
Abstract
Alcoholic steatosis, instead of being innocuous, plays a critical role in liver inflammation and fibrogenesis. The severity of fatty liver is governed by the concerted balance between lipid transport, synthesis, and degradation. Whereas scavenger receptor class B, type I (SR-B1) is critical for reverse cholesterol uptake by the liver, peroxisome proliferator-activated receptor-gamma (PPARγ) coactivator-1α and -β (PGC1α and PGC1β) are critical for lipid degradation and synthesis, respectively. Because betaine is a lipotropic agent, we have evaluated its effects on alcoholic steatosis. Betaine effectively prevented chronic alcohol-mediated (i) impaired SR-B1 glycosylation, plasma membrane localization, and consequent impaired cholesterol transport; and (ii) up regulation of PGC-1β, sterol regulatory element-binding protein 1c and downstream lipogenic genes with concomitant increased liver cholesterol, triglycerides and hepatic lipid score. Similarly, because of its anti-inflammatory and anti-fibrotic effects in other organs, we evaluated the protective effects of thymosin β4 (Tβ4) against carbon tetrachloride (CCl4)-induced hepatotoxicity in rat. Tβ4 prevented CCl4-induced (i) necrosis, inflammatory infiltration and up-regulation of α1(2)collagen, alpha-smooth muscle actin (α-SMA), platelet derived growth factor beta (PDGF-β) receptor and fibronectin mRNA expression; (ii) down-regulation of adipogenic gene, PPARγ and the up-regulation of epigenetic repressor gene, methyl CpG binding protein 2 (MeCP2) mRNA levels, suggesting that the anti-fibrogenic actions of Tβ4 involve the prevention of trans-differentiation of quiescent hepatic stellate cells into myo-fibroblasts largely by up-regulating PPARγ and by down-regulating MeCP2 genes. We therefore conclude that betaine and Tβ4 can effectively protect against alcoholic hepatosteatosis and hepatic fibrogenesis, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hepatic steatosis, inflammation and fibrogenesis
Since hepatotoxins lead to major liver injury, the authors wish to address the following key questions on the current status of (i) How chronic alcohol exposure manifests hepatosteatosis, and how does dietary betaine prevent this alcoholic liver pathology?, and (ii) How does carbon tetrachloride exposure lead to hepatic fibrogenesis and how thymosin-β4 (Tβ4), a small mammalian peptide produced by thymus gland, protects against this injury. Thus, the present review summarizes our ongoing and published studies [1] with special emphasis on the therapeutic potentials of these two natural compounds, namely, betaine, a lipotropic nutrient, and Tβ4 for the treatment of liver injury.
Hepatosteatosis
Numerous studies have established that chronic alcohol exposure leads to: (i) increased adipose fat mobilization into the liver due to increased adipose lipoprotein lipase [2], (ii) increased fat synthesis due to up-regulation of lipogenic genes via PGC-1β and SREBP1c [3, 4], (iii) decreased fat oxidation due to down-regulation of fatty acid oxidation genes via PGC-1α and PPARα [3, 4] and (iv) impaired synthesis of apolipoprotein B and VLDL secretion [5], the major lipoprotein for the export of hepatic lipids to peripheral tissues. Significantly, PPARα and SREBP1c are tightly controlled by two transcription coactivators, PGC-1α and PGC-1β, respectively [6–8]. Silence regulator gene (SIRT) inactivates PGC-1α by deacetylation, whereas histone acetyltransferases (HAT) activate PGC-1α by acetylation [9], which in concert with PPARα increases fatty acid oxidation. On the contrary, SREBP1c is stabilized by HAT by acetylation and destabilized by SIRT by deacetylation. Dietary saturated fat up-regulates PGC-1β and SREBP1c, which coactivates LXR families of transcription factors causing increased lipogenesis, lipid transport and VLDL secretion [10].
Scavenger receptor class B, type I (SR-B1)
It is well known that the liver is the major site for plasma HDL cholesteryl ester uptake and degradation into bile acids [11]. Cholesterol uptake by the liver is crucial for maintaining cholesterol homeostasis in peripheral tissues of mammals as exemplified by early onset of atherosclerosis in familial hypercholesterolemia [12] and Tangier disease [13]. HDL levels are inversely correlated with incidence of atherosclerosis partly because of the ability of HDL to return extra hepatic cholesterol to the liver for conversion into bile acids and secretion into bile in a process termed reverse cholesterol transport [14]. In this process, SR-B1 plays the major role in the direct uptake of HDL cholesterol by the liver [15]. To a limited extent, HDL2 cholesterol can be taken by the liver via the ApoB/E receptor or be transferred to apolipoprotein B containing particles via cholesteryl ester transfer protein (CETP) and subsequent uptake by the LDL receptor (LDLR).
SR-B1 is an approximately 82 kDa membrane glycoprotein belonging to the CD36 family of transmembrane proteins [16]. SR-B1 mediates the cellular uptake of HDL-derived cholesterol and cholesteryl ester in excess of the uptake of HDL-derived apolipoproteins such as ApoA1 and ApoA2 in a process called “selective uptake” [16, 17]. The importance of SR-B1 in the uptake of HDL cholesterol and the antiatherogenic role of SR-B1 has been studied in SR-B1 deficient mouse models [18, 19]. An elegant study [20] using SR-B1 knockout mice showed that SR-B1 is the sole molecule mediating the selective uptake of cholesterol esters from HDL by the liver. Using an in vivo model, van der Velde et al. [21] have confirmed the central role of SR-B1 in reverse cholesterol transport (RCT). Our ongoing finding is that ethanol-mediated decrease in sphingomyelin (SM) composition of HDL [22] may also influence the function of SR-B1.
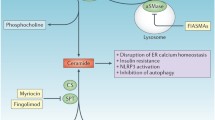
Betaine, a potent lipotropic nutrient, plays an important role in reducing fatty liver [23]. Therefore, it is reasonable that betaine may prevent the deleterious effects of heavy alcohol and high omega-3 polyunsaturated fatty acids (ω-3 PUFA) on SR-B1, plasma lipids and hepatic lipid metabolizing pathway and lipid homeostasis by altering hepatic GSH and reactive oxygen species (ROS) (Fig. 1).

As shown, the vast majority of ethanol is oxidized to acetaldehyde by the hepatocytes of the liver. On the other hand, ethanol-induced cytochrome P4502E1 (CYP2E1) mediated oxidation of ethanol also produces a state of oxidative stress by generating reactive oxygen species (ROS) within the cells that is responsible for the progression of alcoholic fatty liver and liver disease. Chronic ethanol can also activate Kupffer cells to induce TNFα leading to the generation of more ROS in the hepatocytes. One of the key metabolites generated due to oxidative stress is α,β-unsaturated aldehyde, 4-hydroxy-2-nonenal (HNE) that may be more harmful than ROS because it has a longer half-life and can easily diffuse into cellular membranes. Thus, ethanol/ROS mediated down regulation of ST6Gal1 markedly represses SRB1 glycosylation, its cholesterol transport function, as well as depletes liver GSH, the natural intracellular antioxidant. In contrast, betaine, by virtue of its lipotropic property, restores phosphatidyl choline synthesis, and intracellular GSH that attenuates the deleterious effects of ROS
In view of the above, we have explored the possible action of a chronic heavy alcohol/high PUFA diet and the protective role of betaine on (i) hepatic SR-B1 expression, and relative glycosylation rate, and (ii) the expression of various lipogenic genes and hepatic lipid status.
Inflammation and fibrogenesis
Liver injury, regardless of its origin, typically induces hepatocyte necrosis and apoptosis. Necrosis engages classic inflammatory and fibrogenic signals [24]. Liver damage can be caused by viral infection, auto-immune disorders, ischemia, and several xenobiotics, including drugs, alcohol or toxins [25]. Carbon tetra-chloride (CCl4)-induced acute liver injury model is widely used to investigate the mechanisms of liver damage and regeneration [26]. Treatment with CCl4, a known hepatotoxin, stimulates experimental acute liver failure through free radical-mediated wide peroxide injuries [26]. This treatment is accompanied by extensive necrosis and inflammation [27]. Even though during acute liver damage there is no fibrosis, there is activation of hepatic stellate cells (HSC) [28, 29]. HSC are the main fibrogenic cells of the injured liver. In their normal (quiescent) stage they mainly produce an extracellular matrix (ECM) present in basement membranes such as type IV collagen [30]. They store vitamin A and triglycerides and express regulators of the adipocyte phenotype such as peroxisome proliferator-activated receptor (PPARγ), sterol regulatory element binding-protein1 (SREBP-1c) and methyl-CpG binding protein 2 (MeCP2) among others [31, 32]. In the fibrotic liver, HSC undergo trans-differentiation from lipid-storing pericytes to myofibroblastic cells. This activation requires coordinated changes in activity of several growth factors such as the platelet-derived growth factor (PDGF) and the transforming growth factor β1 (TGFβ1) [28, 33]. Specifically, PDGF is the most potent proliferative cytokine acting on HSC [33]. Activated HSC show significant alterations at gene expression, where expression of PPARγ and SREBP-1c is down regulated [34], while expression of MeCP2 is up-regulated [32]. HSC lose the retinoid-binding proteins and their vitamin A stores [35]. The activated HSC are proliferative, proinflammatory and fibrogenic with induced ability to synthesize and deposit large amounts of ECM proteins [29, 30] (Fig. 2). Also, activated HSC overexpress genes that confer the myofibroblastic phenotype such as collagens I and III, fibronectin and the de novo synthesis of α-smooth muscle actin [29, 30, 35]. Thus, a better understanding of the mechanism underlying HSC transdifferentiation is a pivotal step towards identification of molecular targets to develop new liver damage therapeutic treatments.

HSC are perisinusoidal cells of the liver that store vitamin A and undergo phenotypic trans-differentiation characterized as “myofibroblastic activation” during liver fibrogenesis. Activated HSC lose the vitamin A stores and express cytokine receptors like PDGF-β receptor. Moreover, they acquire a contractile cytoskeleton and express α-SMA, which are the markers of HSC trans-differentiation. They also show the down-regulation of the adipogenic PPARγ and up-regulation of its transcriptional repressor MeCP2. The activated myofibroblasts then, migrate and proliferate to the site of injury and form a fibrous scar. In addition, they also deposit ECM proteins such as collagen I, III, IV and fibronectin
Thymosin β4 (Tβ4) is a 43 amino acid polypeptide that was initially isolated from calf thymus [36]. It is a component of a family of approximately 15 members with a highly conserved amino acid sequence [37]. Interestingly, Tβ4 prevents inflammation and fibrosis, promoting healing in the eye, skin and heart [38–41]. In the eye, it promotes corneal re-epithelization after skin injury. It also inhibits the strong inflammatory component that occurs after injury with NaOH [37, 38]. Overall, it prevents inflammation by blocking the secretion of inflammatory cytokines and suppressing the activation of NFκB [42]. In the heart, it prevents the formation of scar tissue after a myocardial infarction by enhancing the survival of myocardial tissue and endothelial cells, thus sustaining cardiac function and preventing scar formation [40, 41]. Recently, it was shown that Tβ4 inhibits the appearance of myofibroblast (Mybs) in a model system of wound healing [43]. Our previous studies have revealed that rat HSC clones derived from cirrhotic rat liver express Tβ4 [44]; moreover, the addition of Tβ4 to HSC/Mybs cultures inhibits PDGF-β receptor expression and prevents binding of AKT to actin and its phosphorylation by PDK1 and mTOR [45]. Based on these findings, we believe Tβ4 could have therapeutic properties to prevent liver injury. Therefore, we have investigated the potential of Tβ4 to inhibit liver damage induced with CCl4 in an in vivo model.
Results and discussion
Betaine and hepatosteatosis
The present review summarizes our ongoing investigation (full manuscript being published in The American Journal of Pathology 2014) demonstrating that chronic ethanol exposure markedly increased liver cholesterol and triglycerides with a concomitant 260 % (p < 0.01) increase in hepatic lipid score that was significantly blunted by betaine. Furthermore, chronic ethanol markedly inhibited the relative glycosylation of SR-B1 with a concomitant impaired hepatic cholesterol uptake that was alleviated by betaine. Since chronic ethanol is known to cause increased ROS [46], especially in the presence of high polyunsaturated fat, the possible mechanism of this protective action of betaine seems to be due to its ability to restore the hepatic intracellular antioxidant, GSH, that is markedly decreased by chronic alcohol, which could induce apoptosis in liver due to stressed mitochondria and endoplasmic reticulum. Again, the fact that betaine treatment essentially corrects these defects suggests that membrane integrity of mitochondria was essentially restored by betaine treatment.
Furthermore, our results on the action of chronic ethanol on the hepatic lipid metabolic signaling pathways clearly showed that, whereas chronic ethanol up-regulated PGC-1β, SREBP1c and the downstream lipogenic genes, it down-regulated PGC-1α and downstream lipid oxidizing genes resulting in impaired hepatic lipid oxidation. Significantly, dietary betaine supplementation markedly reversed the effects of chronic ethanol on these lipid signaling pathways. These mechanistic findings point out that the possible mechanisms of action of betaine in protecting against alcoholic hepatosteatosis involve its ability to not only prevent chronic alcohol-mediated up-regulation of PGC-1β and lipogenic genes, but also the restoration of PGC-1α and lipid oxidizing genes resulting in near-normal hepatic lipid score found in betaine-supplemented chronic ethanol-fed animals in spite of feeding a very high PUFA fat diet.
Thymosin β4, inflammation and fibrogenesis
Acute and chronic liver diseases constitute a global concern. At present, there is no approved therapy to treat these diseases even in the developed world. Therefore, intensive research in finding effective therapeutic agents is highly relevant. CCl4 mediated liver injury is probably the most reproducible model for screening various potentially beneficial compounds for their hepatoprotective activity. Even a single dose CCl4 exposure can lead rapidly to a severe hepatic necrosis, steatosis and portal inflammation [47, 48]. We are pleased to reproduce the pertinent figures describing the results of this portion of this review thanks to the kind permission from the editorial office of our recent publication on the anti-fibrogenic actions of Tβ4 [1]. Thus, we show that Tβ4 preserved the hepatocellular membrane and suppressed CCl4-induced liver injury by the reduction of the infiltration of inflammatory cells, necrosis and microvascular steatosis observed during histological analysis (Fig. 3). We further showed that Tβ4 prevented CCl4,-induced activation of HSC. It is well known that upon liver injury, HSC proliferate and differentiate into myofibroblast-like cells. The activated HSC undergo continuous proliferation and express activation markers such as α-SMA and produce large amounts of ECM proteins, including type I collagen [29, 30]. One of the key events in the activation of HSC is the expression of the PDGF-β receptor [33]. We clearly showed that Tβ4 effectively blocked CCl4-induced up-regulation of α-SMA, PDGF-β receptor and collagen type I expression showing thereby it prevented the activation of HSC and consequent liver injury. Moreover, Tβ4 also inhibits the CCl4-induced down-regulation of PPARγ and the up-regulation of MeCP2 mRNA levels, indicating that the mechanism of action of Tβ4 in reducing liver damage may be through the inactivation of HSC (Fig. 4). We also demonstrated in vitro that Tβ4 prevented HSC/Mybs transdifferentiation, proliferation and migration [45].

Hematoxylin and eosin staining of liver sections from a rats treated with CCl4 with or without Tβ4 at a and b 0, c–f 24, and g and h 48 h, respectively. Panels c and e show the presence of portal inflammation, centrizonal necrosis and distortion of liver around portal triads, vacuole generation and microvascular steatosis, 24 h after CCl4 treatment. As shown in panels d and f, Tβ4 prevented histological changes in CCl4-treated rat livers

Quantitative RT-PCR analysis of a PPARγ, b MeCP2, c PDGF-β receptor, d α-SMA, e collagen 1α2, and f fibronectin mRNA. Total RNA was extracted from whole livers of rats treated with either CCl4 or CCl4 plus Tβ4 at 1 mg/kg body weight in various time points indicated in the figure. All the values are means of triplicate experiments and they were corrected with GAPDH mRNA expression
Conclusions
Based on our ongoing and published studies [1] on the possible protective actions of betaine and Tβ4 on liver injury caused by hepatotoxins such as ethanol and carbon tetrachloride, we draw the following conclusions: (1) chronic alcohol leads to impaired cholesterol homeostasis in the liver resulting in hepatosteatosis. The possible mechanisms of action of alcohol involve the (i) impaired cholesterol uptake by the liver due to decreased relative glycosylation and localization on the liver plasma membrane of the mature SR-B1, the key liver receptor for reverse-cholesterol uptake from plasma HDL, (ii) up-regulation of PGC-1β, SREBP1c and downstream lipogenic genes, and (iii) down-regulation of SIRT1, PGC-1α and downstream lipid oxidation pathway genes and fatty acid oxidation. (2) Betaine counteracts the above actions of ethanol, presumably by quenching the ROS by restoring reduced GSH, the endogenous antioxidant and lipogenic and lipid oxidizing signaling genes, and thus prevents hepatosteatosis as well as maintains normal reverse-cholesterol transport. (3) Carbon tetrachloride markedly causes hepatic fibrogenesis in vivo by activating hepatic stellate cells essentially by down-regulating adipogenic transcription factor PPARγ expression and up-regulating the epigenetic repressor, MeCP2. Additionally, Tβ4 also seems to exert anti-inflammatory actions. Thus, we suggest that betaine and Tβ4 can effectively protect against alcoholic hepatosteatosis and hepatic fibrogenesis, respectively.
References
Reyes-Gordillo K, Shah R, Arellanes-Robledo J, Rojkind M, Lakshman MR. Protective effects of thymosin β4 on carbon tetrachloride-induced acute hepatotoxicity in rats. Ann NY Acad Sci 2012;1269:61–68
Baraona E, Leo MA, Borowsky SA, Lieber CS. Pathogenesis of alcohol-induced accumulation of protein in the liver. J Clin Invest 1977;60:546–554
Lieber CS, Leo MA, Wang X, Decarli LM. Effect of chronic alcohol consumption on hepatic SIRT1 and PGC-1alpha in rats. Biochem Biophys Res Commun 2008;370:44–48
You M, Cao Q, Liang X, Ajmo JM, Ness GC. Mammalian sirtuin 1 is involved in the protective action of dietary saturated fat against alcoholic fatty liver in mice. J Nutr 2008;138:497–501
Lakshman MR, Chirtel SJ, Chambers LC, Campbell BS. Hepatic synthesis of apoproteins of very low density and high density lipoproteins in perfused rat liver: influence of chronic heavy and moderate doses of ethanol. Alcohol Clin Exp Res 1989;13:554–559
Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 2009;20:98–105
Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002;109:1125–1131
Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interact with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem 2005;280:16456–16460
Lakshman MR. Some novel insights into the pathogenesis of alcoholic steatosis. Alcohol 2004;34:45–48
Sozio M, Crabb DW. Alcohol and lipid metabolism. Am J Physiol Endocrinol Metab 2008;295:E10–E16
Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci USA 1983;80(17):5435–5439
Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1992;1(6):445–466
Attie AD, Kastelein JP, Hayden MR. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J Lipid Res 2001;42:1717–1726
Tall AR. An overview of reverse cholesterol transport. Eur Heart J 1998;19(Suppl A):A31–A35
Silver DL, Jiang XC, Arai T, Bruce C, Tall AR. Receptors and lipid transfer proteins in HDL metabolism. Ann NY Acad Sci 2000;902:103–111; discussion 111–112
Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996;271(5248):518–520
Hauser H, Dyer JH, Nandy A, Vega MA, Werder M, et al. Identification of a receptor mediating absorption of dietary cholesterol in the intestine. Biochemistry 1998;37(51):17843–17850
Arai T, Wang N, Bezouevski M, Welch C, Tall AR. Decreased atherosclerosis in heterozygous low density lipoprotein receptor-deficient mice expressing the scavenger receptor BI transgene. J Biol Chem 1999;274(4):2366–2371
Braun A, Trigatti BL, Post MJ, Sato K, Simons M, et al. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res 2002;90(3):270–276
Out R, Hoekstra M, Spijkers JAA, Kruijt JK, van Eck M, Bos IST, et al. SRB1 is solely responsible for selective uptake of cholesteryl esters from HDL by the liver and the adrenals in mice. J Lipid Res 2004;45:2088–2095
van der Velde AE, Groen AK. Shifting gears: liver SRB1 drives reverse cholesterol transport in macrophages. J Clin Invest 2005;115:2699–2701
Marmillot P, Munoz J, Patel S, Garige M, Rosse RB, Lakshman MR. Long-term ethanol consumption impairs reverse cholesterol transport function of high density lipoproteins by depleting HDL sphingomyelin in both rats and humans. Metabolism 2007;56:947–953
Barak AJ, Beckenhauer HC, Badakhsh S, Tuma DJ. The effect of betaine in reversing alcoholic steatosis. Alcohol Clin Exp Res 1997;21:1100–1102
Cohen-Naftaly M, Friedman SL. Current status of novel antifibrotic therapies in patients with chronic liver disease. Ther Adv Gastroenterol 2011;4:391–417
Kim HY, Kim JK, Choi JH, Jung JY, Oh WY, et al. Hepatoprotective effect of pinoresinol on carbon tetrachloride-induced hepatic damage in mice. J Pharmacol Sci 2010;112(1):105–112
Muriel P. Peroxidation of lipids and liver damage. In Baskin S, Salem H, editors. Oxidants and Free Radicals. Washington: Taylor & Francis; 1997. pp 237–257
Reyes-Gordillo K, Segovia J, Shibayama M, Vergara P, Moreno MG, Muriel P. Curcumin protects against acute liver damage in the rat by inhibiting NF-kappaB, proinflammatory cytokines production and oxidative stress. Biochim Biophys Acta 2007;1770(6):989–996
Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol 1999;30(1):77–87
Safadi R, Friedman SL. Hepatic fibrosis—role of hepatic stellate cell activation. MedGenMed 2002;4:27
Rojkind M, Reyes-Gordillo K. Hepatic stellate cells. In: Arias IMWAW, Boyer JE, Cohen DE, Shafritz DA, Fausto N, Alter HJ, editors. The Liver Biology and Pathobiology. Wiley Blackwell: Oxford; 2009. pp 407–432
Tsukamoto H, Zhu NL, Asahina K, et al. Epigenetic cell fate regulation of hepatic stellate cells. Hepatol Res 2011;41:675–682
Mann J, Oakley F, Akiboye F, et al. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ 2007;14:275–285
Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol 2011;6:425–456
She H, Xiong S, Hazra S, et al. Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem 2005;280:4959–4967
Mann J, Mann DA. Transcriptional regulation of hepatic stellate cells. Adv Drug Deliv Rev 2009;61:497–512
Badamchian M, Damavandy AA, Damavandy H, et al. Identification and quantification of thymosin beta4 in human saliva and tears. Ann NY Acad Sci 2007;1112:458–465
Goldstein AL, Hannappel E, Kleinman HK. Thymosin beta4: actin-sequestering protein moonlights to repair injured tissues. Trends Mol Med 2005;11:421–429
Dunn SP, Heidemann DG, Chow CY, et al. Treatment of chronic nonhealing neurotrophic corneal epithelial defects with thymosin beta 4. Arch Ophthalmol 2010;128:636–638
Philp D, Goldstein AL, Kleinman HK. Thymosin beta4 promotes angiogenesis, wound healing, and hair follicle development. Mech Ageing Dev 2004;125:113–115
Bock-Marquette I, Saxena A, White MD, et al. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature 2004;432:466–472
Smart N, Risebro CA, Melville AA, et al. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007;445:177–182
Sosne G, Qiu P, Kurpakus-Wheater M, et al. Thymosin beta4 and corneal wound healing: visions of the future. Ann NY Acad Sci 2010;1194:190–198
Ehrlich HP, Hazard SW 3rd. Thymosin beta4 enhances repair by organizing connective tissue and preventing the appearance of myofibroblasts. Ann NY Acad Sci 2010;1194:118–124
Barnaeva E, Nadezhda A, Hannappel E, et al. Thymosin beta4 upregulates the expression of hepatocyte growth factor and downregulates the expression of PDGFbeta receptor in human hepatic stellate cells. Ann NY Acad Sci 2007;1112:154–160
Reyes-Gordillo K, Shah R, Popratiloff A, et al. Thymosin-beta4 (Tbeta4) blunts PDGF-dependent phosphorylation and binding of AKT to actin in hepatic stellate cells. Am J Pathol 2011;178:2100–2108
Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol 2009;83:519–548
Manibusan MK, Odin M, Eastmond DA. Postulated carbon tetrachloride mode of action: a review. J Environ Sci Health 2007;25:185–209
Recknagel RO, Glende EA Jr, Dolak JA, et al. Mechanisms of carbon tetrachloride toxicity. Pharmacol Ther 1989;43:139–154
Acknowledgements
This work is supported by the NIH grants R21 AA017965 (MRL), RO1 AA020720 (MRL) and RO1 10541 (MRL). The synthetic Tβ4 used for this work was a kind gift from RegeneRx Biopharmaceuticals, Inc.
Compliance with ethical requirements and Conflict of interest
This article does not contain any studies with human or animal subjects performed by any of the authors without the Institutional Human or Animal studies subcommittees’ approval. M. Raj Lakshman, Karina Reyes-Gordillo, Ravi Varatharajalu, Jaime Arellanes-Robledo, Leslie C. Leckey, Mamatha Garige, and Ruchi Shah declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
It is important to point out that results of our ongoing investigations on the potential benefits of betaine in preventing alcoholic hepatosteatosis are not described in detail in this review article because a complete manuscript of this investigation is in the process of being published in the American Journal of Pathology. Since our article has not yet been published the Editorial Office of this journal is unable to grant permission to reproduce some of the actual figures pertaining to these results. Therefore, we have highlighted the salient findings of this portion of our ongoing study in the present review.
Rights and permissions
About this article
Cite this article
Lakshman, M.R., Reyes-Gordillo, K., Varatharajalu, R. et al. Novel modulators of hepatosteatosis, inflammation and fibrogenesis. Hepatol Int 8 (Suppl 2), 413–420 (2014). https://doi.org/10.1007/s12072-014-9526-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-014-9526-8