
Abstract
Hypertension is one of the major risk factors for central nervous system (CNS) disorders like stroke and Alzheimer’s disease (AD). On the other hand, CNS diseases like AD have been associated with gliosis and impaired neurogenesis. Further, renin angiotensin system (RAS) is intricately associated with hypertension; however, the accumulating evidences suggest that over-activity of RAS may perpetuate the brain inflammation related with AD. Therefore, in the present study, we examined the effect of hypertension and RAS on glial (astrocytes and microglia) activation and hippocampal neurogenesis in a rat model of chronic hypertension. We used Candesartan [angiotensin type 1 receptor (AT1R) blocker (ARB)] both at a low dose (0.1 mg/kg) and anti-hypertensive dose (2 mg/kg) to explore whether their effect on astrocyte and microglial activation, neuroinflammation, and neurogenesis is blood pressure (BP) dependent or independent. Our data revealed that hypertension induces robust microglial and astrocyte activation, neuroinflammation, and cripples hippocampal neurogenesis. Importantly, AT1R blockade by Candesartan, even at low dose (0.1 mg/kg), prevented astrocyte and microglial activation and neuroinflammation in the brain of hypertensive rats. Mechanistically, AT1R blockade prevented the activation of NADPH oxidase, reactive oxygen species (ROS) generation, suppression of MAP kinase and NFкB signaling. Importantly, we, for the first time to our knowledge, provided the evidence that AT1R blockade by activating Wnt/β-catenin signaling, promotes neurogenesis during hypertensive state. We conclude that AT1R blockade prevents astrocyte and microglial activation and improves hippocampal neurogenesis in hypertensive state, independent of BP lowering action.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A number of experimental and clinical studies demonstrate that hypertension is one of the major risk factors for central nervous system (CNS) diseases like stroke and Alzheimer’s disease (AD; the predominant form of dementia) [1,2,3,4]. The relationship between hypertension and dementia is further strengthened when anti-hypertensive agents like angiotensin-converting enzyme inhibitors and angiotensin type receptor (AT1R) blockers (ARBs), improved memory functions in hypertensive subjects [2,3,4]. However, the pathology of AD is intricately associated with neuroinflammation that is mediated by the activation of glia (microglia and astrocytes) [4, 5]. Astrocytic and microglial activation results in the release of various inflammatory mediators like tumor necrosis factor-α (TNF-α), IL-1β, IL-6, and reactive oxygen species (ROS) production [6,7,8], leading to the neuronal injury and neurodegeneration [4, 7]. Further, increased level of TNF-α has also been reported in the cerebrospinal fluid (CSF) and serum of AD patients [8] indicating the role of inflammatory mediators in the progression of AD.
Renin angiotensin system (RAS) plays a key role in blood pressure (BP) regulation; however, over-activity of central RAS has been associated with neuroinflammatory changes in AD brains [4, 9]. Presence of RAS in CNS has been well established and its components are detected in several brain regions like hippocampus, cortex, and in different CNS cells like neurons and glia (astrocytes and microglia) [4, 5, 9,10,11]. Angiotensin II (Ang II), considered as the principal effector RAS component, acts mainly via activation of the AT1R [4, 5, 9], known to regulate the neuroinflammation, central sympathetic activity, behavior, and cognition [4, 5, 9]. Importantly, in several clinical and pre-clinical studies, ARBs showed neuroprotection and prevented cognitive decline [3, 12,13,14,15]. Further, the neuroprotective effects of ARBs like candesartan and telmisartan have been demonstrated in primary neuronal, astrocyte, and microglia cell cultures and in various rodent models of brain disease, like stroke, AD, and neuroinflammation [4, 5, 14,15,16,17,18]. Moreover, the studies from our lab also demonstrated the improvement in the cognitive functions by candesartan [19, 20]. Conclusively, these studies suggested that ARB-mediated neuroprotection might be independent of their anti-hypertensive actions; however, the underlying mechanisms are still unknown.
Recently, AD has been associated with the impaired adult hippocampal neurogenesis in addition to the progressive loss of neurons and gliosis [21, 22]. Of the neurogenic zones in the adult brain, hippocampal neurogenesis draws the most attention, because of its involvement in cognitive functions [23]. However, the modulation of adult hippocampal neurogenesis in hypertension is highly debated. Studies by Pietranera and others showed hypertension impaired hippocampal neurogenesis [24,25,26]. On the contrary, it has been shown that hypertension promoted the hippocampal neurogenesis [27, 28]. Thus, it remains ambiguous how neurogenesis is affected in hypertension and needs further exploration.
We previously have reported that hypertension increases the susceptibility to neurodegeneration [9]; however, the contribution of glia (astrocytes and microglia) mediated inflammatory changes in the brain during hypertensive state is still elusive. Further, RAS is over-activated in hypertension; however, its involvement in glial cell activation and neurogenesis is still not completely explored. Therefore, in the present work, we carried out a mechanistic study to demonstrate the effect of hypertension and RAS on glial (astrocytes and microglia) activation and hippocampal neurogenesis in a rat model of chronic hypertension. We used candesartan (ARB) both at a low dose (0.1 mg/kg) [19] and anti-hypertensive dose (2 mg/kg) [29] to explore the blood pressure dependent or independent actions of ARB.
Materials and Methods
Materials
Candesartan cilexetil; BrdU; 2′,7′-dichlorofluorescin diacetate (DCF-DA); 3,3′,5,5′-tetramethylbenzidine (TMB); collagenase; protease inhibitor; sodium chloride; potassium chloride; HEPES; magnesium chloride; and all other chemicals used in the study were purchased from Sigma-Aldrich (USA). Primary antibodies, anti-GFAP (Sigma, USA), anti-p65NFкB, anti-IкB-α, anti-Iba-1, anti-phospho-GSK-3 (Ser-9), anti-phospho-GSK-3 (Y214), total-GSK-3 (Abcam, USA), phospho-p38 mitogen-activated protein kinase (p38MAPK), total p38MAPK, phospho-ERK1/2, total-ERK1/2, phospho-β-catenin, total β-catenin, anti-DCX, and anti-Nestin antibodies (Cell Signaling Technology, USA), anti-p22phox, anti-p40phox goat polyclonal anti-gp91phox, anti-p47phox, anti-Histone H3 (Santa Cruz Biotechnology, USA) and Alexa Fluor 594 and Alexa Fluor 488 IgG conjugate (Invitrogen), secondary HRP-conjugated antibodies were purchased from Santa Cruz Biotechnology (USA). PVDF membrane was purchased from Millipore (USA). Primers used in the study were purchased from Integrated DNA Technology (USA). ELISA kits for interleukin-10 (IL-10) and tumor necrosis factor-α (TNF-α), were purchased from Peprotech (USA).
Animals
The experiments were carried out with adult male Sprague-Dawley (SD) rats (RGD, 10395233) procured from the Laboratory Animal Services Division of CSIR-Central Drug Research Institute (CDRI), Lucknow, India. Experiments were performed according to internationally followed ethical standards and approved by Institutional Animal Ethics Committee (IAEC) of CSIR-CDRI and Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India. Rats were maintained under standard housing conditions (room temperature 24–27 °C and humidity 60–65%) with a 12-h light and dark cycle. Food and water were available ad libitum.
Model of Chronic Hypertension
Chronic hypertension was induced in male SD rats (180–200 g), by partial occlusion of left renal artery, while in sham animals, renal artery was only exposed but not occluded as described previously [30]. Briefly, animals were anesthetized with pentobarbital (50 mg/kg i.p.). After a midline abdominal incision, the left renal artery was exposed and isolated over a short segment by a blunt dissection. After isolation, renal artery was constricted by making the loop of folded-in-two cotton thread tunneled through a plastic tube (0.5-mm internal diameter, 1.5–2.0-mm long). The artery was slowly inserted into the tube by pulling the ends of thread. The thread was tied such that it did not constrict the artery but prevented its slippage from the plastic tube [30]. Sham control rats underwent the same surgical procedure without the partial ligation of renal artery. After surgery, neosporin, an anti-fungal powder and betadine, a topical antiseptic, were applied at the site of incision. The animals had access to food and water ad libitum.
Experimental Plan and Drug Administration
After recovery of 1 week, animals were randomly distributed in following experimental groups: Group 1—sham (normotensive); Group 2—hypertensive (HTN); Group 3—HTN + candesartan (2 mg/kg, p.o.); Group 4—HTN + candesartan (0.1 mg/kg, p.o.).
Candesartan was dissolved in a vehicle solution (1 mg in 950 μl of phosphate-buffered saline and 50 μl 1 M Na2CO3) [20]. Candesartan at 0.1 and 2.0 mg/kg was administered orally daily for 35 days starting from the first day after recovery.
Measurement of Hemodynamic Parameters
Blood pressure (BP) of the rats was measured weekly starting from 0 up to 5th week after the surgery. Briefly, the rats were anesthetized with urethane (1.25 g/kg, i.p.) and placed on an isothermal pad to maintain normal body temperature during surgical procedures. The hemodynamic parameters like systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial blood pressure (MAP), and heart rate (HR) were recorded by inserting fluid-filled catheters in the carotid artery. The catheter was attached to the pressure transducer coupled to the Data Acquisition System. All animals were allowed to stabilize for 20 min before the final readings.
After taking hemodynamic parameters, the animals were sacrificed by transcardiac perfusion with ice-cold saline. The brain was immediately removed and kept on ice-cold plate to dissect cerebral cortex and hippocampus.
Estimation of Cytokines
Inflammatory cytokines in the brain tissue were measured by ELISA, following the protocol provided by the manufacturer (Peprotech, USA), as described previously [30]. In brief, brain tissues were homogenized in ice-cold Tris buffer (pH 7.2) containing 50 mM Tris, 1 mM EDTA, 6 mM MgCl2, and 5% (w/v) protease inhibitor cocktail (Sigma-Aldrich, USA). After homogenization, samples were sonicated for 10 s using an ultrasonic processor (Heat systems-Ultrasonic Inc.) at a setting of 20 duty cycles and then centrifuged at 20,800g for 20 min at 4 °C. Supernatants were collected and level of the pro-inflammatory cytokine (TNF-α) and anti-inflammatory cytokine (IL-10) was determined using commercially available ELISA kit, following the protocol provided by the manufacturer (Peprotech, USA). The concentration of protein in the sample was determined by comparison with a standard curve of known protein concentrations. Cytokines level (TNF-α and IL-10) were expressed as picogram per milligram of protein [30].
Estimation of ROS
For ROS determination, rat brains were isolated and homogenous suspension was prepared in HEPES–Tyrode solution (145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM glucose, 5 mM HEPES, pH 7.4) by treatment of the finely chopped forebrain tissue with collagenase (750 unit/ml) for 45 min at 37 °C. After enzymatic treatment, the dissociated cells were passed through a filter to remove large tissue fragments. Viability test was done by trypan blue and 90% of the viable population of cells was obtained. Generation of ROS was estimated by fluorescent dye DCF-DA using flow cytometer (Becton Dickinson, UK). Data was analyzed using Cell Quest Software (Becton Dickinson, USA). DCF-positive cells, 10,000 counts per condition, were quantified using the FACS Calibur (BD) with the Cell Quest software, as described previously [5].
Western Blot Analysis
For the western blot analysis, nuclear and cytosolic fractions were prepared as described previously [5, 30]. Briefly, the brain of rats were homogenized in five volumes of ice-cold hypotonic buffer (20 mM HEPES (pH 7.5), 250 mM sucrose, 1 mM dithiothreitol, 1.5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.1 mM PMSF, 1× protease inhibitor cocktail) using a Teflon homogenizer. The cell lysate was then centrifuged at 800g for 10 min at 4 °C. Supernatants containing cytosolic fractions were removed and stored at − 70 °C. Pelleted nuclei were resuspended in hypertonic buffer (hypotonic buffer + 350 mM NaCl). Debris was removed by centrifugation at 13,000g and nuclear fractions were stored at − 70 °C. Protein estimation in the supernatants was done by Lowry method. Samples were mixed with the 4× loading buffer containing 100 mM Tris–HCl (pH 6.8), 200 mM dithiothreitol (DTT), 4% sodium dodecyl sulfate (SDS), 0.2% bromophenol blue, and 20% glycerol and were boiled for 5 min at 95 °C.
An equal amount of proteins was separated on 10 or 12% SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked with blocking buffer (5% BSA, 10 mM Tris pH 7.5, 100 mM NaCl, and 0.1% tween-20) for 2 h. Membranes were then incubated overnight at 4 °C with primary antibodies rabbit polyclonal anti-p22phox, goat polyclonal anti-gp91phox, anti-p47phox (1:500; Santa Cruz Biotechnology, USA), rabbit polyclonal anti-p65NFкB, anti-IкB-α, anti-Iba-1 (1:1000; Abcam, UK), anti-phospho-p38 mitogen-activated protein kinase (p38MAPK), total p38MAPK, rabbit polyclonal anti-ERK1/2, and mouse polyclonal anti-p-ERK1/2 (1:1000; Cell Signaling Technology, USA) and mouse monoclonal anti-GFAP (1:1000), anti-histone H3 (1:1000), anti-p-S-9-GSK-3β (1:1000), anti-p-Y-216-GSK-3β (1:1000), goat anti- GSK-3β (1:1000; Abcam, UK) and β-actin (1:10,000; Sigma, St. Louis, MO, USA). After washing, membranes were incubated with respective HRP-conjugated secondary antibodies (1:5000) for 2 h at room temperature. Blots were then developed by the ECL chemiluminescence detection system (Millipore, USA). The band intensity was measured using spot densitometry analysis by MY IMAGE ANALYSIS software (Thermo Scientific). β-actin protein was used as an internal control.
Bromodeoxyuridine Injection
We used bromodeoxyuridine (BrdU) to label proliferating cells. On day 35, all groups of rats were treated with four intraperitoneal injections of BrdU (50 mg/kg in saline, Sigma-Aldrich, St. Louise, MO, USA) in a 4-h interval, because the half-life of BrdU is about 2 h [31] and the number of BrdU+ cells peaks at about 15 h after BrdU injection and decreases afterward [32]. Two hours after the last injection, the hemodynamic parameters were measured and rats were transcardially perfused with ice-cold 0.1 M PBS (pH 7.4) followed by 4% paraformaldehyde in 0.1 M PBS. The brains were quickly removed for immunohistochemical studies as discussed below.
Immunohistochemistry Analysis
Brains were isolated, fixed overnight in 4% paraformaldehyde, and cryoprotected by 30% sucrose in PBS [30]. The serial coronal sections of cortical and hippocampal tissue of 20-μm thickness, corresponding to the coordinates of − 2.75 to − 3.6 mm from bregma [30, 33] were collected by using cryostat (Micron International, GmbH, Germany) on the poly-l-lysine coated slides and stored at − 20 °C till used for immunohistochemical studies. To ensure the homogeneity in the tissue collected from rats across different groups, sections corresponding to these coordinates were included and quantified, ensuring that the regions of interest were similar among different groups and experiments [30]. For BrdU staining, the sections were treated with 2 N HCl for 30 min, followed by neutralization with borate buffer. Then, the sections were post-fixed and permeabilized by ice-cold acetone for 10 min. Then, sections were blocked with 1% bovine serum albumin to block non-specific binding. The sections were washed with PBS and then incubated with primary antibodies, mouse anti-BrdU (1:100; Sigma, USA), rabbit anti-GFAP (1:100; Sigma, USA), rabbit anti-DCX (1:100; CST, USA) and goat anti-Iba-1 (1:200; Abcam, USA) overnight. After rinsing again with PBS, sections were incubated with Alexa Fluor 594 goat anti-rabbit, Alexa Fluor 488 goat anti-mouse and Alexa Fluor 488 donkey anti-goat IgG conjugate (1:400) for 2 h. Finally, the sections were counterstained with DAPI and mounted with mounting media (Invitrogen). Brain sections were visualized using a fluorescence microscope (Leica Microsystems, Germany). BrdU+ cells were counted by the experimenter who was blind to the experimental conditions during quantification procedures. The number of BrdU+ cells was counted in the dentate gyrus of hippocampus as described previously [34]. In brief, the magnified fluorescent images (×400) of immunoreactive BrdU+/DAPI+ cells were counted and the area of the dentate gyrus was determined using free image analysis software ImageJ (NIH, MD, USA). Similarly, the number of GFAP+ and Iba-1+ cells in cortex and hippocampus were counted. Finally, cell counts were divided by the area of the section and expressed as cells per square millimiter [34]. The mean fluorescent intensity of GFAP or Iba-1 immunoreactivity images was also measured by image analysis software ImageJ (NIH, MD, USA) the after subtracting background levels [30]. For comparison between different groups, immunofluorescence intensity was quantified within fixed areas of the cortex and hippocampus for each animal by two different examiners that were blind to experimental parameters [30].
A skeleton analysis method was used to quantify the microglia morphology in immunofluorescent images as described previously [30, 35]. In brief, the maximum intensity projection of the Iba-1 positive channel and the image was converted first to a binary and then skeletonized using ImageJ software (Supplementary Fig. 1). The AnalyzeSkeleton plugin (http://imagejdocu.tudor.lu/) was then applied to all skeletonized images to collect data on the number of endpoints per frame (Supplementary Fig. 1, blue) and process length (Supplementary Fig. 1, orange) as reported previously [30, 35]. The data was quantified by an experimenter who was blind to the experimental groups. The morphological analysis of astrocytes was performed on the immunofluorescent images by Leica Qwin software (Version 3.1) using interactive measurement application tool as described previously [30]. Soma area was calculated by an experimenter blind to the experimental groups and the data were analyzed by a second experimenter. For quantitative analysis, soma area was determined and presented as the soma area (in μM2) [30].
RNA Preparation and RT-PCR
Quantitative gene expression analysis was performed by using SYBR Green technology as described previously [36]. Briefly, the total RNA was extracted from different groups using TRIzol (Sigma, USA) isolation procedure and complementary DNA (cDNA) was synthesized using RevertAid™ H Minus first-strand cDNA synthesis kit following the manufacturer’s protocol (Invitrogen, USA). Messenger RNA (mRNA) expression of key genes associated with the neuronal fate determination was quantified using the following primers as described previously [37]. Nestin (Forward-CCCTTAGTCTGGAGGTGGCTA and Reverse-GGTGTCTGCAACCGAGAGTT), Neurogenin (Forward-CTCGGGGAGCACCCTTAC and Reverse-CAGGTATCCCTCCCCTCTTTA), Neuro-D1 (Forward-ACGCAGAAGGCAAGGTGT and R-TTTGGTCATGTTTCCACTTCC), Axin (Forward-GCAGGACCCACATCCTTCT and Reverse-AGCCAGTCTCTTTGGCTCTTT), Cyclin D1 (Forward-GCACAACGCACTTTCTTTCC and Reverse-TCCAGAAGGGCTTCAATCTG), Wnt-3 (Forward-GCTACTCGGCCTCCTGCT and Reverse-GGCCAGAGACGTGTACTGCT), LRP-5 (Forward-CATCCATGCTGTGGAGGA and Reverse-TGTCTCGGGCACAAGGAT), BMP-2 (Forward-GGAAAACTTCCCGACGCTTCT and Reverse-CCTGCATTTGTTCCCGAAAA) and β-actin (Forward-CCCGCGAGTACAACCTTCT and Reverse-CGTCATCCATGGCGAACT). Real-time RT-PCR was carried out in QuantStudio 12 K Flex Real-Time PCR System (Applied Biosciences Indianapolis, USA). Relative mRNA expression was calculated by using comparative cycle threshold (2 − ΔΔ Ct) method using β-actin as an internal standard and the relative amount of mRNA was presented in the form of fold change over control [36].
Statistical Analysis
Statistical analysis was done by using Prism software version 5.0 (Graph Pad Software, San Diego, CA, USA). All Results are expressed as mean ± SEM. Statistical significance was evaluated by student’s t test and one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. A value of p < 0.05 was considered to be statistically significant.
Results
Candesartan Prevented the Increase in the BP in Established Model of Hypertension
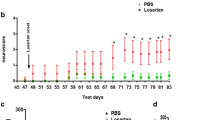
The temporal (weekly) changes in the hemodynamic parameters like SBP and HR in rat model of renovascular hypertension are shown in Fig. 1. The significant increase in SBP was observed after week 2 which continued to rise till the end of week 5. Further, HR significantly increased after week 4 as compared to sham (normotensive) rats. On the contrary, treatment with Candesartan at anti-hypertensive dose (2 mg/kg) prevented the development of hypertension in renovascular rats. However, candesartan at a dose of 0.1 mg/kg had no effect on the BP, validating that the dose of 0.1 mg/kg as low one (Fig. 1).

Effect of candesartan on hemodynamic parameters in hypertensive rats. Candesartan at 0.1 mg/kg had no effect on SBP or HR, validating the dose of 0.1 mg/kg as non-anti-hypertensive. However, candesartan at 2 mg/kg reduced the SBP and HR in hypertensive rats. Data values are expressed as mean ± S.E.M (n = 6). *Significant increase (**p < 0.01) in comparison to sham. #Significant decrease (# p < 0.05 and ## p < 0.01) in comparison to hypertension
AT1R Blockade Ameliorated Hypertension Induced Astrocyte and Microglial Activation in Brain
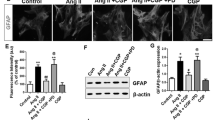
With the rise in the BP, we observed the marked astrocyte and microglial activation (gliosis) as revealed by increased immunostaining of GFAP (astroglia marker) and Iba-1(microglia marker) in both hippocampus and cortex (Figs. 2 and 3; Supplementary Figs. 2, 4). Further, astrocytes in sham rats (normotensive rats) had thin and fine processes with small cell body (Table 1); however, at 3rd, 4th, and 5th week of hypertension, the astrocytic GFAP immunostaining was more intense and hypertrophied astrocytes, i.e., thick and dense processes and enlarged cell body were observed (Table 1). Similarly, microglia displayed non-activated/resting phenotype, i.e., ramified and profusely branched processes in sham rats compared to the activated phenotype evidenced by decreased branch length, thick processes, and large cell body (Table 2) at 3rd week and ultimately amoeboid phenotype at 4th and 5th week of hypertension in both hippocampus and cortex (Fig. 3a, b; Table 2; Supplementary Fig. 4) of hypertensive rats. However, the most prominent astrocyte and microglial and morphological change were observed by 5th week of hypertension. Treatment with candesartan at either low (0.1 mg/kg) or anti-hypertensive (2 mg/kg) doses, in a time-dependent manner, prevented the activation of astrocytes (decreased GFAP immunostaining) and microglia (decreased Iba-1 immunostaining) along with change in cell morphology as evident from decreased hypertrophy in astroglia (Fig. 2; Table 1) and increased ramification in microglia (Fig. 3; Table 2) in the brain of hypertensive rats. However, the cell quantification of GFAP+ and Iba-1+ cells revealed that there is an increase (though not significant) in the number of GFAP+ cells, but not in Iba-1+ cells in hypertensive rats as compared to the sham that was reversed by Candesartan at both the doses (Supplementary Fig. 3a, b and 5a, b).

AT1R blockade prevented hypertension induced astrogliosis in brain. Representative fluorescent images of astrocytes (GFAP+) in the (a) hippocampus and (b) cortex of sham, hypertensive and Candesartan treated rats (n = 5). Representative higher magnification images showing morphological changes in astrocytes during hypertension. The resting astrocytes, with thin fine processes and lightly stained cell body, as observed in sham (arrows) were hypertrophied (arrows), having thick and bushy processes with intensely stained cell body during hypertension that was reversed by Candesartan treatment, even at low dose. Scale bar represents 50 or 20 μm

AT1R blockade ameliorated hypertension induced microgliosis in brain. Representative fluorescent images of Iba-1 positive microglia along with magnified skeletonized inset from (a) hippocampus and (b) cortex of sham, hypertensive and Candesartan treated rats (n = 5). Microglia process endpoints/cell and process length/cell were significantly decreased in hypertension as compared to the sham that was mitigated by Candesartan treatment even at low dose. Scale bar represents 50 or 20 μm
AT1R Blockade Prevented Hypertension Induced Imbalance in Inflammatory Cytokines in Brain
Consistent with the activation of glia, we observed a time-dependent increase in the level of pro-inflammatory TNF-α and decrease in level of anti-inflammatory cytokine IL-10 in hypertensive rats (Table 3). However, treatment with Candesartan both at low (0.1 mg/kg) or anti-hypertensive (2 mg/kg) dose, in a time-dependent manner, prevented the imbalance in the inflammatory cytokines as evident from decrease in TNF-α and increase in IL-10 levels in the hippocampus and cortex (Table 3) of hypertensive rats.
AT1R Blockade Prevented the Increased Expression of NADPH Oxidase Subunits and ROS Generation in Hypertension
Since robust activation of astrocyte and microglia and inflammatory response were observed at 5 weeks of hypertension, therefore, the mechanistic studies were carried out at this time point. Since, NADPH oxidase activation results in ROS generation in brain, therefore, we assessed the expression of NADPH oxidase in hypertensive rats. Hypertension induced a significant increase in the expression of major NADPH oxidase subunits, gp91phox, p47phox, p40phox, and p22phox in the cortex and hippocampus regions of brain as compared to the sham (Fig. 4a, b). On the contrary, candesartan at either low (0.1 mg/kg) or anti-hypertensive (2 mg/kg) dose blunted the increased expression of NADPH oxidase subunits in both cortex and hippocampus (Fig. 4a, b).

AT1R blockade even at low dose prevented NADPH oxidase activity and ROS generation in hypertensive rats. Immunoblot analysis of NADPH oxidase subunits in (a) cortex and (b) hippocampus. c Representative flow cytometry images for the DCF-DA positive cells in cortex and hippocampus. Data values are expressed as mean ± S.E.M (n = 5). *Significant increase (**p < 0.01) in comparison to sham. #Significant decrease (# p < 0.05 and ## p < 0.01) in comparison to hypertension
Further hypertension by increasing the NADPH oxidase activity (major ROS generating system) resulted in increased generation of ROS (Fig. 4c) in both the brain regions of hypertensive rats. However, candesartan at both doses attenuated the hypertension induced ROS generation in both the brain regions (Fig. 4c).
AT1R Blockade Prevented Hypertension Induced IκB-α Degradation and Nuclear Translocation of NFкB in Brain
Activation of NFкB is central to astrocyte and microglial activation, by regulating the expression of various inflammatory mediators and is usually held in an inactive state by IκB-α (physiological NFкB inhibitor) [5, 30]. In the present study, hypertension induced degradation of IκB-α and resulted in the nuclear translocation of NFкB (increased expression of nuclear p65-NFkB expression) in both cortex and hippocampus (Fig. 5a). However, candesartan at both doses (0.1 mg/kg and 2 mg/kg) prevented the activation of NFкB signaling cascade as evident from the stabilization of IκB-α and suppression of nuclear translocation of NFкB in both the brain regions (Fig. 5a).

AT1R blockade prevented NFкB and MAPK inflammatory signaling in hypertensive rats. a Representative immunoblots and quantification of IκB-α and nuclear p65-NFkB subunit in cortex and hippocampus. b Representative immunoblots and quantification pERK1/2 and pP38 MAPK in cortex and hippocampus. The IκB-α expression was normalized by β-actin; p65-NFκB expression was quantified by normalization with histone H3 expression and pP38 and pERK1/2 by total P38 and ERK1/2 expression, respectively. Data values are expressed as mean ± S.E.M (n = 5). *Significant increase (**p < 0.01) in comparison to sham. #Significant decrease (# p < 0.05 and ## p < 0.01) in comparison to hypertension
AT1R Blockade Ameliorated Hypertension Induced Activation of MAPK Signaling in Brain
Astrocyte and microglial activation has been associated with the activation of MAP kinases. Therefore, we assessed the activation of p38 and ERK1/2 in the hypertensive rats. Hypertension significantly increased the phosphorylation of p38 MAPK and ERK1/2 in both cortex and hippocampus regions of the brain (Fig. 5b). However, candesartan at either low (0.1 mg/kg) or anti-hypertensive (2 mg/kg) doses attenuated the hypertension induced phosphorylation of p38 and ERK1/2 MAPK kinases in both brain regions (Fig. 5b).
AT1R Blockade Promoted Hippocampal Neurogenesis in the Hypertensive Rats
We observed an increase in the number of BrdU+ cells in the hippocampus of hypertensive rats as compared to normotensive rats (Fig. 6a). Further, AT1R blockade by Candesartan promoted the proliferation of BrdU+ cells in the hippocampus of hypertensive rats (Fig. 6a). However, we observed that hypertension decreased the population of immature neurons as evidenced by the decrease in the DCX expression and number of DCX+ (immature neuronal marker) cells in the hippocampus of hypertensive rats (Fig. 6b). On the contrary, Candesartan, independent of its BP regulating action, increased the DCX expression and number of DCX+ cells in the hippocampus of hypertensive rats (Fig. 6b).

AT1R blockade promoted proliferation of neuronal stem cells and neurogenesis in brain of hypertensive rats. a Representative images and quantification of BrdU+ cells in the hippocampus. b Representative fluorescent and immunoblot images and quantification of DCX+ cells in the hippocampus. Scale bars: 50 and 100 μM. c qPCR measurement of mRNA expression of neurogenic genes NeuroD1 and Neurogenin and gliogenic BMP-2. Data values are expressed as mean ± S.E.M (n = 4). *Significant increase (*p < 0.05) in comparison to sham and #Significant increase/decrease (### p < 0.001) in comparison to hypertension
Further, hypertension decreased the expression of neurogenic genes like NeuroD1 and Neurogenin (important for neuronal fate and differentiation), while increased the gliogenic BMP2 expression in the hippocampus of hypertensive rats (Fig. 6c). However, we did not find any change in the neuronal precursor cell marker Nestin in the hypertensive state (Fig. 6c). In contrast, candesartan even at low dose (0.1 mg/kg) significantly increased the expression of neurogenic genes NeuroD1 and Neurogenin, while strongly suppressing the gliogenic BMP2 expression in the hippocampus of hypertensive rats (Fig. 6c). These data clearly indicated that AT1R blockade promoted the hippocampal neurogenesis by suppressing gliogenic while enhancing neurogenic modulators.
AT1R Blockade Promoted the Activation of Wnt/β-Catenin Signaling in the Hypertensive Rats
Recently, Wnt/β-Catenin pathway has been demonstrated to be involved in cell survival, proliferation, and differentiation of neuronal precursors [38]. Further, the characteristic of the Wnt/β-catenin signaling is the stabilization of cytosolic β-catenin by the inhibition of GSK-3β which targets β-catenin for ubiquitination and degradation by the proteasome [39]. We therefore addressed the link between hypertension and Wnt signaling. We observed that hypertension impaired Wnt/β-Catenin signaling as evident from the decreased expression of Wnt specific genes like Wnt-3A, LRP-5, Axin, and Cyclin D1 (Fig. 7a). Further, hypertension increased the GSK-3β activation (significant increase in phosph-Y-216/phosph-S-9 GSK-3β ratio) (Fig. 7b), β-catenin degradation (increased phosphorylation of β-catenin) and decreased nuclear translocation of β-catenin (Fig. 7c) in the hippocampus of hypertensive rats. On the contrary, candesartan even at low dose (0.1 mg/kg) promoted the activation of Wnt/β-Catenin specific genes, inhibited GSK-3β activation and stabilized and promoted the nuclear translocation of β-Catenin in the hippocampus of hypertensive rats (Fig. 7a–c).

AT1R blockade promoted the Wnt/β-catenin target genes in hypertensive rats. a qPCR of gene expression of Wnt-3A, LRP-5, Axin, and Cyclin D1. b Representative immunoblot images and quantification of p-Y216-GSK-3 β, p-S9-GSK-3 β, and GSK-3 β. c Representative immunoblot images and quantification of p-β-catenin in cytosol and β-catenin in nucleus. Data represent mean ± S.E.M (n = 4). *Significant increase (*p < 0.05) in comparison to sham. #Significant increase/decrease (## p < 0.01) in comparison to hypertension
Discussion
Hypertension is reported to increase the susceptibility to neurodegeneration [9], however, the involvement of glia (astrocytes and microglia) and AT1R in neuroinflammation and neurogenesis in hypertensive state is still elusive. Our study revealed that hypertension induced astrocytes and microglial activation, neuroinflammation and impaired hippocampal neurogenesis. On the contrary, AT1R blockade by candesartan, independent of BP-lowering action, attenuated astrocytes and microglial activation, neuroinflammatory changes and promoted hippocampal neurogenesis in hypertensive rats. Mechanistically, AT1R blockade prevented the activation of NADPH oxidase, MAP kinase, and NFкB, while promoting the Wnt/β-catenin signaling in hypertensive rats.
Although often neglected, the brain is one of the main target organs subjected to detrimental dysfunctions caused by high blood pressure [9]. Importantly several studies have revealed that hypertension could modulate the initiation and development of cognitive impairment by inducing neuroinflammatory processes in brain [2,3,4, 40, 41]. Since CNS is considered an immune privileged tissue, therefore, in brain, the inflammatory responses are mainly orchestrated by the activation of astrocytes and microglia [42,43,44]. In the present study, we also observed that hypertension induced activation of astrocyte (increased GFAP expression) and microglial (increased Iba-1 expression) along with the morphological transformation from resting to active state. Similarly, the previous studies have demonstrated that astrocytes and microglial activation is associated with the change in morphology during CNS inflammatory disease [45,46,47,48,49]. Consistent with the temporal activation of astrocytes and microglia, we witnessed a shift towards pro-inflammatory TNF-α and decrease in anti-inflammatory IL-10 in both cortex and hippocampus of hypertensive rats. Similarly, previous reports have demonstrated an imbalance in inflammatory cytokines following activation of astrocytes and microglia in both in vitro and in vivo studies [4, 50, 51]. Of note, AT1R blockade by candesartan, even at low (0.1 mg/kg) dose, prevented astrocyte and microglial activation and subsequent imbalance in inflammatory cytokines in the hypertensive rats. These results suggest that AT1R blockade shows anti-inflammatory action, independent of its BP-lowering effect.
Several reports have implicated the NADPH oxidase-mediated ROS generation and activation of NFкB and MAPK as a pathological hallmark of glial activation during neuroinflammatory or neurodegenerative diseases like AD and PD [9, 40, 52,53,54,55]. Similarly, in the present study, we found that hypertension induced astrocyte and microglial activation was associated with the increased activation of NADPH oxidase, ROS production, NFкB, and MAPK activation (evidenced by the IκB-α degradation, NFкB nuclear translocation, and p38 and ERK1/2 MAP Kinase activation). However, AT1R blockade by candesartan, even at low dose of 0.1 mg/kg prevented the hypertension induced NADPH oxidase activation, ROS generation, NFкB nuclear translocation and p38 and ERK1/2 MAP kinase activation in the brain of hypertensive rats. Likewise, previous studies have reported that inhibition of NADPH oxidase or NFкB prevents astrocyte and microglial activation in rodent models of dementia and in vitro culture of astrocytes and microglia [4, 52,54,55,56,57,58,59]. Conclusively, these results suggest that AT1R blockade, independent of its BP lowering effect, prevents the activation of NADPH oxidase, ROS production, NFкB and MAPK signaling during hypertensive state.
Besides neuroinflammatory changes, impaired neurogenesis has been implicated in pathogenesis of AD [60]. In the present study, we also observed that hypertension impaired adult hippocampal neurogenesis as apparent from the decreased DCX+ cells and neurogenic genes NeuroD1 and Neurogenin (important for neuronal commitment, differentiation and neurogenesis). Notably, the gliogenic BMP-2 (important for glial commitment and gliogenesis) was upregulated in hypertensive rats. On the contrary, AT1R blockade by candesartan reversed hypertension induced impaired neurogenesis by increasing the number of DCX+ cells and the expression of neurogenic genes Neurogenin and Neuro-D1, while decreasing gliogenic BMP-2 gene expression. Similarly, previous reports demonstrated that decreased expression of neurogenic genes and neuroinflammatory conditions impaired hippocampal neurogenesis [61,61,62]. However, we observed the expression of Nestin did not change during the hypertensive state and Candesartan treatment slightly increased its expression. Therefore, we conclude that AT1R blockade promotes neurogenesis by activating neurogenic genes and decreasing gliogenic genes, without affecting the neuronal precursor population in hypertensive state.
Apart from neurogenic factors, Wnt/β-catenin signaling plays an indispensable role in neurogenesis [63, 64]. Importantly, attenuation of canonical Wnt signaling has been reported to promote the gliogenesis in neural stem cells (NSCs) [38]. In the present study, we observed attenuation of Wnt signaling specific genes like Wnt-3a, LRP-5, Axin and Cyclin D1 in the hippocampus of hypertensive rats. Further, activation of GSK-3β (increased p-Y216-GSK-3β/GSK-3β and decreased p-S9-GSK-3β/GSK-3β ratio), a central component of Wnt/β-catenin pathway, promoted the degradation of β-catenin in the hypertensive rats. In contrast, AT1R blockade, even at low dose (0.1 mg/kg) attenuated GSK-3β activation, while promoting the expression of Wnt signaling genes in the hypertensive rats. Our results are backed by the previous studies which reveal that ARBs, by attenuating GSK-3β activation, show neuroprotection in neurodegenerative diseases [56, 65]. Taken together, it can be inferred that AT1R blockade, through activation of Wnt/β-catenin pathway, improves hippocampal neurogenesis in hypertension.
In summary, we show that hypertension induces robust astrocyte and microglial activation, neuroinflammation and cripples neurogenesis in brain. Of note, AT1R blockade by candesartan, independent of its BP lowering effect, prevented astrocyte and microglial activation and associated neuroinflammation in the brain of hypertensive rats. Mechanistically, AT1R blockade prevented the activation of NADPH oxidase, MAP kinase and NFкB. Importantly, we, for the first time to our knowledge; provide the evidence that AT1R blockade, by activating Wnt/β-catenin signaling, promotes hippocampal neurogenesis in hypertension.
References
Stumpf C, John S, Jukic J, Yilmaz A, Raaz D, Schmieder RE, Daniel WG, Garlichs CD (2005) Enhanced levels of platelet P-selectin and circulating cytokines in young patients with mild arterial hypertension. J Hypertens 23:995–1000
Fogari R, Mugellini A, Zoppi A, Marasi G, Pasotti C, Poletti L, Rinaldi A, Preti P (2004) Effects of valsartan compared with enalapril on blood pressure and cognitive function in elderly patients with essential hypertension. Eur J Clin Pharmacol 59:863–868
Fogari R, Mugellini A, Zoppi A, Lazzari P, Destro M, Rinaldi A, Preti P (2006) Effect of telmisartan/hydrochlorothiazide vslisinopril/hydrochlorothiazide combination on ambulatory blood pressure and cognitive function in elderly hypertensive patients. J Hum Hypertens 20(3):177–185
Saavedra JM (2012) Angiotensin II AT(1) receptor blockers as treatments for inflammatory brain disorders. Clin Sci (Lond) 123:567–590
Bhat SA, Goel R, Shukla R, Hanif K (2016) Angiotensin receptor blockade modulates NFкB and STAT3 signalling and inhibits glial activation and neuroinflammation better than angiotensin converting enzyme inhibition. Mol Neurobiol 53(10):6950–6967
Tansey MG, McCoy MK, Frank-Cannon TC (2007) Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208(1):1–25
Guadagno J, Xu X, Karajgikar M, Brown A, Cregan SP (2013) Microglia-derived TNFalpha induces apoptosis in neural precursor cells via transcriptional activation of the Bcl-2 family member puma. Cell Death Dis 4:e538
Wang L, Hagemann TL, Kalwa H, Michel T, Messing A, Feany MB (2015) Nitric oxide mediates glial-induced neurodegeneration in Alexander disease. Nat Commun 6:8966
Goel R, Bhat SA, Rajasekar N, Hanif K, Nath C, Shukla R (2015) Hypertension exacerbates predisposition to neurodegeneration and memory impairment in the presence of a neuroinflammatory stimulus: protection by angiotensin converting enzyme inhibition. Pharmacol Biochem Behav 133:132–145
Stornetta RL, Hawelu-Johnson CL, Guyenet PG, Lynch KR (1988) Astrocytes synthesize angiotensinogen in brain. Science 242:1444–1446
McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen et al (2003) The brain renin-angiotensin system: location and physiological roles. Int J Biochem Cell Biol 35(6):901–918
Hajjar I, Brown L, Mack WJ, Chui H (2012) Impact of angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch Neurol 69:1632–1638. https://doi.org/10.1001/archneurol.2012.1010
Kume K, Hanyu H, Sakurai H, Takada Y, Onuma T, Iwamoto T (2012) Effects of telmisartan on cognition and regional cerebral blood flow in hypertensive patients with Alzheimer’s disease. Geriatr Gerontol Int 12:207–214. https://doi.org/10.1111/j.1447-0594.2011.00746.x
Mogi M, Li JM, Tsukuda K, Iwanami J, Min LJ, Sakata A, Fujita T, Iwai M et al (2008) Telmisartan prevented cognitive decline partly due to PPAR-gamma activation. Biochem Biophys Res Commun 375(3):446–449
Tsukuda K, Mogi M, Iwanami J, Min LJ, Sakata A, Jing F, Iwai M, Horiuchi M (2009) Cognitive deficit in amyloid-β-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-γ activation. Hypertension 54:782–787
Saavedra JM (2016) Evidence to consider angiotensin ii receptor blockers for the treatment of early Alzheimer’s disease. Cell Mol Neurobiol 36(2):259–279. https://doi.org/10.1007/s10571-015-0327-y
Danielyan L, Lourhmati A, Verleysdonk S, Kabisch D, Proksch B, Thiess U, Umbreen S, Schmidt B et al (2007) Angiotensin receptor type 1 blockade in astroglia decreases hypoxia-induced cell damage and TNFa release. Neurochem Res 32:1489–1498
Wu X, Kihara T, Hongo H, Akaike A, Niidome T, Sugimoto H (2010) Angiotensin receptor type 1 antagonists protect against neuronal injury induced by oxygen-glucose depletion. Br J Pharmacol 161:33–50. https://doi.org/10.1111/j.1476-5381.2010.00840.x
Tota S, Hanif K, Kamat PK, Najmi AK, Nath C (2012) Role of central angiotensin receptors in scopolamine-induced impairment in memory, cerebral blood flow, and cholinergic function. Psychopharmacology 222(2):185–202
Tota S, Kamat PK, Awasthi H, Singh N, Raghubir R, Nath C, Hanif K (2009) Candesartan improves memory decline in mice: involvement of AT1 receptors in memory deficit induced by intracerebral streptozotocin. Behav Brain Res 199(2):235–240
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140(6):918–934
Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia et al (2006) Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A 103:17501–17506
Kempermann G, Song H, Gage FH (2015) Neurogenesis in the adult hippocampus. Cold Spring Harb Perspect Biol 7:a018812
Pietranera L, Saravia F, Deniselle MCG, Roig P, Lima A, De Nicola AF (2006) Abnormalities of the hippocampus are similar in deoxycorticosterone acetate-salt hypertensive rats and spontaneously hypertensive rats. J Neuroendocrinol 18:466–474
Pietranera L, Lima A, Roig P, De Nicola AF (2010) Involvement of brain-derived neurotrophic factor and neurogenesis in oestradiol neuroprotection of the hippocampus of hypertensive rats. J Neuroendocrinol 22:1082–1092
Hwang IK, Yoon YS, Choi JH, Yoo KY, Yi SS, Chung et al (2008) Doublecortin-immunoreactive neuronal precursors in the dentate gyrus of spontaneously hypertensive rats at various age stages: Comparison with Sprague-Dawley rats. J Vet Med Sci 70:373–377
Perfilieva E, Risedal A, Nyberg J, Johansson BB, Eriksson PS (2001) Gender and strain influence on neurogenesis in dentate gyrus of young rats. J Cereb Blood Flow Metab 21:211–217
Kronenberg GL, Lippoldt A, Kempermann G (2007) Two genetic rat models of arterial hypertension show different mechanisms by which adult hippocampal neurogenesis is increased. Dev Neurosci 29(1–2):124–133
Kim S, Zhan Y, Izumi Y, Iwao H (2000) Cardiovascular effects of combination of perindopril, candesartan, and amlodipine in hypertensive rats. Hypertension 35(3):769–774
Bhat SA, Goel R, Shukla R, Hanif K (2016) Platelet CD40L induces activation of astrocytes and microglia in hypertension. Brain Behav Immun 59:173–189. https://doi.org/10.1016/j.bbi.2016.09.021
Deng W, Aimone JB, Gage FH (2010) New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11:339–350
Mandyam CD, Harburg GC, Eisch AJ (2007) Determination of key aspects of precursor cell proliferation, cell cycle length and kinetics in the adult mouse subgranular zone. Neuroscience 146:108–122
Paxinos G, Watson C (1982) The rat brain in stereotaxic coordinates. Academic Press, San Diego
Nixon K, Crews FT (2004) Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. J Neurosci 24(43):9714–9722
Morrison HW, Filosa JA (2013) A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation 10:4
Khanna V, Jain M, Singh V, Kanshana JS, Prakash P, Barthwal MK et al (2013) Cholesterol diet withdrawal leads to an initial plaque instability and subsequent regression of accelerated iliac artery atherosclerosis in rabbits. PLoS One 8:e77037
Tiwari SK, Agarwal S, Seth B, Yadav A, Nair S, Bhatnagar P et al (2014) Curcumin-loaded nanoparticles potently induce adult neurogenesis and reverse cognitive deficits in Alzheimer’s disease model via canonical Wnt/beta-catenin pathway. ACS Nano 8:76–103
Kalani MY, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL et al (2008) Wnt-mediated self-renewal of neural stem/progenitor cells. Proc Natl Acad Sci U S A 105:16970–16975
L’Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Deleidi M et al (2012) Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson’s disease involves cross talk between inflammatory and Wnt/beta-catenin signaling pathways: functional consequences for neuroprotection and repair. J Neurosci 32:2062–2085
Frishman WH (2002) Are antihypertensive agents protective against dementia? A review of clinical and preclinical data. Heart Dis 4:380–386
Manolio TA, Olson J, Longstreth WT (2003) Hypertension and cognitive function: pathophysiologic effects of hypertension on the brain. Curr Hypertens Rep 5:255–261
Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, Doolittle ND et al (2013) Immunologic privilege in the central nervous system and the blood–brain barrier. J Cereb Blood Flow Metab 33(1):13–21
Cerbai F, Lana D, Nosi D, Petkova-Kirova P, Zecchi S, Brothers HM et al (2012) The neuronastrocyte- microglia triad in normal brain ageing and in a model of neuroinfl ammation in the rat hippocampus. PLoS One 7(9):e45250
Jensen CJ, Massie A, De Keyser J (2013) Immune players in the CNS: the astrocyte. J Neuroimmun Pharmacol 8(4):824–839
Patro IK, Pathak S, Patro N (2005) Central responses to peripheral nerve injury: role of non-neuronal cells. Molecular and Cellular Neurobiology 217
Patro N, Nagayach A, Patro IK (2010) Iba1 expressing microglia in the dorsal root ganglia become activated following peripheral nerve injury in rats. Indian J Exp Biol 48:110–116
Nagayach A, Patro N, Patro I (2014) Astrocytic and microglial response in experimentally induced diabetic rat brain. Metab Brain Dis 29:747–761
Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T (2001) Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol 60(9):906–916
Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodríguez JJ (2010) Astrocytes in Alzheimer’s disease. Neurotherapeutics 7:399–412
Shi P, Diez-Freire C, Jun JY, Qi Y (2010) Brain microglial cytokines in neurogenic hypertension. Hypertension 56:297–303
Sriramula S, Cardinale J, Pariaut R, Francis J (2008) Central nervous system blockade of tumor necrosis factor attenuates angiotensin II induced hypertension. Circulation 118:S383
Uttara B, Singh AV, Zamboni P, Mahajan RT (2009) Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 7(1):65–74
Kim GH, Kim JE, Rhie SJ, Yoon S (2015) The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol 24(4):325–340
Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, Chui D, Yu AC (2011) Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 8(1):67–80
Zhang ZH, Yu Y, Wei SG, Felder RB (2010) Centrally administered lipopolysaccharide elicits sympathetic excitation via NAD(P)H oxidase-dependent mitogen-activated protein kinase signaling. J Hypertens 28(4):806–816
Pang T, Wang J, Benicky J, Sánchez-Lemus E, Saavedra JM (2012) Telmisartan directly ameliorates the neuronal inflammatory response to IL-1β partly through the JNK/c-Jun and NADPH oxidase pathways. J Neuroinflammation 9:102
Dong YF, Kataoka K, Tokutomi Y, Nako H, Nakamura T, Toyama K, Sueta D, Koibuchi N et al (2011) Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J 25(9):2911–2920
Shi P, Raizada MK, Sumners C (2010) Brain cytokines as neuromodulators in cardiovascular ontrol. Clin Exp Pharmacol Physiol 37(2):e52–e57
McCarthy CA, Facey LJ, Widdop RE (2014) The protective arms of the renin-angiontensin system in stroke. Curr Hypertens Rep 16(7):440
Varela-Nallar L, Inestrosa NC (2013) Wnt signaling in the regulation of adult hippocampal neurogenesis. Front Cell Neurosci 7:100. https://doi.org/10.3389/fncel.2013.00100
Gerlach J, Donkels C, Münzner G, Haas CA (2016) Persistent gliosis interferes with neurogenesis in organotypic hippocampal slice cultures. Front Cell Neurosci 10:131
Belarbi K, Rosi S (2013) Modulation of adult-born neurons in the inflamed hippocampus. Front Cell Neurosci 7:145. https://doi.org/10.3389/fncel.2013.00145
Russo I, Barlati S, Bosetti F (2011) Effects of neuroinflammation on the regenerative capacity of brain stem cells. J Neurochem 116:947–956
Kunke D, Bryja V, Mygland L, Arenas E, Krauss S (2009) Inhibition of canonical Wnt signaling promotes gliogenesis in P0- NSCs. Biochem Biophys Res Commun 386:628–633
Agrawal A, Shukla R, Tripathi LM, Pandey VC, Srimal RC (1996) Permeability function related to cerebral microvessels enzymes during ageing in rats. Int Jr Devl Neuroscience 14:87–91
Acknowledgments
The authors are highly thankful to Mr. A. L. Vishwakarma, Mrs. M. Chaturvedi and Mr. Dhananjay Sharma for their help with the flow cytometry and confocal microscopy procedures, respectively. We are extremely thankful to Ms. Anika Sood and Ms. Zoya Fatima for quantification of immunohistochemical data in the study. We are highly thankful to Mr. Jitender Singh Kanshana and Mr. Anant Jaiswal for help in real-time PCR studies. We also acknowledge THUNDER (BSC0102) and MoES (GAP0118) for the confocal facility. The CSIR-CDRI Communication number of this article is 9554.
Funding
The study was supported by a financial grant to Kashif Hanif from Department of Biotechnology (DBT, grant No. BT/PR4021/MED/30/676/2011) and CSIR Network Project MIND (BSC0115). Award of research fellowships to SAB from Indian Council of Medical Research (ICMR), and RG from UGC, New Delhi, are greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOC 53475 kb)
Rights and permissions
About this article

Cite this article
Bhat, S.A., Goel, R., Shukla, S. et al. Angiotensin Receptor Blockade by Inhibiting Glial Activation Promotes Hippocampal Neurogenesis Via Activation of Wnt/β-Catenin Signaling in Hypertension. Mol Neurobiol 55, 5282–5298 (2018). https://doi.org/10.1007/s12035-017-0754-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0754-5