
Abstract
Judiciously designed cyanine based fluorogenic probe (L) can exhibit interesting solvent polarity induced isomerization. The probe displayed a highly selective TURN-ON fluorescence response towards hypochlorite among various reactive oxygen species (ROS) and analytes in a mixed aqueous medium. The sensing process was attributed to the formation of a probe-OCl adduct which results in restricting the donor-acceptor extended conjugation. The detection limit was found to be as low as 3 \(\upmu \hbox {M}\). The proposed sensing mechanism is supported by mass spectrometric analysis and HPLC study.
Graphical abstract
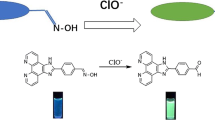
Cyanine based fluorogenic probe (L) selectively detect hypochlorite among various ROS with a detection limit of 3 \(\upmu \hbox {M}\) in mixed aqueous medium through TURN-ON fluorescence response.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Reactive oxygen species (ROS) such as \(^{1}\hbox {O}_{{2}}\), \(^{\bullet }\hbox {OH}\), \(\hbox {O}_{{2}}^{-}\), \(\hbox {H}_{{2}}\hbox {O}_{{2}}\), t-BuOOH and HOCl/\(\hbox {ClO}^{-}\) cause oxidative stress and play vital roles in toxicology as well as pathology.[1, 2] Reactive oxygen species (ROS) have very close involvement in diverse biological processes including its alleged role in many diseases like cancer and neurodegenerative disorders.[3,4,5,6] Hypochlorous acid (HClO) or its conjugate base hypochlorite (ClO\(^-\)) is a well-known ROS which is commonly used as bleaching agents.[7,8,9] Hypochlorous acid (HClO)/hypochlorite (ClO\(^-\)) can be generated endogenously from the peroxidation reaction of hydrogen peroxide and chloride ion, catalyzed by the enzyme myeloperoxidase (MPO).[10,11,12,13,14] Importantly, Hypochlorous acid (HClO) can serve as a powerful antimicrobial agent and it exerts a destructive effect on invading bacteria and pathogens to enable a defence mechanism in the immune system.[15, 16] However, unregulated ClO\(^-\) production could lead to the damage of DNA and proteins which might induce several ailments such as cardiovascular diseases, atherosclerosis, osteoarthritis, rheumatoid arthritis, lung injury and even various types of cancer.[17,18,19,20,21,22,23] Hence, the sensitive detection of ClO- with high selectivity is much awaited to understand the biological insight of ClO\(^-\) related diseases.
The fluorescence-based sensing probes[24,25,26,27,28,29,30,31,32,33] are much more attractive compared to other systems owing to the superiority of fluorescence technique in terms of high sensitivity, rapid response and user friendliness along with its ability to provide non-invasive temporal detection of target analytes in living cells. However, till now there are only a few reports which deal with the sensitive detection of (HClO)/hypochlorite (ClO\(^-\)) using fluorescent probe.[5, 34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49] Some mitochondria-targeted fluorescent probes for HClO/ClO\(^-\) have been reported recently which significantly contribute to research related to the role of ClO\(^-\) in physiology and pathology.[50,51,52,53,54,55] Among them, a number of reported fluorescent probes for (HClO)/hypochlorite (ClO\(^-\)) detection are mostly confined to oxime and imine based probes.[5, 34,35,36,37,38,39,40,41,42,43,44,45,46, 52] But oxime or imine based probes might be vulnerable to acid catalyzed hydrolysis which clearly may curb their application potential in complex biological milieu.[19] Especially inside the cancer cells, wherein the lower pH environment exists, the stability of oxime-based fluorescent probe might become a pertinent issue.[56,57,58,59,60] Hence, cyanine based probes[61, 62] with high stability could be an alternative system to develop hypochlorite sensor.
In this context, we would like to report a new cyanine based spiropyran fluorogenic probe which can demonstrate a highly selective TURN-ON fluorescence response towards hypochlorite (ClO\(^-\)) in 1:1 methanol-aqueous medium. The sensing process was explained because of the disruption in extended conjugation due to the addition reaction between L and ClO- anion.
2 Experimental
2.1 General information and materials
All the materials for synthesis were purchased from commercial suppliers and used without further purification. The absorption spectra were recorded on a Perkin-Elmer Lamda-25 UV-Vis spectrophotometer using 10 mm path length quartz cuvettes in the range of 300–800 nm wavelength, whereas fluorescence measurements were performed on a Horiba Fluoromax-4 spectrofluorometer using 10 mm path length quartz cuvettes with a slit width of 3 nm at 298 K. The mass spectrum of the probe L was obtained using Waters Q-ToF Premier mass spectrometer. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 600 MHz NMR instrument. The chemical shifts were recorded in parts per million (ppm) on the scale. The following abbreviations are used to describe spin multiplicities in \(^{1}\hbox {H}\) NMR spectra: s \(=\) singlet; d \(=\) doublet; t \(=\) triplet; q \(=\) quartet, m \(=\) multiplet.
2.2 Synthesis of the probe L
First, 3–ethyl–1,1,2–trimethyl–1H–benzo[e]indol–3–ium was synthesized through the reported procedure as described in our previously published work (first step of Scheme 1).[26, 28] Thereafter, 1.0 mmol of this 3–ethyl–1,1,2–trimethyl–1H–benzo[e]indol–3–ium was taken in a round-bottomed flask and it was dissolved in EtOH by gentle warming. 1.5 mmol of anhydrous sodium acetate was then added to it and the mixture was refluxed for 1 h. Subsequently, 1.1 mmol of 2-hydroxy-1-naphthaldehyde was added to the reaction mixture in the heating condition and it was further refluxed for 6 h (Scheme 1). A colourless crystalline solid product was obtained upon cooling the reaction mixture to the room temperature. The solid product was then filtered, washed thoroughly with cold methanol and dried in a desiccator. Calculated yield: 64%. \(^{1}\hbox {H}\) NMR [600 MHz, \(\hbox {CDCl}_{{3}}\), TMS, J (Hz), \(\delta \) (ppm)]: 8.06 (1H, d, \(J = 8.4\)), 7.95 (1H, d, \(J = 8.4\)), 7.81 (1H, d, \(J = 8.4\)), 7.76 (2H, m), 7.65 (2H, m), 7.53 (1H, t, \(J = 7.2\)), 7.41 (1H, t, \(J = 7.2\)), 7.35 (1H, t, \(J = 7.2\)), 7.22 (1H, t, \(J = 7.2\)), 7.02 (1H, d, \(J=9.0\)), 6.93 (1H, d, \(J=9.0\)), 5.87 (1H, t, \(J = 10.2\)), 3.46 (2H, m), 1.69 (3H, s), 1.37 (3H, s) 1.22 (3H, t, \(J = 7.2\)). \(^{13}\hbox {C}\) NMR [150 MHz, \(\hbox {CDCl}_{{3}}\), TMS, \(\delta \) (ppm)]: 152.80, 145.13, 130.46, 130.18, 129.99, 129.67, 129.21, 128.98, 128.81, 128.77, 126.85, 126.34, 125.25, 123.34, 121.45, 121.39, 120.70, 117.90, 117.68, 110.23, 110.08, 105.81, 53.86, 38.05, 24.16, 21.82, 14.93. ESI-MS (positive mode, m/z) Calculated for \(\hbox {C}_{{28}}\hbox {H}_{{25}}\hbox {NO}\) \((\hbox {M}+\hbox {H}^{+})\): 392.2009. Found: 392.2051.

Design and synthesis of probe L.
2.3 Preparation of ROS and RNS
Various ROS and RNS including \(^{1}\hbox {O}_{{2}}\), \(^{\bullet }\hbox {OH}\), \(\hbox {O}_{{2}}^{-}\), \(\hbox {H}_{{2}}\hbox {O}_{{2}}\), t-BuOOH, \(\hbox {ClO}^{-}\), \(\hbox {NO}_{{2}}^{-}\), \(\hbox {NO}_{{3}}^{-}\), NO and \(\hbox {ONOO}^{-}\) were prepared according to the following procedures. The exact concentration of hypochlorite (\(\hbox {OCl}^{-})\) solution was determined from the extinction coefficient of 350 \(\hbox {M}^{-1}\hbox {cm}^{-1}\) (292 nm) at pH=9.0. Hydroxyl radical (\({\bullet }\hbox {OH}\)) was produced using Fenton reaction, wherein \(\hbox {Fe}(\hbox {NH}_{4})_{2}(\hbox {SO}_{4})_{2}\cdot \hbox {6H}_{{2}}\hbox {O}\) was reacted with 10 equivalents of \(\hbox {H}_{{2}}\hbox {O}_{{2}}\) to generate \({\bullet }\hbox {OH}\). The concentration of the prepared \({\bullet }\hbox {OH}\) was estimated from the concentration of \(\hbox {Fe}^{2+}\). Commercially available stock of \(\hbox {H}_{{2}}\hbox {O}_{{2}}\) was used for the study and concentration of the \(\hbox {H}_{{2}}\hbox {O}_{2}\) solution was estimated by optical absorbance at 240 nm. Singlet oxygen \((^{1}\hbox {O}_{2})\) was generated by adding NaOCl to \(\hbox {H}_{{2}}\hbox {O}_{{2}}\) according to the reported procedure.[63] The source of \(\hbox {NO}_{{2}}^{-}\) and \(\hbox {NO}_{{3}}^{-}\) were \(\hbox {NaNO}_{2}\) and \(\hbox {NaNO}_{{3}}\) respectively. Nitric oxide (NO) was prepared from sodium nitro-ferricyanide(III) dihydrate. Peroxynitrite (ONOO\(^-\)) was prepared following the reported method[64] and the concentration of it was estimated by using an extinction coefficient of 1670 \(\hbox {M}^{-1}\hbox {cm}^{-1}\) (302 nm).[65] Superoxide (\(\hbox {O}_{{2}}^{-})\) is prepared from \(\hbox {KO}_{{2}}\). t-BuOOH was obtained from commercial suppliers.
2.4 UV-Vis and fluorescence spectroscopic studies
Stock solutions of various anions (\(1 \times 10^{-1} \hbox { mol.L}^{-1})\) were prepared in methanol or Millipore water depending upon the preferred solubility of the analytes chosen. A stock solution of L (\(5 \times 10^{-3}\hbox { mol.L}^{-1})\) was prepared in DMSO. The solution of L was then diluted to \(10 \times 10^{-6}\hbox { mol.L}^{-1}\) for spectral studies by taking only \(4.0\, \upmu \hbox {L}\) stock solution of L and making the final volume to 2.0 mL by adding 1:1 methanol-water mixed solvent. In fluorescence selectivity experiment, the test samples were prepared by placing appropriate amounts of the stock solutions of the respective anions/analytes into 2.0 mL of probe solution (\(10 \times 10^{-6}\hbox { mol.} \hbox {L}^{-1})\). For UV-visible and fluorescence titration experiments, another set of \(\hbox {OCl}^{-}\) standard solution having 5.0 mM concentration was prepared by diluting the earlier prepared stock solutions (\(1 \times 10^{-1}\hbox { mol.L}^{-1})\) in Millipore water. Quartz optical cells of 1.0 cm path length were filled with 2.0 mL solutions of L for UV-visible and fluorescence titration experiments respectively, to which the newly prepared stock solutions of \(\hbox {OCl}^{-}\) (5.0 mM) were gradually added using a micropipette as necessary. For fluorescence measurements, excitation was provided at 360 nm and emission was acquired from 380 nm to 650 nm. Spectral data were recorded within 1 min after addition of the analytes except for kinetic studies.
2.5 Detection limit
The detection limit was calculated on the basis of the fluorescence titration. The fluorescence emission spectrum of L was measured 10 times, and the standard deviation of blank measurement was estimated (\(\uplambda _{\mathrm{em}} = 435\hbox { nm}\)). To measure the slope, the fluorescence emission at 435 nm was plotted as a function of the concentration of \(\hbox {OCl}^{-}\) from the titration experiment. The detection limit was then calculated using the following equation:
Where \(\upsigma \) is the standard deviation of blank measurement, and k is the slope between the fluorescence emission intensity versus [\(\hbox {OCl}^{-}\)].
2.6 Crystallization of L
A small amount of brown-red solid of L was taken in a test tube containing DCM-EtOH mixture and sonicated for 5 min to obtain a clear solution. Then the solution was allowed to evaporate slowly at room temperature and after one week, X-ray mountable plate-shaped orange-red colored crystals appeared at the bottom of the test tube.
2.6.1 Crystallographic refinement details
Herein, a suitable crystal was selected from the mother liquor and immersed into silicone oil, then mounted on the tip of glass fibre and cemented using epoxy resin. Intensity data for all crystals were collected \(\hbox {Mo-K}\upalpha \) radiation (\(\uplambda = 0.71073{\AA }\)) at 298(2) K, with increasing \(\upomega \) (width of 0.3 \(^{\circ }\) per frame) at a scan speed of 6 s/ frame on a Bruker SMART APEX diffractometer equipped with CCD area detector. The data integration and reduction were processed with SAINT[66] software. Empirical absorption correction was applied to the collected reflections with SADABS.[67] The structures were solved by direct methods using SHELXTL-2014 and were refined on F2 by the full-matrix least-squares technique using the SHELXL-2014 program package.[68] Graphics are generated using MERCURY 2.3.[69] In all the cases, non-hydrogen atoms are treated anisotropically. The hydrogen atoms are located on a difference Fourier map and refined. In other cases, the hydrogen atoms are geometrically fixed.
CCDC number – 1572275, formula – \(\hbox {C}_{{28}}\hbox {H}_{{25}}\hbox {NO}\), Fw – 391.49, crystal system – monoclinic, space group – P 21/c, \(a = 22.441(4)\) Å, \(b = 11.222\) (14) Å, \(c = 18.966(2)\) Å, \(\alpha = 90.00\), \(\beta = 114.981(4)\), \(\gamma = 90.00\), \(V = 4329.5(10)\) Å\(^{3}\), \(Z = 4\), \(D_{\mathrm{c}} = 0.601\hbox { g cm}^{-3}\), \(\mu \) \((\hbox {Mo K}\upalpha ) = 0.036 \hbox { mm}^{-1}\), \(F(000) = 832.0\), \(T = 298(2)\hbox { K}\), \(\hbox {Theta}_{\mathrm{max}} = 28.532\), total no. of reflections \( = 75550\), independent reflections \(=\) 10726, observed reflections \( = 8457\), parameters refined \(=\) 274, \(R_{1}\), \(I > 2\upsigma \hbox {(I)} = 0.1438\), \(\hbox {w}R_{2}\), \(I > 2\upsigma \hbox {(I)} = 0.3210\), GOF (\(F^{2}) = 0.993\).
3 Results and Discussions
3.1 Design, synthesis and structural elucidation of the probe L
The probe was synthesized by following a procedure summarized in Scheme 1. Condensation of an indole derivative and 2-hydroxy-1-naphthaldehyde in the presence of anhydrous sodium acetate led to the formation of the probe L with about 64% product yield. The synthesized probe was well-characterized by NMR and MS analysis. The structure of L was further reaffirmed by its crystal structure analysis. The X-ray crystal structure reveals that L exists in spiropyran (SP) ring form rather than merocyanine (MC) form in the solid state. The X-ray single-crystal structure of L showed that it has adopted a non-centrosymmetric molecular arrangement with the polar space group of P 21/c (experimental methods). The crystal packing of the probe L suggested that two different types of conformers are present in a unit cell of L (Figure 1a). Both the types of conformers of L are subjected to eleven short-range \(\hbox {C}-\hbox {H}\cdots \uppi \) intermolecular interactions each (Figure 1b–1c). The aromatic naphthyl planes of the two conformers are found to have almost perpendicular orientations (87.7 \(^{\circ })\) (Figure 1d). The packing diagrams of L have been presented in Figure 1e and Figure 1f.

(A) ORTEP molecular structure at 30% probability level of two conformers, Intermolecular \(\hbox {C}-\hbox {H}\cdots \uppi \) interactions within the range of 2.814–2.899 Å in (B) conformer 1, (C) conformer 2, (D) Almost perpendicular orientations of aromatic naphthyl planes of two conformer, (E) Packing motif along crystallographic b axis depicting wave-like architecture construction by alternative conformer array in symmetry-equivalence (blue-conformer 1, green-conformer 2) and (F) molecular packing in single crystals in symmetry-equivalence as viewed along crystallographic c axis.
3.2 Isomerization of the probe L
It is well known that spiropyran derivatives can exhibit photoisomerization.[70, 71] In the absence of UV irradiation, these types of compounds mainly exist in spiropyran (SP) form and do not show any prominent absorbance in the visible region (Scheme 2). On the contrary, upon UV irradiation it transforms into a merocyanine (MC) form which displays a well-defined absorbance maximum in the visible region.[72] In accordance with this, we found that probe L does not reveal any noticeable absorbance peak in an aqueous medium which clearly indicates that the probe is in the SP form. However, it was noted that when we varied the polarity of the medium by changing the water fraction of a methanol-water mixture, there were interesting changes in the colourimetric behaviour of L (Figure 2). It was observed that when the water fraction of the methanol-water mixture is high (>70%) the probe does not reveal any absorbance peak above 400 nm (Figure 2). However, increasing the methanol content of the mixed solvent leads to the generation of a new absorbance peak at around 583 nm. Interestingly when the methanol content of the mixed solvent is 50% the absorbance maximum of the probe L at 583 nm increases substantially and reaches the highest absorbance value (Figure 2). However, a further increase in the methanol content from 50% to 100% resulted in the gradual decrease in the absorbance maxima at 583 nm (Figure 2). Hence, the study clearly indicates that the probe preferentially attains merocyanine form when there is 50% methanol and 50% water in a methanol-water mixed solvent. As the merocyanine form of the probe could only comprise a potential nucleophilic site, it is important to carry out the sensing studies of L in an optimal condition wherein the conversion to the MC form from SP form will be maximum. Herein, the probe can be easily transformed into MC form without irradiation of UV light just by changing the water fraction of a methanol-water mixed solvent. Hence, we can utilize the probe for sensing purpose with merely adjusting the ratio of methanol and water in a methanol-water mixed solvent. As the maximum conversion to the MC form is attained at 50% methanol and 50% water in a methanol-water mixed solvent, all the subsequent spectral studies of L regarding its sensing pursuit in 1:1 methanol-aqueous solution were carried out at room temperature.

Solvent polarity induced isomerization of the probe L for sensing study.

Changes in the absorbance behavior of L upon varying the water fraction of the methanol-water mixture of the mixed solvent; Bottom: visual changes.
3.3 UV-Vis spectroscopic study of L
Probe L displayed a strong absorption peak at 583 nm (2800 \(\hbox {M}^{-1}\) \(\hbox {cm}^{-1})\) in 1:1 methanol- aqueous solution at room temperature as shown in Figure 3a. The absorption band at 583 nm is possibly originated from the conjugated cyanine moiety present in L in its MC form and the absorption band might be attributed to the \(\uppi -\uppi *\) charge transfer. Initially, UV–Vis spectroscopic studies of L were carried out by recording the absorption spectral changes upon the addition of hypochlorite and other reactive oxygen/nitrogen species. The addition of excess (60 equivalents) of various ROS/\(\hbox {RNS}\) to L induced a negligible change in the colourimetric behaviour of L except for hypochlorite (Figure S6, Supplementary Information). It was noted that among various ROS/RNS only addition of \(\hbox {OCl}^{-}\) could substantially reduce the absorbance maximum of L (Figure 3a) at 583 nm. This spectral change was accompanied by a visible color change as violet (only L) experimental solution turned into almost colorless (L+ \(\hbox {OCl}^{-})\) solution (Figure 3a inset). No other reactive species was able to yield any substantial change in the spectral behaviour when interacted with L, which clearly indicates the high selectivity of the probe towards \(\hbox {OCl}^{-}\). It was also noted that the incremental addition of \(\hbox {OCl}^{-}\) to L (0–60 equivalents) led to a regular decrease in the absorbance maxima of L at 583 nm (Figure 3). So, the UV-Visible spectral studies suggest that the probe L could exhibit good selectivity towards \(\hbox {OCl}^{-}\). A fluorescence study was followed in detail to understand the analytical prospect of L.

UV-Visible spectra of L (\(10\, \upmu \hbox {M}\)) in presence of varying concentration of \(\hbox {OCl}^{-}\) (0–60 equivalents), INSET: Visual change in the color of the solution of L in presence of \(\hbox {OCl}^{-}\) under daylight.

Fluorescence spectra of L (\(10\, \upmu \hbox {M}\)) in 1:1 methanol-aqueous solution in presence of excess (40 equivalents) of various anions, ROS, RNS and bioanalytes.

Fluorescence spectra of L (10 \(\upmu \hbox {M}\)) in the presence of varying concentration of \(\hbox {OCl}^{-}\); INSET: Changes in the emission intensity at 435 nm with the addition of equivalents of \(\hbox {OCl}^{-}\).
3.4 Fluorescence spectral response of L towards \(\hbox {OCl}^{-}\)
The selectivity, sensitivity and response time of L toward \(\hbox {OCl}^{-}\) were investigated using fluorescence spectroscopy. Free probe L was found to be almost non-fluorescent in 1:1 methanol-aqueous solution wherein the merocyanine (MC) form predominates. Hence, upon excitation at 360 nm, L revealed a very low-intensity emission band around 435 nm (Figure 4). However, the fluorescence intensity of the probe L increased more than 10 folds upon addition of 40 equivalents of \(\hbox {OCl}^{-}\) to L and a well-defined emission maximum was found to emerge at 435 nm (Figure 4). However, negligible changes in fluorescence were observed when L interacted with an excess of other ROS and RNS (Figure 4). Especially, the common interfering ROS/RNS like \(^{1}\hbox {O}_{{2}}\), \(^{\bullet }\hbox {OH}\), \(\hbox {O}_{{2}}^{-}\), \(\hbox {H}_{{2}}\hbox {O}_{{2}}\), t-BuOOH, \(\hbox {NO}_{{3}}^{-}\), \(\hbox {NO}_{{2}}^{-}\) and \(\hbox {ONOO}^{-}\) could not bring any noticeable change in the fluorescence intensity of L when added to the solution of L in the similar experimental conditions. Moreover, it was also found that various common anions and bio-analytes including \(\hbox {F}^{-}\), \(\hbox {Cl}^{-}\), \(\hbox {Br}^{-}\), \(\hbox {I}^{-}\), \(\hbox {CH}_{{3}}\hbox {COO}^{-}\), \(\hbox {H}_{{2}}\hbox {PO}_{{4}}^{-}\), \(\hbox {PF}_{{6}}^{-}\), \(\hbox {HCO}_{{3}}^{-}\), \(\hbox {SO}_{{4}}^{2-}\), \(\hbox {HSO}_{{4}}^{-}\), \(\hbox {ClO}_{{4}}^{-}\), \(\hbox {SCN}^{-}\), \(\hbox {S}_{{2}}\hbox {O}_{{3}}^{2-}\), \(\hbox {S}_{{2}}\hbox {O}_{{8}}^{2-}\), pyrophosphate(PPi), cysteine(Cys), homocysteine(Hcy) and glutathione(GSH) hardly induced any change in the fluorescence behavior of L when added to it. Hence, the probe shows an outstanding selectivity towards \(\hbox {OCl}^{-}\). It is also worth mentioning, some recent literature have reported that reactive nucleophilic species like \(\hbox {SO}_{{2}}\) derivatives and \(\hbox {H}_{{2}}\hbox {S}\) can react with various hemi-cyanine dyes containing nucleophilic reaction site.[31, 33, 73] Hence, it was pertinent to check the selectivity of probe L towards \(\hbox {SO}_{{2}}\) derivatives (\(\hbox {SO}_{{3}}^{2-})\) and \(\hbox {H}_{{2}}\hbox {S}\) derivatives (\(\hbox {HS}^{-})\). It was noted that even the presence of 200 equivalents of \(\hbox {SO}_{{2}}\) derivatives and \(\hbox {H}_{{2}}\hbox {S}\) could not bring any noticeable change to the fluorogenic behaviour of the probe L which further reiterated the high selectivity of the probe towards \(\hbox {OCl}^{-}\) (Figure S7, Supplementary Information). Interestingly, the time-dependent fluorescence study of L revealed that it can produce a noticeable change in the fluorescence intensity within seconds of the addition of \(\hbox {OCl}^{-}\) which clearly underlines the rapid sensing aptitude of the probe (Figure S8, Supplementary Information). Hence, the probe can sense hypochlorite selectively with a rapid TURN-ON fluorescence response. However, it is very important to get a quantitative appraisal of the interaction between \(\hbox {OCl}^{-}\) and the probe L to explore the possibility of using the probe in real samples. Thus, a fluorescence titration experiment was performed with varying \(\hbox {OCl}^{-}\) concentration. It was noted that the gradual incremental addition of \(\hbox {OCl}^{-}\) to L in 1:1 methanol-aqueous solution at room temperature resulted in a sequential increase of the emission intensity at 435 nm as evident from Figure 5. It should be mentioned here that the detection limits for \(\hbox {OCl}^{-}\) was determined from the fluorescence titration experiment. The detection limit for \(\hbox {OCl}^{-}\) was found to be \(3.0 \times 10^{-6}\hbox { M}\) (Figure S9, Supplementary Information). In addition, since the Cyanine is sensitive to pH, and the solution of NaOCl is alkaline, the following investigation study was performed to explore the effect of pH on the emission intensity of L at 435 nm upon \(\hbox {OCl}^{-}\) addition (Figure S10, Supplementary Information).

Plausible sensing mechanism.
3.5 Mechanism of L responding to OCl\(^{-}\)
The sensing of the \(\hbox {OCl}^{-}\) by L can be accredited to the nucleophilic addition reaction between L and \(\hbox {OCl}^{-}\). The proposed sensing mechanism is depicted in Scheme 3. Essentially the nucleophilic attack of \(\hbox {OCl}^{-}\) to the nucleophilic site of L resulted in the formation of the product L-OCl adduct which is responsible for the observed change in the optical behaviour of L. It is worth mentioning that the plausible nucleophilic attack of \(\hbox {OCl}^{-}\) led to the rupture in the conjugation in L which is reflected in the observed decolouring of the solution of L upon interacting with \(\hbox {OCl}^{-}\). To validate the proposed sensing mechanism mass spectrometric study was conducted. The mass spectrometric analysis indicated towards the formation of L-OCl adduct when L interacted with \(\hbox {OCl}^{-}\) by showing a prominent peak at \(\hbox {m/z} = 485.6401\) (Figure S11, Supplementary Information). Moreover, a reverse phase HPLC study of L carried out in the presence of \(\hbox {OCl}^{-}\) also further endorsed the formation of L-OCl adduct. It was observed that HPLC of pure L resulted in a single peak with a retention time of 26.93 min. However, when HPLC was run after the addition of excess \(\hbox {OCl}^{-}\) there was a new prominent peak generated with a retention time of 25.67 min apart from the pure ligand peak which is indicative of the formation of L-OCl adduct (Figure S12, Supplementary Information). Hence, these studies supported the premise that nucleophilic addition of \(\hbox {OCl}^{-}\) toward L is key to sensing. However, it was interesting to observe that \(\hbox {SO}_{{3}}^{2-}\), even being a better nucleophile could not produce a similar fluorescence change. This might be explained by the steric effect around the nucleophilic site. Probably owing to the larger size of sulfite compared to hypochlorite, it could not undergo a nucleophilic addition reaction at the specific reaction site.
4 Conclusions
In conclusion, a new fluorogenic probe was successfully designed to demonstrate interesting isomerization. The probe demonstrated a highly selective rapid TURN-ON fluorescence response towards hypochlorite in a mixed aqueous medium. The sensing process was explained as a consequence of the probe-OCl adduct formation; which in turn restricted the donor-acceptor extended conjugation to facilitate the TURN-ON fluorescence response. The sensing mechanism is well-corroborated by mass spectroscopic and HPLC study.
References
Dickinson B C and Chang C J 2011 Chemistry and biology of reactive oxygen species in signalling or stress responses Nat. Chem. Biol. 7 504
Finkel T and Holbrook N J 2000 Oxidants, oxidative stress and the biology of ageing Nature 408 239
Lambeth J D 2007 Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy Free Radic. Biol. Med. 43 332
Bove P F and van der Vliet 2006 A Nitric oxide and reactive nitrogen species in airway epithelial signaling and inflammation Free Radic. Biol. Med. 41 515
Chen X, Wang F, Hyun J Y, Wei T, Qiang J, Ren X, Shin I and Yoon J 2016 Recent progress in the development of fluorescent, luminescent and colorimetric probes for detection of reactive oxygen and nitrogen species Chem. Soc. Rev. 45 2976
Zhang R, Zhao J, Han G, Liu Z, Liu C, Zhang C, Liu B, Jiang C, Liu R, Zhao T, Han M Y and Zhang Z 2016 Real-time discrimination and versatile profiling of spontaneous reactive oxygen species in living organisms with a single fluorescent probe J. Am. Chem. Soc. 138 3769
Aoki T and Munemori M 1983 Continuous flow determination of free chlorine in water Anal. Chem. 55 209
Hidalgo E, Bartolome R and Dominguez C 2002 Cytotoxicity mechanisms of sodium hypochlorite in cultured human dermal fibroblasts and its bactericidal effectiveness Chem.-Biol. Interact. 139 265
Winterbourn C C, Hampton M B, Livesey J H and Kettle A J 2006 Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: Implications for microbial killing J. Biol. Chem. 281 39860
Fiedler T J. Davey C A and Fenna R E 2000 X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 Å resolution J. Biol. Chem. 275 11964
Yap Y W, Whiteman M and Cheung N S 2007 Chlorinative stress: An under appreciated mediator of neurodegeneration? Cell Signal. 19 219
Pattison D I and Davies M J 2006 Evidence for rapid inter- and intramolecular chlorine transfer reactions of histamine and carnosine chloramines: Implications for the prevention of hypochlorous-acid-mediated damage Biochemistry 45 8152
Harrison J and Schultz J 1976 Studies on the chlorinating activity of myeloperoxidase J. Biol. Chem. 251 1371
Klebanoff S J 2005 Myeloperoxidase: Friend and foe J. Leukoc. Biol. 77 598
Prokopowicz Z M, Arce F, Biedron R, Chiang C L L, Ciszek M D, Katz R, Nowakowska M, Zapotoczny S, Marcinkiewicz J and Chain B M 2010 Hypochlorous acid: A natural adjuvant that facilitates antigen processing, cross-priming, and the induction of adaptive immunity J. Immunol. 184 824
Pattison D I and Davies M J 2001 Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds Chem. Res. Toxicol. 14 1453
Pullar J M, Vissers M C and Winterbourn C C 2000 Living with a killer: The effects of hypochlorous acid on mammalian cell IUBMB Life 50 259
Sugiyama S, Okada Y, Sukhova G K, Virmani R, Heinecke J W and Libby P 2001 Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes Am. J. Pathol. 158 879
Steinbeck M J, Nesti L J, Sharkey P F and Parvizi J 2007 Myeloperoxidase and chlorinated peptides in osteoarthritis: Potential biomarkers of the disease J. Orthop. Res. 25 1128
Greig F H, Hutchison L, Spickett C M and Kennedy S 2015 Differential effects of chlorinated and oxidized phospholipids in vascular tissue: Implications for neointima formation Clin. Sci. 128 579
Daugherty A, Dunn J L, Rateri D L and Heinecke J W 1994 Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions J. Clin. Invest. 94 437
Sugiyama S, Kugiyama K, Aikawa M, Nakamura S, Ogawa H and Libby P 2004 Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: Involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis Arterioscler. Thromb. Vasc. Biol. 24 1309
Oushiki D, Kojima H, Terai T, Arita M, Hanaoka K, Urano Y and Nagano T 2010 Development and application of a near-infrared fluorescence probe for oxidative stress based on differential reactivity of linked cyanine dyes J. Am. Chem. Soc. 132 2795
Samanta S, Goswami S, Hoque Md N, Ramesh A and Das G 2014 An aggregation-induced emission (AIE) active probe renders Al(III) sensing and tracking of subsequent interaction with DNA Chem. Commun. 50 11833
S Samanta, T Ray, F Haque and Das G 2016 A turn-on Rhodamine B-indole based fluorogenic probe for selective sensing of trivalent ions J. Lumin. 171 13
Samanta S, Goswami S, Ramesh A and Das G 2015 A new chemodosimetric probe for the selective detection of trivalent cations in aqueous medium and live cells J. Photochem. Photobiol. A 310 45
Samanta S, Goswami S, Ramesh A and Das G 2014 A new fluorogenic probe for solution and intra-cellular sensing of trivalent cations in model human cells Sens. Actuat. B 194 120
Samanta S, Kar C and Das G 2015 Colorimetric and fluorometric discrimination of geometrical isomers (Maleic Acid vs Fumaric Acid) with real-time detection of maleic acid in solution and food additives Anal. Chem. 87 9002
Samanta S, Manna U, Ray T and Das G 2015 An aggregation-induced emission (AIE) active probe for multiple targets: A fluorescent sensor for \(\text{Zn}^{2+}\) and \(\text{ Al }^{3+}\) & a colorimetric sensor for \(\text{ Cu }^{2+}\) and \(\text{ F }^{-}\) Dalton Trans. 44 18902
Samanta S, Datta B K, Boral M, Nandan A and Das G 2016 A multi-responsive turn-on flurogenic probe to sense \(\text{ Zn }^{2+}\), \(\text{ Cd }^{2+}\) and \(\text{ Pb }^{2+}\): left-right-center emission signal swing Analyst 141 4388
Samanta S, Dey P, Ramesh A and Das G 2016 A solo fluorogenic probe for the real-time sensing of \(\text{ SO }_{{3}}^{2-}\) and \(\text{ SO }_{{4}}^{2-}\)/\(\text{ HSO }_{{4}}^{-}\) in aqueous medium and live cells by distinct turn-on emission signals Chem. Commun. 52 10381
Samanta S, Manna U and Das G 2017 White-light emission from simple AIE-ESIPT-excimer tripled single molecular system New J. Chem. 41 1064
Samanta S, Halder S, Dey P, Manna U, Ramesh A and Das G 2017 A ratiometric fluorogenic probe for the real-time detection of \(\text{ SO }_{{3}}^{(2-)}\) in aqueous medium: application in a cellulose paper based device and potential to sense \(\text{ SO }_{{3}}^{(2-)}\) in mitochondria Analyst 143 250
Emrullahoğlu M, Üçüncü M and Karakuş E 2013 A BODIPY aldoxime-based chemodosimeter for highly selective and rapid detection of hypochlorous acid Chem. Commun. 49 7836
Reja S I, Bhalla V, Sharma A, Kaur G and Kumar M 2014 A highly selective fluorescent probe for hypochlorite and its endogenous imaging in living cells Chem. Commun. 50 11911
Zhan Z, Liu R, Chai L, Li Q, Zhang K and Lv Y 2017 Turn-on fluorescent probe for exogenous and endogenous imaging of hypochlorous acid in living cells and quantitative application in flow cytometry Anal. Chem. 89 9544
Zhang Y R, Liu Y, Feng X and Zhao B X 2017 Recent progress in the development of fluorescent probes for the detection of hypochlorous acid Sens. Actuat. B 240 18
Qiao L, Nie H, Wu Y, Xin F, Gao C, Jing J and Zhang X 2017 An ultrafast responsive BODIPY-based fluorescent probe for the detection of endogenous hypochlorite in live cells J. Mater. Chem. B 5 525
Wang X, Wang X, Feng Y, Zhu, Yin H, Guo Q and Meng X 2015 A two-photon fluorescent probe for detecting endogenous hypochlorite in living cells Dalton Trans. 44 6613
Chen W C, Venkatesan P and Wu S P 2015 A turn-on fluorescent probe for hypochlorous acid based on HOCl-promoted removal of the C=N bond in BODIPY-hydrazone New J. Chem. 39 6892
Kang J, Huo F, Yue Y, Wen Y, Chao J, Zhang Y and Yin C 2017 A solvent depend on ratiometric fluorescent probe for hypochlorous acid and its application in living cells Dyes Pigm. 136 852
Zhao Y, Li H, Xue Y, Ren Y and Han T 2017 A phenanthroimidazole-based fluorescent probe for hypochlorous acid with high selectivity and its bio-imaging in living cells Sens. Actuat. B 241 335
Xu X and Qian Y 2017 A novel pyridyl triphenylamine-BODIPY aldoxime: Naked-eye visible and fluorometric chemodosimeter for hypochlorite Spectrochim. Acta A 183 356
Wang E, Qiao H, Zhou Y, Pang L, Yu F, Zhang J and Ma T 2015 A novel “turn-on” fluorogenic probe for sensing hypochlorous acid based on BODIPY RSC Adv. 5 73040
Wang B, Chen D, Kambam S, Wang F, Wang Y, Zhang W, Yin J, Chen H and Chen X 2015 A highly specific fluorescent probe for hypochlorite based on fluorescein derivative and its endogenous imaging in living cells Dyes Pigm. 120 22
Guo B, Nie H, Yang W, Tian Y, Jing J and Zhang X 2016 A highly sensitive and rapidly responding fluorescent probe with a large Stokes shift for imaging intracellular hypochlorite Sens. Actuat. B 236 459
Chen H, Shang H, Liu Y, Guo R and Lin W 2016 Development of a unique class of spiro-type two-photon functional fluorescent dyes and their applications for sensing and bioimaging Adv. Funct. Mater. 26 8128
Xie X, Wu T, Wang X, Li Y, Wang K, Zhai Z, Jiao X and Tang B 2018 A two-photon fluorescent probe for ratiometric visualization of hypochlorous acid in live cells and animals based on a selenide oxidation/elimination tandem reaction Chem. Commun. 54 11965
Huang Y, Zhang P, Gao M, Zeng F, Qin A, Wu S and Tang B Z 2016 Ratiometric detection and imaging of endogenous hypochlorite in live cells and in vivo achieved by using an aggregation-induced emission (AIE)-based nanoprobe Chem. Commun. 52 7288
Zhou X D and Yu X Q 2015 A ratiometric fluorescent probe for in situ quantification of basal mitochondrial hypochlorite in cancer cells Chem. Commun. 51 6781
Li K, Hou J T, Yang J and Yu X Q 2017 A tumor-specific and mitochondria-targeted fluorescent probe for real-time sensing of hypochlorite in living cells Chem. Commun. 53 5539
Li D, Feng Y, Lin J, Chen M, Wang S, Wang X, Sheng H, Shao Z, Zhu M and Meng X 2016 A mitochondria-targeted two-photon fluorescent probe for highly selective and rapid detection of hypochlorite and its bio-imaging in living cells Sens. Actuat. B 222 483
Xiao H D, Li J H, Zhao J, Yin G, Quan Y W, Wang J and Wang R Y 2015 A colorimetric and ratiometric fluorescent probe for \(\text{ ClO }^{-}\) targeting in mitochondria and its application in vivo J. Mater. Chem. B 3 1633
Shen B X, Qian Y, Qi Z Q, Lu C G, Sun Q, Xia X and Cui Y P 2017 Near-infrared BODIPY-based two-photon \(\text{ ClO }^{-}\) probe based on thiosemicarbazide desulfurization reaction: Naked-eye detection and mitochondrial imaging J. Mater. Chem. B 5 5854
Yuan L, Wang L, Agrawalla B K, Park S J, Zhu H, Sivaraman B, Peng J, Xu Q H and Chang Y T 2015 Development of targetable two-photon fluorescent probes to image hypochlorous acid in mitochondria and lysosome in live cell and inflamed mouse model J. Am. Chem. Soc. 137 5930
Thakur B, Jayakumar S and Sawant S N 2015 Probing extracellular acidity of live cells in real time for cancer detection and monitoring anti-cancer drug activity Chem. Commun. 51 7015
Liu J, Ren J, Bao X, Gao W, Wu C and Zhao Y 2016 pH-Switchable fluorescent probe for spatially-confined visualization of intracellular hydrogen peroxide Anal. Chem. 88 5865
Li S Y, Liu L H, Jia H Z, Qiu W X, Rong L, Cheng H and Zhang X Z 2014 A pH-responsive prodrug for real-time drug release monitoring and targeted cancer therapy Chem. Commun. 50 11852
Raju G S R, Benton L, Pavitra E and Yu J S 2015 Multifunctional nanoparticles: Recent progress in cancer therapeutics Chem. Commun. 51 13248
Negrini R, Fong W K, Boyd B J and Mezzenga R 2015 pH-responsive lyotropic liquid crystals and their potential therapeutic role in cancer treatment Chem. Commun. 51 6671
Wu Y, Wang J, Zeng F, Huang S, Huang J, Xie H, Yu C and Wu S 2016 Pyrene derivative emitting red or near-infrared light with monomer/excimer conversion and its application to ratiometric detection of hypochlorite ACS Appl. Mater. Interfaces 8 1511
Xu J, Yuan H, Qin C, Zeng L and Bao G M 2016 A mitochondria-targeted near-infrared probe for colorimetric and ratiometric fluorescence detection of hypochlorite in living cells RSC Adv. 6 107525
Li X H, Zhang G X, Ma H M, Zhang D Q, Li J and Zhu D B 2004 4,5-Dimethylthio-4’-[2-(9-anthryloxy)ethylthio] tetrathiafulvalene, a highly selective and sensitive chemiluminescence probe for singlet oxygen J. Am. Chem. Soc. 126 11543
Sun Z, Liu F, Chen Y, Tam P K H and Yang D 2008 A highly specific BODIPY-based fluorescent probe for the detection of hypochlorous acid Org. Lett. 10 2171
Yu S Y, Hsu C Y, Chen W C, Wei L F and Wu S P 2014 A hypochlorous acid turn-on fluorescent probe based on HOCl-promoted oxime oxidation and its application in cell imaging Sens. Actuat. B 196 203
Sheldrick G M 1995 SAINT and XPREP 5.1 Siemens Industrial Automation Inc.: Madison, WI
Sheldrick G M 1999-2003 SADABS, software for Empirical Absorption Correction, University of Gottingen, Institute fur Anorganische Chemie der Universitat, Tammanstrasse 4, D-3400 Gottingen, Germany
Sheldrick G M 2015 Crystal structure refinement with SHELXL Acta Crystallogr. Sect C: Struct. Chem. 71 3
Mercury 2.3 Supplied with Cambridge Structural Database; CCDC: Cambridge, UK, 2011–2012
Liu F and Morokuma K 2013 Multiple pathways for the primary step of the spiropyran photochromic reaction: A CASPT2//CASSCF study J. Am. Chem. Soc. 135 10693
Be’nard S and Yu P 2000 New spiropyrans showing crystalline-state photochromism Adv. Mater 12 48
Minkin V I 2004 Photo-, thermo-, solvato-, and electrochromic spiroheterocyclic compounds Chem. Rev. 104 2751
Chen Y, Zhu C, Yang Z, Chen J, He Y, Jiao Y, He W, Qiu L, Cen J and Guo Z 2013 A ratiometric fluorescent probe for rapid detection of hydrogen sulfide in mitochondria Angew. Int. Ed. Chem. 52 1688
Acknowledgements
We thank the Council of Scientific and Industrial Research (01/2727/13/EMR-II) and Science & Engineering Research Board (SR/S1/OC-62/2011), India for research grants and Central Instruments Facility (CIF), IIT Guwahati for providing analytical facilities. SS, SH and UM thank IIT Guwahati for research fellowships.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Samanta, S., Halder, S., Manna, U. et al. Specific detection of hypochlorite: a cyanine based turn-on fluorescent sensor. J Chem Sci 131, 36 (2019). https://doi.org/10.1007/s12039-019-1612-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-019-1612-y