
Abstract
Primary pulmonary lymphoma (PPL) is a rare entity often underdiagnosed due to its non-specific clinical presentation. Our aim is to share our experience in the management of these lesions, which should be considered in the differential diagnosis of nodules affecting the lung parenchyma. We retrospectively studied a total of 14 patients who had undergone surgery between 2013 and 2021. We recorded pre- and post-operative information and conducted survival analyses. Of the 14 patients, seven were males. The mean age was 61.5. Most patients were asymptomatic. PPL presented predominantly as nodules or solid masses (71.4%), followed by consolidations with a pneumonia pattern and ground-glass nodule, with a median uptake on peak standardised uptake value (SUV) positron emission computed tomography of 5.7 mg/m2. There were nine cases (64.3%) of mucosa-associated lymphoid tissue lymphoma (MALT), four cases of diffuse large B cell lymphoma (28.6%) and one case of follicular B cell lymphoma. Fifty percent received adjuvant chemotherapy. Only one patient had disease progression. After a median follow-up of 38.6 months, 11 patients (78.6%) were still alive and disease-free. The overall 5-year survival was 92.9%. In conclusion, the most frequent PPL is the MALT type. Surgery seems to play a key role in diagnosis and treatment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Primary pulmonary lymphoma (PPL) is a very rare entity, and accounts for fewer than 0.5% of neoplasms affecting the lung parenchyma. They are usually seen to have a non-specific clinical presentation and the vast majority of patients with this pathology are diagnosed incidentally on a chest X-ray or via a computed tomography (CT) scan performed for another reason [1].
Currently, the classical definition is from Cordier et al. [2]. They define PPL as a malignant monoclonal lymphoid proliferation originating in the parenchyma or bronchi of one or both lungs that must meet the following criteria: (a) absence of mediastinal and extrathoracic involvement at diagnosis and within three months and (b) no previous history of lymphoma.
Our objective is present our experience with a surgical series of patients diagnosed with PPL over the previous 8 years. We analyse the clinical features, radiological manifestations, treatment and survival.
Material and methods
Between 2013 and 2021, 14 patients underwent surgery with a final diagnosis of PPL. The data were collected retrospectively. We studied the following variables: age, sex, comorbidity, tobacco use and oncological history, clinical manifestations, lesion location and morphology in computed tomography (CT), appearance and size of the lesion in millimetres, standardised uptake value (SUV) in positron emission tomography, surgical approach, type of surgical resection, post-operative stay, post-operative complications, histological analysis and survival.
We conducted statistical analyses using the SPSS Version 24.0. We give the overall results as absolute and relative frequencies for qualitative variables. For quantitative variables, we used the mean, median and range.
Results
Over a period of 8 years, we diagnosed 14 PPL patients (seven females and seven males) in our department. The mean age was 61.5 ± 9. Two patients were smokers at the time of the intervention (14.3%), five patients were ex-smokers (35.7%) and seven had never smoked (50%). One patient had an earlier diagnosis of diffuse interstitial lung disease. Other noteworthy antecedents were five patients (35.7%) suffered from different heart diseases, one had suffered from tuberculosis, one had a rheumatological disease, and four (28.6%) had a history of non-lymphomatous oncology.
Most of the patients (n=10, 71.4%) were asymptomatic at the time of surgery. Two patients (14.3%) presented with cough, one patient consulted for chest pain and another for haemoptysis.
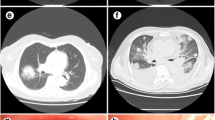
On preoperative CT, five cases (35.7%) showed one nodule/solid mass, another five patients (35.7%) showed multiple nodules/solid masses, three cases (21.4%) showed consolidations with a pneumonia pattern and one patient showed a nodule with a pure ground-glass pattern (Fig. 1). The most frequently affected lobe was the right lower lobe (Table 1). Of the patients with nodules or masses (n= 11), the median nodule size was 2.1 cm (Tables 1 and 2).

Representative images of CT findings. a Right hilar mass. b, c Detail of pneumonia-like consolidations with air bronchogram. d Solid nodule. e Nodule with pure ground-glass pattern
We conducted positron emission tomography computed tomography (PET-CT) in 12 patients (one patient was unable to undergo PET-CT due to morbid obesity and one patient had a pure ground-glass pattern in which we did not consider PET-CT necessary). The lesions had a median maximum SUV of 5.7 mg/m2 (1.6–26.5 mg/m2). No patient had a preoperative diagnosis of pulmonary lymphoma, and cases were referred to our department for diagnostic-therapeutic surgery.
We conducted most of the surgical approaches by videothoracoscopy (11 patients, 78.45%). A thoracotomy was needed in three cases (two patients with a pneumonectomy and one patient who underwent a lobectomy due to blockage of the cavity by severe adhesions). We performed seven wedge resections (50%), five lobectomies (35.7%) and two right pneumonectomies (14.3%). There was only one patient with post-operative complications. It was an haemothorax after a pneumonectomy.
Histological analysis of the samples revealed nine cases of mucosa-associated lymphoid tissue lymphoma (MALT) lymphoma (64.3%), four of diffuse large B cell lymphoma (28.6%) and one of follicular B cell lymphoma. In patients who underwent a major resection, only one case showed lymph node metastasis in peribronchial nodes. All of the patient presented as Ann Arbour stage I, except one of the pneumonectomies that was stage II. We have included an International Prognostic Index score for those patients with diffuse large B lymphoma (Table 3).
At the discretion of the corresponding haematology department, of the 14 patients diagnosed with PPL, seven (50%) received adjuvant chemotherapy, five (35.7%) with R-CHOP therapy, corresponding to diffuse large B cell lymphoma and follicular lymphoma (rituximab-cycophosphamide as alkylating agent, hydroxydaunorubicin, oncovin or vincristine and prednisone). Two patients (14.3%) were treated with rituximab, corresponding to MALT-type lymphomas (Table 2). No cases received radiotherapy (RT). The remaining seven patients were followed up with laboratory and imaging tests. Of the seven patients who received treatment, only one showed disease progression. This was a case with lesions in all lobes of the right hemithorax that had undergone a diagnostic atypical resection. Of the seven patients who did not receive treatment after surgery, none had disease progression. The median follow-up in our series was 38.6 months (7.7–88.6). From the intervention to the present, 11 patients (78.6%) were still alive and three (21.4%) dead, only one of these due to uncontrolled distant disease (Figs. 2, 3).

Survival analysis according to the Kaplan-Meier method

Disease-free survival analysis according to the Kaplan-Meier method
Discussion
PPL is an exceedingly rare disease, accounting for less than 1% of non-Hodgkin’s lymphomas and 3.6% of extranodal lymphomas [3]. It is defined as a disorder in which there is a monoclonal proliferation of B lymphocytes.
In the various articles published, there is a wide age range of patients described with a mean age of 50–60 years old. In our series, we have a similar mean age, although we did not see cases in young adults [4]. In terms of sex, there is a tendency to be more frequent in women, especially in Asian publications. Nevertheless, we did not see variability [3, 4].
A wide variety of initial clinical manifestations are identified, as well as a high incidence of asymptomatic patients at diagnosis. Most of the symptoms and signs collected are non-specific such as cough, expectoration, fever, or haemoptysis. Our results are in line with those reported in the literature, with more than two thirds of patients being asymptomatic (71.4%), and with similar clinical symptomatology when present [4].
Radiological presentation of PPL is highly variable, and there is no imaging pattern that leads us to suspect this pathology. There are four different radiological patterns: single or multiple nodules/masses, pneumonic consolidations, ground-glass opacities, and lesions with a mixed pattern [5]. This fact makes the differential diagnosis of this pathology extremely broad. In the series published by Du et al., pneumonic consolidations with or without air bronchogram predominate, and this can be mistaken for pneumonia [6]. Our results, however, coincide with those published by Shen and Zhou where solid lesions (nodules or masses) predominate in more than 50% of cases [7]. In this case, a differential diagnosis with bronchogenic carcinoma should be made. The ground-glass pattern is the least frequent form of presentation both in our study and in the rest of the literature.
According to the various published series, the most common histological subtype of PPL is MALT lymphoma (70% of cases) and the second subtype is diffuse large B cell lymphoma (about 20% of cases) [6, 7]. Our work coincides with those published to date in terms of the most frequent subtypes of PPL, with MALT accounting for almost 65% of the cases in our series. MALT lymphomas are quite common in the mucosa of the gastrointestinal tract and it seems that chronic inflammatory processes favour their appearance [8, 9]. However, the cause of MALT lymphomas of pulmonary origin is unknown. Various authors link them to certain environmental carcinogens that could cause malignant degeneration of the lymphocytes present in the MALT [9]. Lung MALT lymphoma has also been linked to infectious agents. Traces of deoxyribonucleic acid (DNA) from Chlamydophila pneumoniae, Chlamydia trachomatis, Chlamydia psittaci and Mycoplasma pneumoniae have been detected in biopsies from patients with pulmonary MALT lymphoma [10]. In our series, all patients underwent bronchoscopy during the study, and none had positive bronchioloalveolar lavage cultures for any infectious agent. Finally, there are studies that describe the association of lymphoma with rheumatoid arthritis, giving the possibility of Epstein Barr virus infection, methotrexate treatment or the disease activity itself as a related cause [11]. In our series, as mentioned above, one case had rheumatoid arthritis.
For PPL, surgery, chemotherapy, and radiotherapy are the main therapeutic strategies. These are given either alone or in combination. In our study, half of the series received adjuvant chemotherapy. Analysing the cases, we saw that all patients with diffuse large B cell and follicular lymphoma were treated with R-CHOP. However, we saw that in MALT histology there is variability in post-surgical treatment, only two patients out of nine received chemotherapy with rituximab. This may be because our centre has three haematology departments belonging to different hospitals with different working protocols [9]. There are studies that suggest that surgery does not give benefits to this group of patients. Nevertheless, others consider that it could prevent progression and prolong complete response, especially in young patients [9]. The trend in the most recent studies is to use complete surgical resection in localised stages or, alternatively, radiotherapy [12, 13]. Medical oncological treatment would be reserved for advanced stages or for patients in whom surgical resection cannot be performed. As our study is based on a cohort of exclusively surgical patients, we cannot compare survival rates, although we may conclude that the results are encouraging, as only one patient died of disease progression during follow-up. This patient received adjuvant treatment, had multiple lesions in the same hemithorax at diagnosis and belonged to the histological subtype of large B cell lymphoma.
Long-term follow-up of patients is of foremost importance. The main surgical series we reviewed, which include follow-up and survival, report median follow-ups of around 30–35 months with very wide ranges from less than 1 year to more than ten. Median survival is also quite variable, with numerous studies reporting 55 months and others more than 10 years [6, 7]. In our series, the median follow-up was remarkably similar (38.6 months, with a minimum of 7.7 months and a maximum of 88.6 months). Our median survival of 83.3 months. Three patients died during follow-up, one at 7.7 months due to ischaemic heart disease, another at 75.2 months due to progressive multifocal leukoencephalopathy and the last at 83 months due to uncontrolled distant disease progression.
Conclusions
We can say that PPL is a rare pathology with an anodyne symptomatology and a variable and non-specific radiological presentation. Surgery seems to play a key role in both diagnosis and treatment and yields good survival rates. The two main limitations of our study are that our series is relatively small, largely due to the low prevalence of this pathology, and that it is an entirely surgical series.
References
Borie R, Wislez M, Antoine M, Cadranel J. Lymphoproliferative disorders of the lung. Respiration. 2017;94:157–75.
Cordier J, Chailleux E, Lauque D, Reynaud-Gaubert M, Dietemann-Molard A, Dlaphin JC, et al. Primary pulmonary lymphoma: a clinical study of 70 cases in nonimmunocompromised patients. Chest. 1993;103:201–8.
Yao D, Zhang L, Wu PL, Gu XL, Chen YF, Wang LX, et al. Clinical and misdiagnosed analysis of primary pulmonary lymphoma: A retrospective study. BMC Cancer. 2018;18:1–7.
Wang L, Ye G, Liu Z, Shi L, Zhan C, Gu J, et al. Clinical characteristics, diagnosis, treatment, and prognostic factors of pulmonary mucosa-associated lymphoid tissue-derived lymphoma. Cancer Med. 2019;8:7660–8.
Wislez M, Cadranel J, Antoine M, Milleron B, Bazot M, Mayaud C, et al. Lymphoma of pulmonary mucosa-associated lymphoid tissue: CT scan findings and pathological correlations. Eur Respir J. 1999;14:423–9.
Du C, Zhang J, Wei Y, Bai J, Duan MC, Liu GN, et al. Retrospective analysis of 9 cases of primary pulmonary mucosa-associated lymphoid tissue lymphoma and literature review. Med Sci Monit Basic Res. 2018;24:233–40.
Shen H, Zhou Y. Clinical features and surgical treatment of primary pulmonary lymphoma: a retrospective study. Front Oncol. 2022;12:1–9.
Cadranel J, Wislez M, Antoine M. Primary pulmonary lymphoma. Eur Respir J. 2002;20:750–62.
García Clemente M, Cuétara Suárez P, Rosón Portoc M, López Anglada J, González Martínez M, Seco García A. Linfoma de tejido linfoide asociado al bronquio. Arch Bronconeumol. 2003;39:233–5.
Borie R, Wislez M, Antoine M, Copie-Bergman C, Thieblemont C, Cadranel J. Pulmonary mucosa-associated lymphoid tissue lymphoma revisited. Eur Respir J. 2016;47:1244–60.
Ebeo C, Girish M, Byrd R, Roy T, Metha J. Metrotrexate-induced pulmonary lymphoma. Chest. 2003;123:2150–3.
Di Rocco A, Petrucci L, Assanto GM, Martelli M, Pulsoni A. Extranodal marginal zone lymphoma: pathogenesis, diagnosis and treatment. Cancers (Basel). 2022;14:1742.
Santopietro M, Kovalchuk S, Battistini R, Puccini B, Annibali O, Romano I, et al. Treatment and prognosis of primary pulmonary lymphoma: a long-term follow-up study. Eur J Haematol. 2021;106:49–57.
Funding
None.
Author information
Authors and Affiliations
Contributions
Montserrat Blanco: writing original draft. Conception and design of the study.
Daniel Otero: writing original draft and formal analysis.
Laura Sacristán: data curation.
Carlos Magdalena: final approval of the version.
Rommel Carrasco: final approval of the version.
Milagros Moldes: final approval of the version.
Miguel A Cañizares: final approval of the version.
Eva García Fontán: supervision and final approval version.
Corresponding author
Ethics declarations
Ethics committee approval
The study was approved by the ethic Committee of our institution (ref number 011-21). Date of approval - January 29, 2021.
Informed consent
Taken from all patients.
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Human and animal rights statement
The author(s) state(s) that this research was conducted in accordance with the Helsinki Declaration as revised in 2008. No animals were involved.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Otero Lozano, D., Blanco Ramos, M., Sacristán Robles, L. et al. Primary pulmonary lymphoma: a surgical series. Indian J Thorac Cardiovasc Surg (2024). https://doi.org/10.1007/s12055-024-01774-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12055-024-01774-x