
Abstract
Sphingosine receptors (S1PRs) are implicated in the progression of neurodegenerative diseases and metabolic disorders like obesity and type 2 diabetes (T2D). The link between S1PRs and cognition in type 2 diabetes, as well as the mechanisms that underpin it, are yet unknown. Neuroinflammation is the common pathology shared among T2D and cognitive impairment. However, the interplay between the M1 and M2 polarization state of microglia, a primary driver of neuroinflammation, could be the driving factor for impaired learning and memory in diabetes. In the present study, we investigated the effects of fingolimod (S1PR1 modulator) on cognition in high-fat diet and streptozotocin-induced diabetic mice. We further assessed the potential pathways linking microglial polarization and cognition in T2D. Fingolimod (0.5 mg/kg and 1 mg/kg) improved M2 polarization and synaptic plasticity while ameliorating cognitive decline and neuroinflammation. Sphingolipid dysregulation was mimicked in vitro using palmitate in BV2 cells, followed by conditioned media exposure to Neuro2A cells. Mechanistically, type 2 diabetes induced microglial activation, priming microglia towards the M1 phenotype. In the hippocampus and cortex of type 2 diabetic mice, there was a substantial drop in pSTAT3, which was reversed by fingolimod. This protective effect of fingolimod on microglial M2 polarization was primarily suppressed by selective jmjd3 blockade in vitro using GSK-J4, revealing that jmjd3 was involved downstream of STAT3 in the fingolimod-enabled shift of microglia from M1 to M2 polarization state. This study suggested that fingolimod might effectively improve cognition in type 2 diabetes by promoting M2 polarization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroinflammation is one of the significant factors linking dementia to type 2 diabetes (T2D) [1]. There is mounting evidence that type 1 diabetes and T2D are associated with cognitive deterioration and dementia. Specifically, T2D, in particular, is linked to a 50% increased risk of dementia [2,3,4] with impaired attention, processing, motor speed, executive functioning, and verbal memory. Persistent T2D, when combined with an earlier onset, has been linked to the development of mild cognitive impairment [3]. Several studies emphasize the increased risk of cognitive decline and neurodegeneration in diabetic and metabolic syndrome patients [5]. Yet cognitive decline remains the least deciphered complication of diabetes. Although the pathogenesis of cognitive impairment is complex and uncertain, it is believed that hyperglycemia, inflammation, and poor insulin signaling are some of the contributing factors [1]. Insulin resistance correlates with low brain-derived natriuretic factor (BDNF) levels, ultimately affecting cognition [6]. Cerebral plasticity is also intricately modulated by diet and nutrient-related hormones such as insulin and lipids [7]. T2D has been associated with lipid overload, mainly sphingolipids that mediate insulin resistance [8]. Sphingosine-1-phosphate (S1P) has proven beneficial in various diabetic complications like diabetic nephropathy [9] and wound healing [10].
S1PRs have been recommended as a potential target for several disorders due to their regulation of lymphocyte trafficking, vascular permeability, maintenance of bronchial tone, and cardiac function. S1PR modulators were initially designed to decrease immunological response after renal transplantation; however, the only approved use is for multiple sclerosis [11, 12]. Recently their effect on cognitive decline in neurodegenerative disorders like Alzheimer’s, Parkinson’s, and Huntington’s was explored [13,14,15]. Sphingolipid dysregulation has been observed in peripheral organs in metabolic diseases like diabetes and obesity, but it is less explored in the diabetic brain. T2D is arbitrated by many factors, including consuming a diet high in saturated fatty acids [16]. Therefore, the overabundance of fatty acids, more broadly, the perturbation of lipid metabolism that occurs in T2D, which may contribute to adverse outcomes, has piqued researchers’ interest. The brain sphingolipids like S1P and ceramide imbalance contributes to neurodegenerative disease pathology [17]. S1PR1 agonists like fingolimod and SEW2871 have been proven beneficial in alleviating cognitive impairment in Alzheimer’s [18].
One of the mechanisms involved in the neuroprotection offered by S1PR1 activation is the modulation of microglial polarization [19]. Microglia, the brain’s innate immune cells, are the first responders to insults to the brain. They are the main drivers of neuroinflammation and change their activation states [20]. They continuously monitor their environment through the ramified processes and acquire either the pro-inflammatory M1 or anti-inflammatory M2 phenotype based on the nature of the insult [21]. The M1 phenotype is characterized by the release of pro-inflammatory cytokines like tumor necrosis factor α (TNF-α), interleukin (IL)-6 and with increased expression of cluster of differentiation markers 86, 16/32 (CD86, CD16/32), major histocompatibility complex II (MHCII), and inducible nitric oxide synthase (iNOS). The M2 phenotype is further subdivided into M2a, M2b, and M2c. It is characterized by the expression of anti-inflammatory cytokines like IL-4 as well as markers like arginase-1 (Arg1), CD206, and mouse Chitinase-3-like-3 (Ym-1) [22]. Signal transducer and activator of transcription (STAT)-6 and STAT3 signaling upregulation are involved in sustaining the M2 phenotype [23]. STAT3 phosphorylation (p-STAT3) is linked to microglial polarization and is a putative negative regulator of inflammatory cytokine release. In ischemic stroke, p-STAT3 expression was reduced during pro-inflammatory microglial activation, but this effect was reversed when microglia were polarized towards an anti-inflammatory M2 state [24]. However, numerous genes have been revealed as potential STAT3 targets through knockout and biochemical studies, but only a few are confirmed to be direct STAT3 targets [25]. A recent study has reported that Jumonji domain-containing protein 3 (Jmjd3) works downstream of STAT3 in mediating M2 macrophage polarization [26].
S1PRs effect on microglial polarization is well explored in multiple sclerosis. Therefore, in this study, we evaluated the impact of T2D on the microglial activation state and its modulation by the S1PR1 activator fingolimod. In addition, we further investigated the role of the pSTAT3- jmjd3 axis using GSKJ4 (jmjd3 inhibitor) in BV2 microglial cells.
Materials and Methods
Reagents and Antibodies
Sodium palmitate (P9767), fatty acid-free BSA (A6003), DMEM high glucose (D7777), and GSK-J4 (jmjd3 inhibitor, SML0701) were purchased from Sigma Aldrich (St. Louis, USA), murine IL-4 (DY404), IL-6 (DY406) and BDNF (DY248) from R & D biosystems and murine TNF-α (88–7324-88) from Invitrogen. S-FTY720-P (10,006,408) was purchased from Cayman Chemicals, and Fingolimod from TCI (F1018). p-STAT3 (sc8059), Bcl-2 (sc7382), Bax (sc7480), iNOS (sc7271), histone h3 (sc517576), β-actin (sc47778) and CD206 (sc58986) were purchased from SantaCruz. Arg-1 (ab60176), YM-1 (ab192029), MHCII (ab180779), and Synaptophysin (ab32127) were purchased from Abcam. Tri-methyl histone H3 (9733S), psd95 (36233S), and Iba-1 (17198S) were purchased from Cell Signaling Technologies. Anti- mouse Alexa flour 488 (A21202), anti-rabbit Alexa Flour 647 (A21245), anti-rabbit Alexa fluor 488 (A11008), and anti-mouse Alexa Flour 546 (A11003) were purchased from Invitrogen.
Animal Model
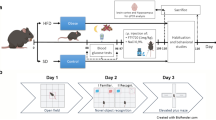
Adult male C57BL6 mice (8 weeks old) were used in this study. The mice were kept in a standard laboratory animal facility with a 12-h light/dark cycle and had free access to food and water ad libitum. All of the studies were carried out in line with the regulations of the Institutional animal ethics committee NIPER-Hyderabad (Protocol number: NIP/10/2020/PC/384). Mice were randomly separated into two groups after a week of acclimation to laboratory settings. For 16 weeks, mice were given either a normal pellet diet (NPD; 13.1% kcal fat) or a high-fat diet (HFD; 60 percent kcal fat). After 1 month on the HFD, the mice were given a single low dosage of streptozotocin (STZ; 100 mg/kg/i.p., dissolved in citrate buffer (pH 4.4)) to shorten the time taken for the establishment of the animal model by inducing partial insulin insufficiency (Fig. 1a) [27]. Mice with fasting serum glucose levels > 200 mg/dl, impaired glucose, and insulin tolerance were classified as T2D mice [28]. NPD mice were divided into two groups: one group was administered saline (NC), and the other group was administered fingolimod at 1 mg/kg i.p. (Perse). T2D mice were further divided into four groups: T2D mice were administered saline (disease control, DC) and T2D mice were administered fingolimod at three doses 0.1 mg/kg, i.p., (low dose, LD), 0.5 mg/kg, i.p., (mid-dose, MD) and 1 mg/kg, i.p. (high dose, HD), dissolved in saline). These groups were administered fingolimod thrice a week for 30 days. Twelve mice were included in each group (a total of 72 mice were used).

The effect of fingolimod on memory consolidation in Morris water maze test. a Study timeline. b Representative track plots of probe trial. c % distance travelled, and time spent in target quadrant (NC N = 5, DC N = 6, LD, MD, HD, Perse N = 5. **p < 0.01, ***p < 0.001 vs NC; ##p < 0.01, ###p < 0.001 vs DC)
Determination of Body Weight and Serum Glucose Levels
Body weights (BW) and fasting plasma glucose levels [29] of all animals were monitored weekly (data not shown). Mice fasted for four hours before measuring fasting glucose levels. Blood glucose concentrations from blood samples taken from the tip of the tail were measured using a glucometer.
Oral Glucose Tolerance Test
The oral glucose tolerance test (OGTT) is a widely used clinical test to diagnose glucose intolerance and T2DM [30, 31]. Mice were fasted overnight and then administered glucose (2 g/kg/p.o., dissolved in saline). Glucose levels from blood samples collected from the tip of the tail were measured using a glucometer at 0, 30, 60, and 120 min after the administration. The area under the concentration versus time curve (AUC glucose 0–120 min, mg/dl * minutes) was calculated. The animals were segregated into different groups based on their blood glucose level.
Intraperitoneal Insulin Tolerance Test (IPITT)
Mice fasted for 4 h before the test. The mice were injected with insulin (0.8 U/kg/i.p., dissolved in saline). Glucose levels from blood samples collected from the tip of the tail were measured using a glucometer at 0, 15, 30, 60, and 120 min after the bolus [32]. The area under the concentration versus time curve (AUC glucose 0–120 min, mg/dl * minutes) was calculated.
Behavioral Tests
Morris Water Maze
The Morris water maze (MWM) consisted of a large circular white pool (diameter: 150 cm; height: 50 cm, filled to a depth of 30 cm with water at 22 ± 2 °C). The tank was in a large, darkened room illuminated by dim light. The pool was divided into four evenly spaced imaginary quadrants and filled with non-fat milk powder. Mice were subjected to trials for 7 days, with 2 days on the visible platform and 5 days on the hidden platform. During the “Visible Platform” trial, mice were given two sessions per day (separated by 3 h) of two trials each. Mice were first trained to locate an “escape” platform submerged 1 cm below the water’s surface using a cue (a colored flag, 2.5-cm radius,8-cm height). If the mice failed to find the platform in 60 s, the experimenter placed them on the platform for 20 s. To avoid procedural bias during each trial in the “Visible Platform,” mice were placed in various positions across the maze. After the “Visible Platform” trials, mice were trained to locate the platform using cues during the “Hidden Platform” trials, which required the mice to rely on extra-maze cues for spatial reference and orientation. Extramaze cues consisted of four large (50 × 50 cm) cues of various shapes and color combinations placed at the quadrant’s borders. During the Hidden Platform trials, the platform was not rotated and remained in the “Target” quadrant. Water was made opaque during the hidden platform trial by adding dry non-fat milk powder. Spatial memory retention was assessed 24 h following the last “Hidden Platform” training session. The parameters of the hidden platform (escape latency and path length) and the probe-trial parameter (number of entries to the target platform zone and time in quadrants) were measured [33, 34].
Object Location test and Novel Object Recognition Test
The object location test (OLT) is a simple and effective test that measures hippocampus-dependent spatial memory. The task depends on an animal’s natural preference for novelty rather than additional external reinforcement and can typically avoid complications associated with differential emotional responses. OLT test comprised three successive trials with an inter-trial interval (ITI) of 60 min. The mice were placed in the center of an empty open field box and allowed to freely explore the box for 5 min in the first trial (i.e., the habituation phase). The second trial involved placing the mice in the center of the open field box with two similar objects on opposite sides and allowing them to freely investigate the objects for 5 min (i.e., the sample phase). Following a 60-min ITI, the mice were placed in the center of the same open field box for a 5-min third trial, with one of the objects remaining in the same location as in second trial and the second object being transferred to a new location in the open field box (i.e., the testing phase of OLT). The first two trials in novel object recognition test (NORT) were the same as in OLT—the habituation phase and the sample phase. Each trial was for 10 min. After 24 h, in the third trial (the testing phase), mice were allowed to investigate objects—the familiar object from second trial and the novel object for 5 min in the same open field box. The percentage time to explore the moved object (in OLT) and novel object (in NORT) was calculated [35, 36].
Y Maze Test
Spontaneous spatial recognition was assessed in the Y-maze and used as a hippocampal-dependent test. The Y maze apparatus comprises three arms, each 21 cm long, 4 cm wide, and 40 cm tall, with a central entry zone. It is made of metal and placed at an angle of 33°. Visual cues were distributed throughout the testing area and remained constant throughout the test. The mice could distinguish between novelty and familiarity based on the diverse environmental characteristics they could perceive from each arm of the Y-maze. The first trial of the test (acquisition) involved closing one arm with a door and allowing mice to visit the other two arms for 5 min freely. After a 60-min ITI, mice were placed in the start arm for the second trial (retrieval) and given free access to all three arms for 5 min. The percent time spent in the novel arm and the number of novel arm entries were recorded. Analyses were based on the time spent exploring the novel and the familiar arms during the retrieval trial [37].
Cell Culture
BV2 microglial cells and Neuro2A (N2A) neuroblastoma cells were purchased from Elabscience, USA, and NCCS Pune, India, respectively. BV2 microglial cells and N2A were cultured in DMEM medium (25 mM glucose, 1 mM pyruvic acid, and 2 mM glutamine) supplemented with 100 IU/ml penicillin/streptomycin and 10% fetal bovine serum (FBS) at 37 °C and 5% CO2. For experiments, cells were seeded in plates at concentrations described for each method. After 24 h, the culture medium was replaced by the serum-free medium (high glucose DMEM without FBS). Cells were exposed to Palmitate (Pal) 200 μM, S-FTY720P (FTY) 100 nM, and 200 nM in serum-free medium for 24 h.
Palmitate-BSA Stock Preparation
The Palmitate-BSA complexes were prepared as previously described [38]. Briefly, 5.56 mg sodium palmitate was dissolved in 200 μL sterile water (100 mM Pal stock solution) by heating (70 °C) and mixing (250 rpm) for 10 min in a thermomixer (Eppendorf). 50 μL of the Pal stock solution was added to 950 μL serum-free DMEM containing 5% fatty acid-free BSA (the 5 mM Pal working solution). For 1 h, the Pal working solution was heated to 40 °C and shaken at 250 rpm. Finally, the working solution was filtered (0.22-μ diameter filter) and immediately used to expose the cells. Serum-free DMEM containing 5% fatty acid-free BSA was used as the vehicle control.
BV2-Conditioned Medium-Exposed N2A Cells
BV2 microglial cells were seeded in a 6-well plate at a density of 1 × 105 cells/well or in 96-well plate at a density of 20 × 104 cells/well and pre-exposed with S-FTY720P or GSK J4, followed by activation with sodium palmitate. After 24 h of respective exposure, the conditioned medium of exposed cells was transferred to N2A cells seeded in a 6-well plate at a density of 1 × 105 cells/well or in a 96-well plate at a density of 20 × 104 cells/well. After 24 h of incubation with a conditioned medium, the cells were used to measure the activated microglia-induced toxicity to N2A cells by MTT assay and western blotting.
Cell Viability Assay
The tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT; Sigma, St. Louis, MO, USA) assay was used to evaluate cell viability [22]. The optical density was measured at 570 nm using Perkin Elmer Envision spectrophotometer Version 1.13.3009.1401, and the results are expressed as a percentage of surviving cells compared with the control.
Reactive Oxygen Species Production
DCFDA was used to evaluate the level of reactive oxygen species (ROS) like H2O2 in BV2 cells, employing fluorescent microscopy. Cells were grown in 12- or 6-well plates (1–2 × 105 cells/well, respectively), exposed as specified, and incubated for 30 min with DCFDA (10 μM) at the end of the exposure. Then, the cells were washed with phosphate-buffered saline (PBS) twice and imaged using a fluorescent microscope (Nikon Eclipse Ti2, Germany). The mean fluorescence intensity of DCF-positive cells was calculated using ImageJ software (NIH) and presented as a percentage change from the control [22].
Mitochondrial Membrane Potential
The dye accumulates in the mitochondrial matrix under physiological conditions, forming J aggregates with distinct absorption and emission spectra that can be measured by fluorescence. After various stimulations, BV2 microglia were incubated with JC-1 dye (2.5 μM) in a culture medium for 30 min at 37 °C. The cells were washed with PBS twice and then imaged using a fluorescent microscope (Nikon Eclipse Ti2, Germany) [22].
Nitrite Estimation
Nitrite estimation in cell culture supernatant from different groups was estimated by the Griess reagent [22]. In brief, an equal amount of culture supernatant (100 μl) from different groups was mixed with an equal amount (100 μl) of Griess reagent (1% p-amino-benzenesulfonamide, 0.01% napthylethylenediamide in 2.5% phosphoric acid), then incubated 20 min in the dark. The absorbance was read at 540 nm. Nitrite release is expressed as a percent increase from the control.
Cytokine Levels
According to the manufacturer’s instructions, the cytokine level in the culture supernatant of BV2 microglia or cortex and hippocampus lysates was estimated using specific ELISA kits (TNF-α, IL-6, IL-4, BDNF) (R and D biosystems).
Immunocytochemistry and Confocal Microscopy
As described previously, immunocytochemical analysis was done in microglia [22]. In a nutshell, cells were grown on coverslips and fixed with ice-cold 4% paraformaldehyde in PBS for 10 min after different stimulations. 1% BSA + 0.3% (v/v) Triton X-100 + 0.3 M glycine (1% BSA) in PBS for 1 h at room was used as blocking. The cells were incubated with primary antibodies, mouse Iba-1 (1:250), rabbit anti-Ym-1 (1:300), anti-MHCII (1:200), mouse anti-CD206, and rabbit Iba-1 (1:250) diluted in 1% BSA at 4 °C overnight. The cells were rinsed in PBS before incubating with the secondary antibody Alexa Fluor 488 and 647 respective IgG conjugates (Invitrogen, USA) for 2 h at room temperature. Coverslips were mounted with DAPI fluoroshield mounting medium (Invitrogen, USA). Images were acquired with a Leica TCS SP8 Laser Scanning Spectral Confocal Microscope.
Immunofluorescence
The mice brains were removed and postfixed in 4% paraformaldehyde overnight at 4 °C. Brains were serially dehydrated in 15 and 30% (w/v) sucrose solutions and embedded in tissue freezing media (Sigma, USA). 25-μm-thick coronal sections were cut on a cryostat (Leica Microsystems, Germany) and mounted to poly-L-lysine-coated slides for immunofluorescence. After 1-h incubation with 1% BSA in PBS, the slices were incubated with primary antibodies rabbit anti-Iba1 (1: 100; CST), mouse anti-CD206 (1:200; Santacruz), mouse psd95 (1:100; CST), and rabbit anti-synaptophysin (1:400, Abcam) at 4 °C overnight in a humidified chamber. After washing the slides in PBS, they were incubated for two hours at room temperature with Alexa Fluor 647 goat anti-rabbit IgG (1:400) or Alexa Fluor 488 donkey anti-mouse IgG (1:400) and mounted using DAPI fluoroshield mounting medium. Images were acquired using a Leica TCS SP8 Laser Scanning Spectral Confocal Microscope. During the quantification procedures of Iba1+ cells, the researcher was blinded to the experimental conditions. As previously described, the number of Iba1+ and CD206+ cells in the cortex and hippocampus were counted [39]. Finally, cell counts were divided by the area of the section and expressed as cells per square millimeter. After subtracting background levels, the mean fluorescent intensity of psd95 or synaptophysin immunoreactivity images was also measured by image analysis software ImageJ (NIH, MD, USA). Immunofluorescence intensity was measured within defined areas of the cortex and hippocampus for each animal by two different examiners blind to the experimental settings for comparison between groups.
Western Blotting
BV2 cells and tissue samples were lysed in RIPA lysis buffer (50 mM Tris–HCl, pH 8.0; 1% NP-40; 0.5% sodium deoxycholate; 150 mM NaCl; 0.1% SDS) containing protease and phosphatase inhibitor cocktails and 1 mM phenylmethylsulfonyl fluoride. Nuclear and cytosolic fractions were prepared using hypotonic and hypertonic lysis buffers [22, 39]. Equal amounts of protein (30 μg) were loaded per lane and separated by 8%, 10%, or 12% SDS-PAGE. Proteins in the gels were transferred to the nitrocellulose (NC) membrane. For 2 h, membranes were blocked with blocking buffer (3% BSA, 10 mM Tris, pH 7.5, 100 mM NaCl, and 0.1% Tween-20). After blocking, the membranes were incubated with respective primary antibodies anti-YM-1, anti-Arg-1, anti-MHCII, anti-iNOS, anti-pSTAT3, anti-STAT3, anti-trimethyl histone H3, anti-histone h3, anti-Bcl-2, anti-Bax, anti-caspase 3, and anti-synaptophysin. Membranes were washed and then incubated with respective HRP-conjugated secondary antibodies (1:10,000) for 2 h at room temperature. Chemiluminescence reagent ECL was used to visualize the protein bands (FusionCaptAdvance). The band intensity was measured using spot densitometry analysis using Image J software.
Statistical Analysis
GraphPad Prism 8.0 software was used to conduct the analyses. Data were expressed as the mean ± SEM and compared using a student’s t-test between two groups and one-way or two-way ANOVA with Bonferroni’s correction for multiple group comparisons.
Results
Fingolimod Effect on Cognitive Impairment was Independent of Blood Glucose Control
The blood glucose of DC was significantly higher than that of the normal control mice. Fingolimod lowered blood glucose levels in HD mice but not significantly. Moreover, poor glycaemic control was seen in DC compared to NC mice. Fingolimod did not affect glycemic control in T2D mice (Supplementary Fig. 1).
Fingolimod Ameliorated Cognitive Dysfunction in T2D Mice
Hyperglycemia has been shown to cause cognitive abnormalities in the MWM test [1, 40]. In this study, we determined the effect of fingolimod on learning and memory in T2D mice using the MWM test. During the hidden platform training days, the mice’s escape latency decreased with time. In the early stages of training, there was no discernible difference between groups. However, the escape latency of mice in the DC groups was significantly longer than that of the NC group on the fourth, fifth, and sixth days of training. Compared to the DC groups, the MD and HD groups had a much lower delay (Supplementary Fig. 2b). The platform was removed on the day of the probe trial to assess the mice’s spatial memory. The swimming track, the distance and time spent in each quadrant of the MWM were recorded. The percentage of time spent and distance travelled in the target quadrant was significantly lower in the DC group than in the NC group. Fingolimod significantly improved the cognitive ability of T2D mice at doses of 0.5 (MD) and 1 mg/kg (HD) (p < 0.001) (Fig. 1b, c).
Further, we evaluated the time spent investigating the novel arm in the Y maze. The mice were allowed to explore two arms for 5 min. Then after 1-h ITI, the mice were subjected to explore all three arms freely. NC, MD, and HD mice spent significant time exploring the novel arm compared to DC (Fig. 2a).

The effect of Fingolimod on recognition memory. a %Time spent in novel arm (Y-maze). b % Investigation time of moved object (OLT). c % Time exploring novel object (NORT). Y-maze: NC vs DC p < 0.001, NC vs LD p = 0.002, DC vs MD p = 0.008, DC vs HD p < 0.001; OLT: NC vs DC p < 0.001, NC vs LD p = 0.006, DC vs MD p = 0.001, DC vs HD p < 0.001; NORT: NC vs DC p < 0.001, NC vs LD p = 0.027, DC vs MD p = 0.004, DC vs HD. p < 0.001 *p < 0.05, ***p < 0.001 vs NC; #p < 0.05, ##p < 0.01, ###p < 0.001 vs DC (n = 6)
We examined the cognitive ability of T2D mice in a novel object recognition test. After acclimatization, mice were exposed to two identical objects and allowed to investigate freely. The time spent exploring the two identical objects was not significantly altered in any group. Mice were reintroduced to one of the previously familiar objects and a novel object 24 h later. NC, MD, and HD mice demonstrated a strong preference for the novel object, while DC and LD mice did not show any preference (Fig. 2c). Similarly, NC, MD, and HD mice showed a strong preference for the shifted object in the object localization test, whereas DC and LD mice showed no preference. These results suggested impaired recognition memory in DC and LD groups (Fig. 2b).
S-FTY720P Suppressed Inflammatory Cytokines, Nitrite, and ROS in Palmitate-Exposed BV2 Cells
To detect the effective exposure concentrations, we first estimated the dose-dependent effects of Pal and S-FTY720P on the viability of BV2 cells. The result showed that S-FTY720P was not cytotoxic to BV2 cells at concentrations up to 800 nM (Supplementary Fig. 3a), while Pal showed a significant decrease in cell viability at 200, 400, and 500 μM (p < 0.001) (Supplementary Fig. 3b). On the other hand, S-FTY720P at 100 nM and 200 nM significantly rescued the cell viability hampered by Pal (p < 0.001) (Supplementary Fig. 3c). Then we further estimated TNF-α, IL-6, and nitrite levels in the culture supernatant of Pal and S-FTY720P-exposed BV2 cells. Based on the results obtained, we chose a 200 μM Pal, 100 nM, and 200 nM S-FTY720P for future experiments.
We then investigated the anti-oxidative effects of S-FTY720P, which showed that S-FTY720P dose-dependently suppressed the ROS generation in Pal-exposed cells (Fig. 3a, c). Further, we studied the effect of S-FTY720P on the mitochondrial membrane potential, which revealed that Pal shifted the mitochondrial membrane potential towards leaky mitochondria, as evident through decreased red/green fluorescence. S-FTY720P significantly reversed at 100 and 200 nM (Fig. 3b, d).

The effect of S-FTY720P on oxidative stress and mitochondrial membrane potential. a Images of DCFDA in palmitate and S-FTY720P-exposed BV2 cells. b Images of Jc-1 staining in palmitate and S-FTY720P-exposed BV2 cells. Graph of fluorescence integrated density of c DCFDA and d Jc-1 (Image J software). Data values are expressed as mean ± S.E.M (n = 3). Con vs Pal (p < 0.001), Pal vs FTY0.1 (p = 0.003 for DCFDA, p = 0.002 for JC-1), Pal vs FTY0.2 (p < 0.001), ***p < 0.001 vs NC, ##p < 0.01, ###p < 0.001 vs Pal
In addition, we evaluated the effect of S-FTY720P on the cytokine and nitrite levels of the Pal-exposed BV2 cells. We observed that S-FTY720P significantly suppressed the Pal-induced pro-inflammatory cytokines and nitrite generation in BV2 cells (Fig. 4a, b, d). In contrast, S-FTY720P elevated the level of anti-inflammatory cytokines like IL-4 in Pal-exposed BV2 cells (Fig. 4c).

The effect of S-FTY720P on cytokine and nitrite levels in BV2 microglia cells. a TNF-α, b IL-6, c IL-4, and d nitrite level in BV2 culture supernatant in various exposed groups. Data values are expressed as mean ± S.E.M (n = 5). ***p < 0.001 vs NC; ###p < 0.001 vs Pal
Fingolimod, via the pSTAT3-jmjd3 Axis, Reduced M1 Microglia Polarization and Enhanced M2 Microglia Polarization in T2D Animals and Palmitate-Exposed BV2 Cells
Microglial activation with an increased surge of inflammatory mediators initiates neuroinflammation in diet-induced obese mice [41]. Here, we found that number of Iba-1 positive cells was significantly increased in the cortex (Fig. 5a, c cortex) and hippocampus CA1, CA3, and DG regions (Fig. 5a CA1, 5b, 5c CA1, and 5d), characterized by their thickened soma and retracted processes. In contrast, fingolimod administered mice showed fewer Iba-1 positive cells with thin soma and ramified processes, indicating that fingolimod may decrease neuroinflammation in diabetes by attenuating microglial activation (Fig. 5a–d).

The effect of fingolimod on microglial activation in cortex and hippocampus of T2D mice. Representative confocal images of coronal sections labeled with Iba-1 (a) cortex and CA1 (b) CA3 and DG, Scale bar 100 μm. Quantitative analysis of Iba-1 is shown in the histogram (c) cortex and CA1 (d) CA3 and DG. Data values are expressed as mean ± S.E.M (n = 4). ***p < 0.001 vs NC; ##p < 0.01, ###p < 0.001 vs DC
Activated microglia have been generalized into two functional states of polarization: pro-inflammatory M1 type and anti-inflammatory M2 type. M1 phenotype microglia display upregulated expression of CD32, CD86, TNF-α, iNOS, and MHCII, whereas M2 phenotype microglia exhibit increased Arg-1, CD206, IL-10, transforming-like growth factor (TGF)-β, and Ym-1 expression [42]. We studied the microglia phenotype in the T2D brain by co-labeling Iba-1 with M2 marker CD206 in the cortex (Fig. 6a, c) and hippocampus DG region (Fig. 6b, d). Further, we checked the expression of M1 markers (iNOS and MHCII) and M2 markers (Arg-1 and YM-1) through western blots of the hippocampus (Fig. 7a, c) and cortex (Fig. 7b, d).

The effect of fingolimod on protein expression of M2 marker CD206 in T2D mice. Representative confocal images of coronal sections of a cortex and b DG region of hippocampus labeled with Iba-1, CD206. Scale bar, 75 μm. Histograms representing quantification of CD206-positive cells in c cortex and d DG region of the hippocampus. Data values are expressed as mean ± S.E.M (n = 4). ***p < 0.001 vs NC; #p < 0.05, ###p < 0.001 vs DC

The effect of Fingolimod on microglial polarization in the cortex and hippocampus of type 2 diabetic mice. The protein expression of M1 markers (iNOS, MHCII), M2 markers (Arg-1, YM-1) in a hippocampus and b cortex were detected by western blots. Quantitative analysis in c hippocampus and d cortex using Image-J software. Data values are expressed as mean ± S.E.M (n = 3). **p < 0.01, ***p < 0.001 vs Con; ##p < 0.01, ###p < 0.001 vs DC
To study the direct effect of fingolimod on microglial polarization, we employed BV2 microglia and induced microglial activation with Pal in vitro. We then evaluated the expression of several M1/M2 markers under different exposures. For in vitro experiments, S-FTY720P (fingolimod’s active metabolite) was used. Immunofluorescence images revealed that Pal significantly suppressed the M2 marker (YM-1) (Fig. 8a), and the M1 marker (MHCII) (Fig. 8b) was significantly increased. These alterations in M2 and M1 markers were diminished considerably by S-FTY720P exposure (Fig. 8a, b). Consistently, western blot results showed similar changes at the protein level as M1 markers (iNOS and MHCII) (Fig. 8c, e) accentuated, and M2 markers (Arg-1 and CD206) decreased by Pal stimulation (Fig. 8c, d). S-FTY720P exposure considerably reduced these changes, showing that it modulates microglial polarization.

The effect of S-FTY720P on microglial polarization in BV2 cells. a Representative images of BV2 microglial cells labeled with Iba-1, MHCII, and YM-1 in response to different stimulation. Scale bar, 25 μm. b Quantitative analysis was performed as percentage of Ym-1+ and MHCII+ in Iba-1+ cells. c The protein expression of M1 markers (iNOS, MHCII) and M2 markers (Arg-1, YM-1, CD206) in exposed BV2 cells by western blotting. Quantitative analysis of d M2 and e M1 markers using Image J software. Data values are expressed as mean ± S.E.M (n = 3). ***p < 0.001 vs Con; ##p < 0.01, ###p < 0.001 vs Pal
Activation of p-STAT3 in M2 polarization has been reported in fingolimod-exposed primary microglial cells and in the brain of ischaemic mice [43]. To further confirm the involvement of pSTAT3, we performed western blotting of pSTAT3 in the cortex and hippocampus of T2D mice. The protein expression of pSTAT3 was significantly reduced in the cortex (Fig. 9a, c) and hippocampus (Fig. 9b, d) of T2D mice and reversed by fingolimod. Previous studies have reported the involvement of Jmjd3, a histone demethylase inhibitor, in the modulation of microglial polarization [44]. We found that the level of H3K27Me3 was increased in the cortex (Fig. 9a, c) and hippocampus (Fig. 9b, d) of T2D mice suggesting the jmjd3 suppression in T2D mice.

The effect of fingolimod on pSTAT3 and H3K27Me3 in cortex and hippocampus of T2D mice. Representative western blots of pSTAT3, H3K27Me3 in a cortex and b hippocampus. Quantitative analysis of pSTAT3 and TMH3 in c cortex and d hippocampus using Image J software. Data values are expressed as mean ± S.E.M (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs NC; #p < 0.05, ##p < 0.01, ###p < 0.001 vs DC
To further confirm the role of pstat3 and jmjd3 in the modulation of microglial polarization, we studied the expression of pSTAT3 and H3K27Me3 in Pal-exposed BV2 cells. We found that Pal reduced the expression of pSTAT3 in microglial cells and increased the expression of H3K27Me3 in microglial cells, as evident through immunofluorescence (Fig. 10a, b) and western blot studies (Fig. 10c, d). Fingolimod significantly attenuated the effect of Pal on the pSTAT3 and jmjd3 protein expression.

Elucidation of pSTAT3-jmjd3 axis in S-FTY720P-exposed BV2 cells. Representative images of BV2 microglial cells labeled with a Iba-1, pSTAT3 and b Iba-1, TMH3 in response to different stimulations. Scale bar, 15 μm. c The protein expression of pSTAT3 and TMH3 in exposed BV2 cells by western blotting. Quantitative analysis of d pSTAT3 and e TMH3 using Image J software. Data values are expressed as mean ± S.E.M (n = 3). **p < 0.01, ***p < 0.001 vs Con; ###p < 0.001 vs Pal
GSKJ4 Attenuated the S-fty720p-Mediated M2 Polarization in Palmitate-Exposed Microglial Cells via jmjd3 Suppression
To further confirm the involvement of jmjd3 downstream of pSTAT3 in the modulation of microglia polarization in diabetes, we pre-exposed BV2 cells with GSKJ4 (5 μM) before S-FTY720P and Pal exposure. GSKJ4 significantly suppressed the protective effect of S-FTY720P on Pal-exposed BV2 microglial cells. GSKJ4 downregulated the expression of M2 markers, as evident through decreased YM-1 and Arg-1 in GSKJ4-exposed cells, and increased the expression of M1 markers like MHCII (Fig. 11a, b). In addition, H3K27Me3 expression was increased by GSKJ4 exposure emphasizing jmjd3 downregulation. The expression of pSTAT3 was unaltered after GSKJ4 exposure suggesting jmjd3 is acting downstream of STAT3 in modulating microglial polarization.

The effect of GSKJ4 on S-FTY720P and palmitate-exposed BV2 cells. a Representative western blots of pSTAT3, H3K27Me3, M1, and M2 markers in BV2 cells. b Quantitative analysis of proteins using Image J software. c IL-4, d TNF-α, e IL-6, and f nitrite level in BV2 culture supernatant in various exposure groups. Data values are expressed as mean ± S.E.M (n = 3). **p < 0.01, ***p < 0.001 vs Con; #p < 0.05, ##p < 0.01, ###p < 0.001 vs Pal; ^p < 0.05, ^^p < 0.01, ^^^p < 0.001 vs FTY0.2 + Pal
Moreover, GSKJ4 suppressed IL-4 (Fig. 11c) and promoted TNF-α (Fig. 11c) and IL-6 (Fig. 11e) levels in exposed microglial cells, suggesting that downregulation of jmjd3 is detrimental. Similarly, the nitrite level (Fig. 11f) was significantly increased by GSKJ4 in fingolimod-exposed microglial cells suggesting that GSKJ4 blocks the protective effect of fingolimod on the microglial polarization. The above results confirm the involvement of the pSTAT3-jmjd3 axis in the fingolimod-mediated modulation of microglial polarization.
Fingolimod Attenuated Apoptosis in T2D Mice and Sustained Synaptic Plasticity
Several studies have reported neuronal apoptosis in the hippocampus and cortex of T2D mice [45, 46]. Here we evaluated the Bcl-2 and Bax expression in the cortex and hippocampus of T2D mice. Fingolimod at 0.5 and 1 mg/kg inhibited apoptosis in the hippocampus (Fig. 12a, c) and cortex (Fig. 12b, d) of T2D mice. Furthermore, we evaluated the effect of fingolimod on synaptic proteins (PSD95 and synaptophysin). T2D significantly attenuated the expression of PSD95 and synaptophysin in the cortex (Fig. 12b, 13a, c), CA1 (Fig, 13a, d), CA3 (Fig. 13b e), and DG (Fig. 13b, f) regions of the hippocampus. As evident through immunoreactivity studies, fingolimod significantly reversed these effects (Figs. 12a–d and 13a–f). Moreover, TNF-α and IL-6 levels were upregulated in the cortex (Supplementary Fig. 4a) and hippocampus (Supplementary Fig. 4b) of T2D mice, which was successfully attenuated by fingolimod. In contrast, BDNF and IL-4 levels were lowered in the T2D brain and upregulated by fingolimod at 0.5 and 1 mg/kg (Supplementary Fig. 4a and 4b).

The effect of Fingolimod on apoptosis and PSD-95 and synaptophysin in T2D mice. Representative western blots of psd-95, synaptophysin, Bcl-2, and bax in a hippocampus and b cortex. Quantitative analysis of proteins in c hippocampus and d cortex using Image J software. Data values are expressed as mean ± S.E.M (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs NC; #p < 0.05, ##p < 0.01, ###p < 0.001 vs DC

The effect of Fingolimod on psd95 and synaptophysin in T2D mice. Representative confocal images of coronal sections labeled with psd95 and synaptophysin in a cortex and CA1 and b CA3 and DG. Quantitative analysis is shown in the histogram c cortex, d CA1, e CA3, and f DG. Data values are expressed as mean ± S.E.M (n = 4). ***p < 0.001 vs NC; ##p < 0.01, ###p < 0.001 vs DC
Conditioned Media from S-FTY720P-Exposed Palmitate-Induced Microglial Cells Rescues Neuronal Apoptosis
We utilized the conditioned media approach to evaluate further the direct effect of S-FTY720P-exposed microglia on neurons. Firstly, we pre-exposed microglia with different stimulations and then collected the conditioned media. Then we added the conditioned media on N2A cells, conducted a cell viability assay, and checked apoptosis markers like Bcl-2 and Bax. We found that the anti-apoptotic protein Bcl-2 expression was decreased in Pal-exposed cells, effectively reversed by S-FTY720P (Fig. 14a, c, d, e). Moreover, the cell viability in the Pal-conditioned media group was significantly decreased in N2A cells which S-FTY720P reversed (Fig. 14f). Similarly, there was decreased expression of Bcl-2 (Fig. 14 b, g, i) and increased expression of Bax in GSKJ4-conditioned media-exposed N2A cells (Fig. 14b, h, i). GSKJ4-conditioned media also inhibited the N2A cell viability (Fig. 14j), suggesting that jmjd3 downregulation is responsible for neuronal apoptosis.

The effect of conditioned media S-FTY720P-exposed palmitate-induced microglial cells on neuronal apoptosis. Representative western blots of apoptotic markers in N2A cells after conditioned media exposure of BV2 a S-FTY720P and Pal exposure b S-FTY720P, Pal, and GSK-J4 exposure. Quantitative analysis of c Bcl-2, d bax, and e Bcl-2/bax in BV2-conditioned media-exposed N2A cells (S-FTY720P and Pal exposure), f cell viability check after conditioned media exposure from S-FTY720P and Pal-exposed BV2 cells on N2A. Quantitative analysis of g Bcl-2, h bax, and i Bcl-2/bax in BV2-conditioned media-exposed N2A cells (S-FTY720P, GSKJ4, and Pal exposure), j cell viability check after conditioned media exposure from S-FTY720P, Pal, and GSK-J4-exposed BV2 cells on N2A. Data values are expressed as mean ± S.E.M (n = 3). **p < 0.01, ***p < 0.001 vs Con; ###p < 0.001 vs Pal; ^^^p < 0.001 vs FTY0.2 + Pal
Discussion
In this study, we demonstrated that fingolimod could ameliorate cognitive deficit by regulating microglial polarization in T2D by suppressing the M1 phenotype and promoting the anti-inflammatory M2 phenotype in p-STAT3 dependent manner. Fingolimod administration (at 0.5 mg/kg and 1 mg/kg) improved impairments in learning and memory functions in T2D mice (Figs. 1 and 2). According to prior reports, T2D affects cognitive processes, such as memory and executive function [47, 48]. A high-fat diet, along with streptozotocin, increases neuroinflammation, as evident through increased pro-inflammatory cytokines, decreased BDNF levels, and anti-inflammatory cytokines (Supplementary Fig. 4). This ultimately leads to defects in synaptic plasticity, as evidenced by reduced psd95 and synaptophysin expression (Figs. 12 and 13), resulting in cognitive impairment. The MWM test is used to evaluate spatial memory and reconsolidation. In our study, T2D mice displayed spatial memory impairment (Fig. 1), consistent with the previous findings [49]. Insulin resistance, hyperglycemia, oxidative, and nitrosative stress in the diabetes are the prime initiators of cognitive impairment [50]. Therefore, improving insulin resistance can be a therapeutic strategy to rectify cognitive impairment [51]. Interestingly, in our study, we observed that fingolimod did not have a significant hypoglycaemic effect. Still, it improved the cognitive deficit in T2D mice, suggesting its effect is independent of the hyperglycaemic control.
Neuroinflammation is the major contributor to cognitive decline in diabetic mice [1, 52]. Glial activation is one of the prime events in neuroinflammation [21]. Consistent with this, our results suggest that diabetes-associated cognitive decline was related to neuroinflammation and apoptosis. These effects are induced by microglial activation, as evidenced by increased Iba1+ cells in the T2D brain (Fig. 5). Even the Pal-stimulated BV2-conditioned media-exposed N2A cells showed increased apoptotic proteins and decreased cell viability (Fig. 14). S-FTY720P decreased neuronal apoptosis, evidenced by increased anti-apoptotic markers and improved cell viability. Notably, fingolimod attenuates the cognitive deficit by inhibiting the M1 phenotype (Fig. 7) and promoting M2 polarization (Figs. 6 and 7) via the pSTAT3-jmjd3 (Fig. 9) axis, subsequently inhibiting neuronal apoptosis (Fig. 12). Fingolimod has already been reported to improve memory and decrease astrocyte and microglial activation in the AD mouse model [53].
Palmitic acid induces lipotoxicity to mimic lipid dysregulation in diabetes [41]. Moreover, the studies linking microglia to Pal are limited. The effect and underlying mechanisms of fingolimod in Pal-induced lipotoxicity are unexplored. Lipotoxicity activates the caspase activity and decreases Bcl-2 (anti-apoptotic protein), leading to neuronal death and neurodegeneration [54]. Our results revealed that type 2 diabetes and Pal could induce neuronal apoptosis, and fingolimod could reverse the changes induced by Pal and type 2 diabetes (Figs. 12 and 14). Moreover, fingolimod improved the synaptic plasticity altered in T2D mice (Figs. 12 and 13).
In the present study, the microenvironment of type 2 diabetes accentuated the pro-inflammatory M1 phenotype as evident through increased protein expression of MHCII and iNOS and decreased the anti-inflammatory phenotype evidenced by reduced YM-1 and Arg-1 expression (Fig. 7). Significantly, in this study, fingolimod administration prevented the pro-inflammatory M1 phenotype and improved neuroinflammation by inhibiting pro-inflammatory cytokines (TNF-α, IL-6), promoting anti-inflammatory cytokines (IL-4, BDNF) (Supplementary Fig. 4) and improving synaptic plasticity (Figs. 12 and 13). Furthermore, hippocampal inflammation characterized by increased TNF-α and IL-6 levels inhibits synaptic plasticity, long-term potentiation, and neurogenesis leading to cognitive deficit [55].
We found that exposure to S-FTY720P, an S1PR1 modulator under lipotoxic conditions with Pal, stimulated the anti-inflammatory M2 alternative polarization in BV2 cells, representing microglia (Fig. 8). The increased oxidative (Fig. 3), pro-inflammatory cytokines (Fig. 4a, b), and nitrosative stress (Fig. 4d) were also suppressed by S-FTY720P in BV2 cells (Fig. 3). The underlying mechanisms for neuroprotection by S-FTY720P were probably attributed to STAT3-mediated modulation (Fig. 10a, c, d) of microglia towards M2 polarization. Our results demonstrated that fingolimod significantly increased the phosphorylation level of STAT3, which was decreased in the T2D brain (Fig. 9). Previous in vitro data suggested that Sphingosine-1-phosphate could activate JNK/STAT3 signaling cascades [56], which was associated with M2 polarization [57]. Fingolimod protects against ischaemic white matter damage by modulating microglia towards the M2 phenotype via the STAT3 pathway [43].
The downstream factors involved in STAT3 modulation of microglial polarization are still unclear. Jmjd3 is one of the direct targets of pSTAT3 and is involved in the demethylation of trimethyl histone H3 [25]. In this study, we found that the trimethyl histone H3 expression was increased in T2D mice (Fig. 9) and Pal-induced lipotoxicity, suggesting the role of jmjd3 (Fig. 10b, e). Further, to confirm the role of jmjd3 downstream of STAT3, we exposed BV2 cells to GSK-J4 (jmjd3 inhibitor). GSK-J4 attenuated the protective effect of fingolimod by promoting the pro-inflammatory M1 phenotype and decreasing the anti-inflammatory M2 phenotype (Fig. 11). Moreover, GSK-J4-conditioned media from BV2 cells accentuated neuronal apoptosis as evident through cell proliferation assay (Fig. 14b, j) and decreased Bcl-2/ Bax ratio (Fig. 14i). On the other hand, GSK-J4 did not affect the pSTAT3 expression confirming that jmjd3 works downstream of STAT3 (Fig. 11). These results suggest that fingolimod exerts its anti-inflammatory and anti-apoptotic effect via the pSTAT3-jmjd3 axis. The mechanism for fingolimod-mediated amelioration of cognitive impairment in type 2 diabetes is summarized in Fig. 15.

Schematic diagram showing the mechanistic basis for fingolimod-mediated amelioration of cognitive impairment in type 2 diabetes. In type 2 diabetes and palmitate lipotoxicity, microglial activation promotes the pro-inflammatory microglial M1 phenotype and suppresses the M2 phenotype. Under physiological conditions, phosphorylation of STAT3 and its nuclear translocation promotes the jmjd3 transcription factor, subsequent demethylation of H3K27Me3, and its decreased expression. H3K27Me3 is responsible for increased microglial M1 phenotype and decreased M2 phenotype. Type 2 diabetes inhibits pSTAT3 translocation and the switch of microglial M2 to M1 phenotype. The pro-inflammatory M1 phenotype releases detrimental cytokines like TNF-α, IL-6 and increases apoptosis (decreased Bcl-2/bax ratio). This further inhibits the synaptic proteins psd95 and synaptophysin, impairing cognition. Fingolimod rescues neuronal apoptosis and improves cognition by increasing nuclear translocation of pSTAT3 and promoting the M2 phenotype. GSK-J4 pre-exposure in vitro abolishes fingolimod’s protective effect, confirming the epigenetic regulation of microglial phenotype by jmjd3. (S1PR1: sphingosine-1-phosphate receptor 1, T2D: type 2 diabetes, STAT3: signal transducer and activator of transcription 3, jmjd3: Jumonji domain-containing protein-3, H3K27Me3: trimethyl histone h3, Arg-1: arginase 1, CD206: mannose receptor, YM-1: mouse chitinase like 3, MHCII: major histocompatibility complex II, iNOS: inducible nitric oxide synthase, IL-6: interleukin 6, IL-4: interleukin 4, TNF-α: Tumor necrosis factor α, BDNF: brain-derived natriuretic factor, Bcl-2: B-cell lymphoma 2, bax: Bcl-2 associated X, psd95: postsynaptic density protein 95, GSK-J4: selective jmjd3 inhibitor)
However, some limitations of our work should be mentioned. In the present study, the effect of fingolimod in the female mice was not explored. There are gender differences in diabetes and cognitive dysfunction [58]. Therefore, the effect of fingolimod on gender-dependent differences in cognitive decline should be explored. Moreover, the effect of fingolimod was independent of the hyperglycaemic control. Thus, analyzing the role of fingolimod along with anti-diabetic drugs could be more beneficial. This study provides a novel approach for treating diabetes-associated cognitive dysfunction by modulating fingolimod’s microglial phenotype.
Conclusion
Our study demonstrates that the microglial activation of the M1 phenotype contributes to cognitive impairment in type 2 diabetic mice, which is associated with decreased synaptic plasticity and increased apoptosis. Fingolimod via the pstat3-jmjd3 axis provides neuroprotection in diabetic mice by promoting the anti-inflammatory microglial phenotype.
Data Availability
The data that support the findings of this study is available in the manuscript as well as supplementary information file. In addition, data can be available from the corresponding author upon reasonable request.
References
Rom S, Zuluaga-Ramirez V, Gajghate S et al (2019) Hyperglycemia-driven neuroinflammation compromises BBB leading to memory loss in both diabetes mellitus (DM) Type 1 and type 2 mouse models. Mol Neurobiol 56:1883–1896. https://doi.org/10.1007/s12035-018-1195-5
McMillan JM, Mele BS, Hogan DB, Leung AA (2018) Impact of pharmacological treatment of diabetes mellitus on dementia risk: systematic review and meta-analysis. BMJ Open Diabetes Res Care 6:e000563. https://doi.org/10.1136/bmjdrc-2018-000563
Mallorquí-Bagué N, Lozano-Madrid M, Toledo E et al (2018) Type 2 diabetes and cognitive impairment in an older population with overweight or obesity and metabolic syndrome: baseline cross-sectional analysis of the PREDIMED-plus study. Sci Rep 8:16128. https://doi.org/10.1038/s41598-018-33843-8
Biessels GJ, Staekenborg S, Brunner E et al (2006) Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 5:64–74. https://doi.org/10.1016/S1474-4422(05)70284-2
Marseglia A, Darin-Mattsson A, Skoog J et al (2021) Metabolic Syndrome is associated with poor cognition: a population-based study of 70-year-old adults without dementia. J Gerontol Ser A 76:2275–2283. https://doi.org/10.1093/gerona/glab195
Davarpanah M, Shokri-mashhadi N, Ziaei R, Saneei P (2021) A systematic review and meta-analysis of association between brain-derived neurotrophic factor and type 2 diabetes and glycemic profile. Sci Rep 11:13773. https://doi.org/10.1038/s41598-021-93271-z
Mainardi M, Fusco S, Grassi C (2015) Modulation of hippocampal neural plasticity by glucose-related signaling. Neural Plast 2015:1–10. https://doi.org/10.1155/2015/657928
Holland WL, Summers SA (2008) Sphingolipids, Insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev 29:381–402. https://doi.org/10.1210/er.2007-0025
Awad AS, Rouse MD, Khutsishvili K et al (2011) Chronic sphingosine 1-phosphate 1 receptor activation attenuates early-stage diabetic nephropathy independent of lymphocytes. Kidney Int 79:1090–1098. https://doi.org/10.1038/ki.2010.544
Kawanabe T, Kawakami T, Yatomi Y et al (2007) Sphingosine 1-phosphate accelerates wound healing in diabetic mice. J Dermatol Sci 48:53–60. https://doi.org/10.1016/j.jdermsci.2007.06.002
Pérez-Jeldres T, Alvarez-Lobos M, Rivera-Nieves J (2021) Targeting sphingosine-1-phosphate signaling in immune-mediated diseases: beyond multiple sclerosis. Drugs 81:985–1002. https://doi.org/10.1007/s40265-021-01528-8
McGinley MP, Cohen JA (2021) Sphingosine 1-phosphate receptor modulators in multiple sclerosis and other conditions. Lancet 398:1184–1194. https://doi.org/10.1016/S0140-6736(21)00244-0
Wang X, Yang G (2021) Bone marrow mesenchymal stem cells-derived exosomes reduce Aβ deposition and improve cognitive function recovery in mice with Alzheimer’s disease by activating sphingosine kinase/sphingosine-1-phosphate signaling pathway. Cell Biol Int 45:775–784. https://doi.org/10.1002/cbin.11522
Miguez A, García-Díaz Barriga G, Brito V et al (2015) Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75 NTR up-regulation and astrocyte-mediated inflammation. Hum Mol Genet 24:4958–4970. https://doi.org/10.1093/hmg/ddv218
Fagan SG, Bechet S, Dev KK (2022) Fingolimod Rescues memory and improves pathological hallmarks in the 3xTg-AD model of Alzheimer’s disease. Mol Neurobiol 59:1882–1895. https://doi.org/10.1007/s12035-021-02613-5
Guitton J, Bandet CL, Mariko ML et al (2020) Sphingosine-1-Phosphate metabolism in the regulation of obesity/type 2 diabetes. Cells 9:1682. https://doi.org/10.3390/cells9071682
Czubowicz K, Jęśko H, Wencel P et al (2019) The role of ceramide and sphingosine-1-phosphate in Alzheimer’s disease and other neurodegenerative disorders. Mol Neurobiol 56:5436–5455. https://doi.org/10.1007/s12035-018-1448-3
Li C, Li J, Kays J et al (2015) Sphingosine 1-phosphate enhances the excitability of rat sensory neurons through activation of sphingosine 1-phosphate receptors 1 and/or 3. J Neuroinflammation 12:70. https://doi.org/10.1186/s12974-015-0286-8
Gaire BP, Bae YJ, Choi JW (2019) S1P 1 Regulates M1/M2 polarization toward brain injury after transient focal cerebral ischemia. Biomol Ther (Seoul) 27:522–529. https://doi.org/10.4062/biomolther.2019.005
Bilbo S, Stevens B (2017) Microglia: the brain’s first responders. Cerebrum 2017. http://www.ncbi.nlm.nih.gov/pubmed/30210663. Accessed 15 Oct 2022
Sood A, Preeti K, Fernandes V et al (2021) Glia: a major player in glutamate–GABA dysregulation-mediated neurodegeneration. J Neurosci Res 99:3148–3189. https://doi.org/10.1002/jnr.24977
Bhat SA, Sood A, Shukla R, Hanif K (2019) AT2R Activation prevents microglia pro-inflammatory activation in a NOX-dependent manner: inhibition of PKC activation and p47phox phosphorylation by PP2A. Mol Neurobiol 56:3005–3023. https://doi.org/10.1007/s12035-018-1272-9
Yang X, Xu S, Qian Y, Xiao Q (2017) Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav Immun 64:162–172. https://doi.org/10.1016/j.bbi.2017.03.003
Liu Z, Ran Y, Qie S et al (2019) Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci Ther 25:1353–1362. https://doi.org/10.1111/cns.13261
Przanowski P, Dabrowski M, Ellert-Miklaszewska A et al (2014) The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia. J Mol Med (Berl) 92:239–254. https://doi.org/10.1007/s00109-013-1090-5
Liang X, Luo M, Shao B et al (2022) Phosphatidylserine released from apoptotic cells in tumor induces M2-like macrophage polarization through the PSR-STAT3-JMJD3 axis. Cancer Commun 42:205–222. https://doi.org/10.1002/cac2.12272
Kang S, Kim C-H, Jung H et al (2017) Agmatine ameliorates type 2 diabetes induced-Alzheimer’s disease-like alterations in high-fat diet-fed mice via reactivation of blunted insulin signalling. Neuropharmacol 113:467–479. https://doi.org/10.1016/j.neuropharm.2016.10.029
Tabák AG, Herder C, Rathmann W et al (2012) Prediabetes: a high-risk state for diabetes development. Lancet 379:2279–2290. https://doi.org/10.1016/S0140-6736(12)60283-9
Neis VB, Moretti M, Bettio LEB et al (2016) Agmatine produces antidepressant-like effects by activating AMPA receptors and mTOR signaling. Eur Neuropsychopharmacol 26:959–971. https://doi.org/10.1016/j.euroneuro.2016.03.009
American Diabetes Association (2006) Diagnosis and classification of diabetes mellitus. Diabetes Care 29(Suppl 1):S43–S48. https://doi.org/10.1016/B978-0-12-801238-3.65822-1
Muniyappa R, Lee S, Chen H, Quon MJ (2008) Current approaches for assessing insulin sensitivity and resistance in vivo: advantages, limitations, and appropriate usage. Am J Physiol Metab 294:E15–E26. https://doi.org/10.1152/ajpendo.00645.2007
Cui Y, Yang M, Wang Y et al (2021) Melatonin prevents diabetes-associated cognitive dysfunction from microglia-mediated neuroinflammation by activating autophagy via TLR4/Akt/mTOR pathway. FASEB J 35:e21485. https://doi.org/10.1096/fj.202002247RR
Feng Y, Chu A, Luo Q et al (2018) The protective effect of astaxanthin on cognitive function via inhibition of oxidative stress and inflammation in the brains of chronic T2DM rats. Front Pharmacol 9:748. https://doi.org/10.3389/fphar.2018.00748
Johnson LA, Zuloaga KL, Kugelman TL et al (2016) Amelioration of metabolic syndrome-associated cognitive impairments in mice via a reduction in dietary fat content or infusion of non-diabetic plasma. EBioMedicine 3:26–42. https://doi.org/10.1016/j.ebiom.2015.12.008
Denninger JK, Smith BM, Kirby ED (2018) Novel object recognition and object location behavioral testing in mice on a budget. J Vis Exp. https://doi.org/10.3791/58593
Lueptow LM (2017) Novel Object recognition test for the investigation of learning and memory in mice. J Vis Exp 2017:e55718. https://doi.org/10.3791/55718
Sarnyai Z, Sibille EL, Pavlides C et al (2000) Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin 1A receptors. Proc Natl Acad Sci 97:14731–14736. https://doi.org/10.1073/pnas.97.26.14731
Ardid-Ruiz A, Ibars M, Mena P et al (2019) Resveratrol treatment enhances the cellular response to leptin by increasing OBRb content in palmitate-induced steatotic HepG2 cells. Int J Mol Sci 20:6282. https://doi.org/10.3390/ijms20246282
Bhat SA, Goel R, Shukla S et al (2017) Angiotensin receptor blockade by inhibiting glial activation promotes hippocampal neurogenesis via activation of Wnt/β-catenin signaling in hypertension. Mol Neurobiol 556(55):5282–5298. https://doi.org/10.1007/S12035-017-0754-5
Tian Z, Ren N, Wang J et al (2018) Ginsenoside ameliorates cognitive dysfunction in type 2 diabetic Goto-Kakizaki rats. Med Sci Monit 24:3922–3928. https://doi.org/10.12659/MSM.907417
Li X, Cai Y, Luo J et al (2021) Metformin attenuates hypothalamic inflammation via downregulation of RIPK1-independent microglial necroptosis in diet-induced obese mice. Cell Death Discov 7:338. https://doi.org/10.1038/s41420-021-00732-5
Orihuela R, McPherson CA, Harry GJ (2016) Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 173:649–665. https://doi.org/10.1111/bph.13139
Qin C, Fan W-H, Liu Q et al (2017) Fingolimod protects against ischemic white matter damage by modulating microglia toward M2 polarization via STAT3 pathway. Stroke 48:3336–3346. https://doi.org/10.1161/STROKEAHA.117.018505
Tang Y, Li T, Li J et al (2013) Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ 2014 (part of journal title) 213(21):369–380. https://doi.org/10.1038/cdd.2013.159
He X, Pei S, Meng X et al (2022) Punicalagin attenuates neuronal apoptosis by upregulating 5-hydroxymethylcytosine in the diabetic mouse brain. J Agric Food Chem 70:4995–5004. https://doi.org/10.1021/acs.jafc.2c00863
Zhou W, Yao Y, Li J et al (2019) TIGAR attenuates high glucose-induced neuronal apoptosis via an autophagy pathway. Front Mol Neurosci 12:193. https://doi.org/10.3389/fnmol.2019.00193
Hazari MAH, Ram Reddy B, Uzma N, Santhosh Kumar B (2015) Cognitive impairment in type 2 diabetes mellitus. Int J Diabetes Mellit 3:19–24. https://doi.org/10.1016/j.ijdm.2011.01.001
Rucker JL, McDowd JM, Kluding PM (2012) Executive Function and type 2 diabetes: putting the pieces together. Phys Ther 92:454–462. https://doi.org/10.2522/ptj.20100397
Yermakov LM, Griggs RB, Drouet DE et al (2019) Impairment of cognitive flexibility in type 2 diabetic db/db mice. Behav Brain Res 371:111978. https://doi.org/10.1016/j.bbr.2019.111978
Mastrocola R, Restivo F, Vercellinatto I et al (2005) Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J Endocrinol 187:37–44. https://doi.org/10.1677/joe.1.06269
Benedict C, Grillo CA (2018) Insulin Resistance as a therapeutic target in the treatment of Alzheimer’s disease: a state-of-the-art review. Front Neurosci 12:215. https://doi.org/10.3389/fnins.2018.00215
Takechi R, Lam V, Brook E et al (2017) Blood-Brain barrier dysfunction precedes cognitive decline and neurodegeneration in diabetic insulin resistant mouse model: An implication for causal link. Front Aging Neurosci 9:399. https://doi.org/10.3389/fnagi.2017.00399
Carreras I, Aytan N, Choi J-K et al (2019) Dual dose-dependent effects of fingolimod in a mouse model of Alzheimer’s disease. Sci Rep 9:10972. https://doi.org/10.1038/s41598-019-47287-1
Montero ML, Liu J, Orozco J et al (2020) Docosahexaenoic acid protection against palmitic acid-induced lipotoxicity in NGF-differentiated PC12 cells involves enhancement of autophagy and inhibition of apoptosis and necroptosis. J Neurochem 155:559–576. https://doi.org/10.1111/jnc.15038
Ardid-Ruiz A, Ibars M, Mena P et al (2019) Resveratrol treatment enhances the cellular response to leptin by increasing OBRb content in palmitate-induced steatotic HepG2 Cells. Int J Mol Sci 20:49–60. https://doi.org/10.3390/ijms20246282
Garris CS, Wu L, Acharya S et al (2013) Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol 14:1166–1172. https://doi.org/10.1038/ni.2730
Jha MK, Lee W-H, Suk K (2016) Functional polarization of neuroglia: Implications in neuroinflammation and neurological disorders. Biochem Pharmacol 103:1–16. https://doi.org/10.1016/j.bcp.2015.11.003
Espeland MA, Carmichael O, Yasar S et al (2018) Sex-related differences in the prevalence of cognitive impairment among overweight and obese adults with type 2 diabetes. Alzheimer’s Dement 14:1184–1192. https://doi.org/10.1016/j.jalz.2018.05.015
Acknowledgements
All authors are thankful to the Department of Pharmacology and Toxicology, NIPER Hyderabad and Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India.
Funding
This work is supported by National Institute of Pharmaceutical Education and Research, Hyderabad and Department of Pharmaceuticals, Ministry of Chemical and Fertilizers, Govt. of India.
Author information
Authors and Affiliations
Contributions
All authors take responsibility for the integrity and accuracy of this manuscript. Anika Sood, Dharmendra Kumar Khatri, and Shashi Bala Singh contributed to the conceptualization designing and execution of the research; Anika Sood performed studies, evaluated data, and wrote manuscript; Valencia Fernandes, and Kumari Preeti performed studies, evaluated data and reviewed the manuscript; Dharmendra Kumar Khatri, and Shashi Bala Singh reviewed, editied the manuscript, and supervised the research work.
Corresponding authors
Ethics declarations
Ethics Approval
This study was performed in line with the regulations of the Institutional animal ethics committee (IAEC) NIPER-Hyderabad (Protocol number: NIP/10/2020/PC/384).
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sood, A., Fernandes, V., Preeti, K. et al. Fingolimod Alleviates Cognitive Deficit in Type 2 Diabetes by Promoting Microglial M2 Polarization via the pSTAT3-jmjd3 Axis. Mol Neurobiol 60, 901–922 (2023). https://doi.org/10.1007/s12035-022-03120-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-03120-x