Trends Dev Biol 7:59–72
Chen CC, Mo FE, Lau LF (2001) The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J Biol Chem 276:47329–47337
Chen N, Leu SJ, Todorovic V, Lam SC, Lau LF (2004) Identification of a novel integrin alphavbeta3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J Biol Chem 279:44166–44176
Chintala H, Krupska I, Yan L, Lau L, Grant M, Chaqour B (2015) The matricellular protein CCN1 controls retinal angiogenesis by targeting VEGF, Src homology 2 domain phosphatase-1 and notch signaling. Development 142:2364–2374
Choi J, Lin A, Shrier E, Lau LF, Grant MB, Chaqour B (2013) Degradome products of the matricellular protein CCN1 as modulators of pathological angiogenesis in the retina. J Biol Chem 288:23075–23089
Dace DS, Khan AA, Kelly J, Apte RS (2008) Interleukin-10 promotes pathological angiogenesis by regulating macrophage response to hypoxia during development. PLoS One 3:e3381
Darland DC, Cain JT, Berosik MA, Saint-Geniez M, Odens PW, Schaubhut GJ, Frisch S, Stemmer-Rachamimov A, Darland T, D'Amore PA (2011) Vascular endothelial growth factor (VEGF) isoform regulation of early forebrain development. Dev Biol 358:9–22
Guillon-Munos A, Oikonomopoulou K, Michel N, Smith CR, Petit-Court S, Canepa P, Reverdiau N, Heuze-Vourc'h EPD, Courty Y (2011) Kallikrein-related peptidase 12 hydrolyzes matricellular proteins of the CCN family and modifies interactions of CCN1 and CCN5 with growth factors. J Biol Chem 286:25505–25518
Haigh JJ, Gerber HP, Ferrara N, Wagner EF (2000) Conditional inactivation of VEGF-A in areas of collagen2a1 expression results in embryonic lethality in the heterozygous state. Development 127:1445–1453
Koziol A, Gonzalo P, Mota A, Pollan A, Lorenzo C, Colome N, Montaner D, Dopazo J, Arribas J, Canals F, Arroyo AG (2012) The protease MT1-MMP drives a combinatorial proteolytic program in activated endothelial cells. FASEB J 26:4481–4494
Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ (2007) Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci U S A 104:3219–3224
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L (2006) VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 7:359–371
Trindade A, Kumar SR, Scehnet JS, Lopes-da-Costa L, Becker J, Jiang W, Liu R, Gill PS, Duarte A (2008) Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 112:1720–1729
Tung JJ, Tattersall IW, Kitajewski J (2012) Tips, stalks, tubes: notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. Cold Spring Harb Perspect Med 2:a006601
Yan L, Lee S, Lazzaro DR, Aranda J, Grant MB, Chaqour B (2015) Single and compound knock-outs of MicroRNA (miRNA)-155 and its angiogenic Gene target CCN1 in mice Alter vascular and neovascular growth in the retina via resident microglia. J Biol Chem 290:23264–23281
Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K (2015) VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci U S A 112:761–766
Zhou D, Herrick DJ, Rosenbloom J, Chaqour B (2005) Cyr61 mediates the expression of VEGF, alphav-integrin, and alpha-actin genes through cytoskeletally based mechanotransduction mechanisms in bladder smooth muscle cells. J Appl Physiol 98:2344–2354
Acknowledgments
This work was supported by grant from the National Eye Institute of the National Institutes of Health EY022091-01 and Research for the Prevention of Blindness Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chaqour, B. Regulating the regulators of angiogenesis by CCN1 and taking it up a Notch. J. Cell Commun. Signal. 10, 259–261 (2016). https://doi.org/10.1007/s12079-016-0328-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-016-0328-8