
Abstract
Neurodegenerative diseases (NDs) are a cluster of diseases marked by progressive neuronal loss, axonal transport blockage, mitochondrial dysfunction, oxidative stress, neuroinflammation, and aggregation of misfolded proteins. NDs are more prevalent beyond the age of 50, and their symptoms often include motor and cognitive impairment. Even though various proteins are involved in different NDs, the mechanisms of protein misfolding and aggregation are very similar. Recently, several studies have discovered that, like prions, these misfolded proteins have the inherent capability of translocation from one neuron to another, thus having far-reaching implications for understanding the processes involved in the onset and progression of NDs, as well as the development of innovative therapy and diagnostic options. These misfolded proteins can also influence the transcription of other proteins and form aggregates, tangles, plaques, and inclusion bodies, which then accumulate in the CNS, leading to neuronal dysfunction and neurodegeneration. This review demonstrates protein misfolding and aggregation in NDs, and similarities and differences between different protein aggregates have been discussed. Furthermore, we have also reviewed the disposal of protein aggregates, the various molecular machinery involved in the process, their regulation, and how these molecular mechanisms are targeted to build innovative therapeutic and diagnostic procedures. In addition, the landscape of various therapeutic interventions for targeting protein aggregation for the effective prevention or treatment of NDs has also been discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the biggest challenges in the field of neuroscience is the disease-modifying treatment which include several neuroprotective and neurorestorative interventions that focus on slowing the progression of NDs. NDs are a plethora of debilitating and degenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), frontotemporal dementia (FTD), traumatic encephalopathy, dementia with Lewy bodies, and prion diseases [1]. Despite having a difference in pathogenesis and clinical manifestation, these NDs share several common attributes, such as increased prevalence with age, progressive nature, selective neuronal loss, and synaptic dysfunction [2,3,4,5,6]. Importantly, the most prevalent and common feature of these NDs is the occurrence of misfolded proteins and their gradual accumulation, which is thought to be the main cause of these disorders [7,8,9,10,11,12,13,14]. Alpha-synuclein (α-Syn), amyloid-beta (Aβ), huntingtin (HTT), TAR DNA-binding protein 43 (TDP-43), Tau, and superoxide dismutase (SOD) are some of the proteins that are misfolded and accumulate in NDs [10,11,12,13,14]. In this review, we focused on Aβ, tau, α-Syn, HTT, and TDP-43, as they are the most common protein aggregates involved in the pathogenesis and progression of NDs. The distribution of the misfolded proteins involved in several diseases throughout the brain is depicted in Fig. 1, and Table 1 lists the different NDs caused by protein misfolding.

Distribution of unfolded protein in the brain in neurodegenerative diseases: Specific misfolded proteins are distributed through particular regions of the brain
One of the key components of these misfolded proteins is that during misfolding from their native states, they form intermolecular β-sheet-rich structures, ranging from small oligomers to large fibrillar aggregates, in NDs [7]. Protein misfolding results either in the loss of protein function or a gain of toxic function (via aggregation), resulting in cellular responses (for instance, synaptic dysfunction, inhibition of axonal transport, mitochondrial damage, and membrane disruption) that are toxic to cells leading to the progression of NDs [3, 15,16,17,18]. Alternatively, to deal with the accumulation of misfolded proteins, the cell adopts multiple mechanisms, including their synthesis, degradation, and clearance from the cell, to prevent their misfolding and aggregation [19, 20]. Multiple therapeutic strategies that can prevent or reverse the progression of NDs are currently being studied. The primary target of these studies is misfolded proteins and the aggregates formed by these proteins [21,22,23,24,25,26,27]. However, due to the dynamic nature of these proteins, targeting them remains a challenge [28]. This review discusses the common misfolded proteins, cellular mechanisms to deal with the generation of misfolded proteins, and degradation and clearance of these misfolded proteins as a therapeutic target for NDs.
Misfolded Protein Aggregates in NDs
Numerous clinical and experimental studies have provided comprehensive and compelling evidence that the key events that cause pathogenic abnormalities in the brain are protein misfolding, oligomerization, and accumulation during NDs [9,10,11, 29, 30]. The misfolded protein aggregates commonly implicated in NDs include Aβ (in AD); tau (in AD, frontotemporal dementia, chronic traumatic encephalopathy, etc.); α-Syn (in PD, multiple system atrophy, and dementia with Lewy bodies); TAR DNA-binding protein 43 (TDP-43) (in ALS and frontotemporal dementia); and HTT (in HD and prion proteins in Creutzfeldt–Jakob disease (CJD), bovine spongiform encephalopathy, chronic wasting disease, and scrapie) (Table 1). The commonly implicated protein aggregates are discussed below.
Amyloid-beta
AD is a progressive degenerative disease in elderly individuals characterized by the predominant neuropathological feature of the extracellular deposition of misfolded proteins in the brain [15, 21, 37]. There are two major types of aggregates found in AD: (1) intracellular aggregates formed of Tau protein (neurofibrillary tangles) and (2) extracellular aggregates of Aβ (senile plaques) [16, 38, 39]. Both neurofibrillary tangles and plaques formed by the aggregation of misfolded proteins increase the severity of dementia [2, 16, 38]. Increased production or downregulation of the clearing pathway (via ubiquitin proteosome pathway) of misfolded proteins leads to their deposition and accumulation within the organism. The accumulation of such proteins within the organism is toxic and affects many vital physiological pathways, such as lipid metabolism, mitochondrial dynamics, synaptic functions, and the release of neurotransmitters [40, 41]. Increasing clinical and experimental data suggest that oligomers, rather than fibers, are the main species causing toxicity and are involved in disease seeding and spread [42, 43]. Physiologically, the accumulation of Aβ induces oxidative stress and the generation of ROS resulting in dysfunctional mitochondria, the characteristic hallmark of AD [7, 44]. Aβ also induces the activation of the IkB a/NF-kB inflammatory pathway, resulting in a vicious cycle of neuroinflammation and oxidative stress culminating in mitochondrial dysfunction, neurodegeneration, and cognitive impairment [44,45,46].
Aβ commonly occurs in two variants, Aβ40 and Aβ42. Aβ is formed from amyloid precursor protein (APP), a membrane-bound protein, by the action of β- and γ-secretase, known as the amyloidogenic pathway (Fig. 2) [47, 48]. In amyloidogenesis, APP is cleaved by β-secretase, generating APPsβ and β-C-terminal fragments followed by the action of γ-secretase, generating Aβ and amyloid precursor protein intracellular domain (AICD) by acting on the transmembrane domain [47, 48]. However, in the non-amyloidogenic pathway, α-secretase cleaves APP within the sequence of Aβ and thereby limits its production [49]. Briefly, α-secretase cleaves the ectodomain of APP, generating the APPsα fragment and α-CTF. α-CTF is then cleaved by γ-secretase into AICD and P3 peptides [38, 49]. Most of the Aβ in the human brain under normal conditions contains Aβ40, while in diseased conditions, excess Aβ42, the major constituent of amyloid plaques, is formed. The presence of 42-residue Aβ (1–42) results in severe neurotoxicity and aggregates faster than the 40-residue peptide Aβ (1–40) [29]. Although the exact function of Aβ (1–42) is not clear, few studies report that these monomers are neuroprotective in nature [50]. Monomeric Aβ lacks toxicity, and thus, the prevention of monomer aggregation can prevent toxic species formation. The primary structure of Aβ comprises an N-terminal hydrophilic region, a central region containing hydrophilic and hydrophobic residues, and a C-terminal hydrophobic end. The N-terminal region contains a β-sheet region [7, 51, 52]. The partially folded protein units combine by hydrophobic and hydrogen bonds, resulting in the formation of the perinucleus, which has the potential to associate with other Aβ molecules to form a structure called the protofibril. Protofibrils can also self-assemble to form long fibrillar aggregates [51, 52]. The formation of long fibrillar aggregates depends on the concentration of monomeric units and oligomers. Aβ monomers aggregate to form oligomers that act as intermediates that, upon further aggregation, lead to the formation of fibrils by primary nucleation. Once a particular concentration of the fibrils is attained, they catalyze the formation of oligomers on the surface (secondary nucleation) [53].

Processing of APP: (i) In amyloidogenic pathway, the transmembrane protein secretase act on Amyloid Precursor Protein (APP) and cleave to release APPsβ and β-CTF (C-terminal fragment). The latter is then cleaved by γ-Secretase and produce P3 and AICD (amyloid precursor protein intracellular domain). (ii) In non-amyloidogenic pathway, the transmembrane protein α-Secretase act on Amyloid Precursor Protein (APP) and cleave to release APPsα and α-CTF (C-terminal fragment). The latter is then cleaved by γ-Secretase and produce P3 and AICD (amyloid precursor protein intracellular domain)
Tau Protein
Tau protein, a microtubule-associated protein, represents another misfolded protein found commonly in NDs such as AD, PD, HD, and frontotemporal dementia with parkinsonism (FTDP) [15, 39, 54]. Under physiological conditions, tau is located in the axon; however, disruption of either the structure or function of the tau results in the accumulation of tau in the soma or dendrites of neurons [55, 56]. The presence of hyperphosphorylated forms of tau within the neurons that aggregate to form NFTs is another hallmark of AD [13]. The accumulation of tau increases with age and is correlated with cognitive decline, forebrain atrophy, and neuronal loss in the hippocampus and neocortex region of the brain [57]. It is believed that the amount of tau present in the brain increases approximately 10–15 years before the onset of the symptom of the disease [58].
Humans express six different Tau isoforms between 50 and 70 kDa in size, the shortest of which is present within the fetal brain, while the adult brain contains all six isoforms [33]. The variable number of specific inserts at the N-terminal region and in the microtubule-binding region (MTBR) is responsible for the presence of 6 different tau isoforms. Although all 6 isoforms are present in diseased condition such as AD, they may be present in variable quantity [33]. These tau isoforms are encoded by the microtubule-associated protein tau (MAPT) gene, which is present on chromosome 17 [8]. Tau mRNA contains 16 exons comprising an N-terminal domain, 3 or 4 microtubule-binding domains (3R or 4R), a proline-rich domain, and the C-terminal region [59]. The tau inclusions are ultrastructurally made of paired helical filaments (PHFs) and straight filaments (SFs). The core of these filaments is made of identical subunits that comprise nearly 350 tau proteins. These are known to adopt a cross β/βhelix structure [60]. Tau contains approximately 90 phosphorylation sites in the longest isoform that is known to be phosphorylated by 20 kinases [61, 62]. Tau binds to microtubules and aids in the anterograde and retrograde transport of biomolecules. Enhanced expression of tau obstructs kinesin-dependent trafficking in neuronal cells. It enhances dynein binding to the microtubule by binding to the dynein-activator complex. A low level of tau promotes kinesin binding to microtubules, which facilitates transport along the axon [63]. At the synaptic junction, increased concentrations promote kinesin release, thereby facilitating the binding of dynein to microtubules. Hyperphosphorylation results in loss of biological activity of tau, microtubule destabilization, and damage to protein transport, thus resulting in the aggregation of toxic molecules [62].
Tauopathy is also observed in HD patients. A study conducted by Nogales et al. (2014) found that the amount of the 3R binding domain isoforms in the cortex of normal and HD patients was similar; however, a marked increase in the 4R binding domain in HD patients was observed. Similarly, the 4R binding domain was found to be much more elevated than 3R isoforms in the striatum of HD patients [64]. An increase in 4R/3R is also observed in the postmortem patients who recently had undergone transplantation surgeries [65]. These experiments prove that an imbalance of 4R/3R tau binding domains results in HD.
Tau is secreted from damaged neurons in the extracellular medium (CSF; blood serum). Extracellular Tau is responsible for toxicity and can also result in damage to other neuronal cells. Extracellular tau expression increases phosphotidylserine signal on the neuronal cells that induce neuronal loss. Furthermore, extracellular Tau protein activates microglia’s phagocytic activity and enhances its proliferation. Since, in cultures where microglia were depleted, neuronal loss was not observed, microglial cells are considered to be accountable for tau-induced neurotoxicity [39]. This finding indicates that neuronal loss caused by tau is due to the activation of microglia. It is also believed that microglia-mediated inflammation causes tau pathology [66]. Co-existence of microglia activation along with tau accumulation indicates that microglial cell activation that is considered as a part of the repertoire of immune response not only acts as an inflammatory epiphenomenon but also drives tau pathology [67].
Alpha-Synuclein Protein
Alpha-synuclein is a key component that is found in the aggregates of misfolded proteins in PD [31]. Three members of the synuclein family, alpha, beta, and gamma, are present. Alpha and beta are concentrated in the neuronal terminals, while gamma is present throughout the neuron [31]. Alpha and beta can inhibit phospholipase D224, which is involved in cytoskeletal regulation and endocytosis. Thus, these forms are responsible for vesicular transport.
Alpha-synuclein is approximately 140 amino acids long and is known to play a vital role in vesicular transport. α-syn consists of 3 regions — the central hydrophobic terminal flanked by an acid carboxyl-terminal and the N-terminal that contains seven repeats of KTKEGV, which forms the membrane-binding domain [68,69,70]. The central region is responsible for fibrillization, and any deletion in this area explains fibrillization under stress conditions [71]. The circular dichroism (CD) spectra of mutant E46 monomers were found to be randomly coiled, while it exists in the β-pleated structure under assembly conditions indicating that in the oligomeric form, the structure of the protein changes to a β-pleated structure similar to Aβ [72]. The polymerization of α-syn is similar to that of Aβ and is nucleation dependent. Figure 3 depicts the polymerization of α-syn in the Lewy body. When misfolded, α-syn monomers form a β-pleat structure that shows a high affinity for the other monomers, thus resulting in the formation of oligomers. These oligomers act as intermediates and are later formed into protofibrils, fibrils, and finally Lewy bodies via nucleation. Lewy bodies are the insoluble fibrillar species containing intracellular globular cytoplasmic inclusions (consists of proteins, lipids, and membranous organelles). The process of polymerization can be divided into 3 phases. The first phase is slow that allows α-syn monomers to form partially folded oligomers ad ordered oligomers. During the second phase (elongation phase), the ordered proteins cause misfolding of other monomers resulting in formation of larger fibrils. The last phase is fast and fibrils attain their maximum size, beyond which they do not grow [73]. Postmortem brain reports show a similarity between α-syn and Aβ. Like amyloid fibrils, these are unbranched, contain an anti-parallel β-sheet structure, and are resistant to proteolysis, which is even seen in prion proteins. Alpha-synuclein also binds to thioflavin-T, which is a characteristic feature of Aβ [72]. Under acidic conditions, the net charge is neutral, decreasing the intramolecular repulsions, which makes it prone to aggregation [70, 74]. Furthermore, postmortem reports of patients suffering from PD suggest that α-syn undergoes posttranslational modifications such as ubiquitination, phosphorylation, and nitration [75]. However, Bartels et al. (2011) demonstrated that α-syn purified from the brain could form aggregates and that posttranslational modification did not alter the process of aggregation [76]. This indicates that modifications of the protein do not affect the polymerization of the protein.

Mechanism of formation of large sized aggregates in Parkinson disease: Under normal conditions, the protein (α-synuclein) is folded properly. However, environmental and genetic factors trigger misfolding of protein which leads to the formation of aggregates which results in formation of Lewy body. Formation of Lewy body causes neuronal damage which results in neurodegenerative disease, Parkinson
Alpha-synuclein has been found to directly promote SNARE (Soluble NSF Attachment protein Receptor) complex assembly (plays an important role in vesicle fusion) that involves the binding of the N-terminal of α-syn to phospholipids and the C-terminal to synaptobrevin. The SNARE complex assembly formation promotes the vesicle fusion. Loss of SNARE-mediated fusion leads to loss of neuronal function [77]. Thus, loss of function of α-syn is directly related to loss of neuronal function, and these functions of the protein are accomplished by interacting with the lipid bilayer. Pfefferkorn et al. (2010) studied the effect of tryptophan (Trp) mutation on the membrane-binding ability of α-syn. Trp contains an indole ring in its side chain, which is sensitive to the local environment; thus, Trp mutation is ideal to study the membrane-binding property of α-syn [78]. In the study, they substituted Trp and observed that although the membrane-binding affinity was not affected to a greater extent, Trp-4 and Trp-94 showed high membrane affinity. It was also observed that the membrane affinity increased under acidic conditions.
Huntingtin Protein
Guo and his colleague (2018) described the structure of HTT by cryo-electron microscopy. According to them, HTT is an α-helical protein weighing approximately 350 kDa. It consists of three domains: the N-terminal, central, and C-terminal regions. The N-terminal bears 2 membrane-binding regions and contains repeats arranged in a solenoid. The C-terminus is also believed to contain multiple HEAT repeats. The terminals are joined by the salt bridge domain. They also identified HAP-40 (huntingtin-associated protein 40) as the central regulator of proteins that stabilize HTT protein upon binding [9]. However, Guo believed that the heterogeneity in the conformation of HTT prevents a better understanding of its structure. Costanzo (2013) also demonstrated that transfer is cell_cell mediated [79]. The aggregation of these proteins is due to the expansion of polyglutamine residues in the N-terminal. The length of the repeat determines the tendency of aggregation [80]. It is believed that when mutant HTT contains more than 35 polyQ repeats, it forms oligomers, which are responsible for collapsing proteostasis, such as autophagy and the ubiquitin_proteasome system (UPS) [35]. A major mode of regulation of several cellular processes is through posttranslational modifications. These modifications include phosphorylation and acetylation, which affect the 3D structure of the protein and modulate the physiology of the protein. The cytoplasmic and nuclear localization of HTT is mediated by phosphorylation at serine and threonine residues, particularly Ser-13, Ser-16, and Thr-3 [81, 82]. Acetylation of lysine residues within the N-terminal modulates aggregation of the protein and affects membrane binding [83].
TAR DNA-Binding Protein 43
TDP-43 (TAR DNA-binding protein 43) is a versatile RNA/DNA-binding protein that is involved in RNA metabolism [84, 85]. In 2006, TDP-43 was identified as one of the key components of insoluble inclusions isolated from ALS patients. The TDP-43 protein is an approximately 400 amino acid long peptide that encodes the TARDBP gene and is located on the 1st chromosome. TDP-43 has an N-terminal domain, two RNA recognition motifs, a nuclear export signal, and a C-terminal domain. The N-terminal domain has a nuclear localization signal that allows the protein to enter the nucleus. TDP-43 is primarily localized in the nucleus but can export to the cytoplasm under specific conditions [86]. The C-terminal region has a glutamine/asparagine-rich domain and a glycine-rich region [18, 87,88,89]. This protein has multiple functions and is involved in RNA metabolism (transcription, translation, mRNA stabilization, etc.) [4, 18]. TDP-43 proteinopathies are distinguished by cytoplasmic localization of the protein, the presence of hyperphosphorylated and ubiquitinated forms of the protein, the deposition of these forms, the formation of inclusion bodies, the formation of toxic C-terminal TDP-43 fragments, and protein aggregation.
Prions Such as the Nature of Misfolded Proteins and NDs
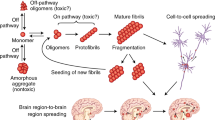
The seeding property was first established for infectious prion particles [90]; however, recent studies have demonstrated that the seeding property is inherent to all misfolded protein aggregates involved in NDs [90,91,92,93]. Prion is composed of misfolded prion protein (PrPSc) aggregates that self-replicate and spread (seed) in the infected brain. The fact that seeding is a common attribute of the misfolded protein aggregates involved in NDs suggests that such protein aggregates have the potential to be infectious and result in conversion from the normal state to a misfolded state [90, 94]. Recently, scientists have also found prion-like proteins in other NDs that are not infectious [80, 91, 93]. These proteins are known to seed by the formation of tunneling nanotubes [92] or by the process of endocytosis [95]. The process of misfolded protein seeding during NDs is supported by the fact that inoculating brain tissue homogenates from ND patients or transgenic animals containing Aβ, tau, and α-Syn protein aggregates resulted in disease induction in the recipient in vitro cellular model system or in vivo in animal models [95,96,97]. Of note, the transmission of synthetic or recombinant misfolded protein aggregates prepared in vitro has also been established. However, more efficient seeding has been witnessed with tissue homogenates than with purified or recombinant proteins, signifying the role of other cellular mediators in the pathological transmission of NDs [97,98,99]. Last, the pathogenic spreading of ND has even been witnessed with the systemic administration of seeds. However, whether spreading or seeding of protein misfolding is restricted to the protein aggregate accumulation of tissue damage and disease progression is an active area of research.
Cellular Response to Misfolding of Proteins
Misfolded proteins can have three fates — they may be degraded, refolded, or delivered to the quality control center that can sequester harmful misfolded proteins [20, 100]. The misfolded proteins present in the cytosol and the endoplasmic reticulum (ER) are recognized by cellular sensors that generate a response that blocks translation, recruits’ chaperones to refold the proteins, and if necessary, degrades the proteins or causes apoptosis [20, 101].
The unfolded protein response (UPR) is a process that maintains homeostasis within a cell. The UPR is activated under stress conditions, as in AD [102]. The ER membrane contains three proteins that act as sensors: PERK (protein kinase R-like endoplasmic reticulum kinase), IRE1 inositol requiring kinase 1), and ATF6 (activating transcription factor 6). These proteins are bound to another protein known as BiP that inactivates the proteins [100]. When BiP dissociates under stress, these proteins are activated and cause modification of the transcription and translation process and prevent cellular damage, while BiP acts as a molecular chaperone and refolds proteins (Fig. 4) [103]. The protein level of BiP/GRP78 is increased in AD, where they assist in refolding of the proteins [104]. The transmembrane protein PERK dimerizes and phosphorylates the translation initiation factor eIF2α, which blocks translation. PERK is also responsible for the activation of ATF4 which increases the expression of chaperones [105]. The expression of ATF4 for a longer duration is linked with apoptosis [100]. IRE1 homodimerizes and undergoes a conformational change that activates the RNase domain in the cytosolic region of the protein and modulates the expression of XBP-1, a transcription factor that transactivates genes related to UPR proteins [106]. Duran et al. (2017) demonstrated that IRE1 deletion resulted in reduced expression of APP in a mouse model, indicating the role of IRE1 in the pathogenesis of NDs [107]. Under stressful conditions, ATF6 is transported to the Golgi body via vesicles where its cytoplasmic domain is cleaved in the cytosol. The domain is then transported to the nucleus, where it acts as a transcription factor. It is believed that ATF6 responds to stress caused by lipids and proteins via separate mechanisms [108]. The UPR is activated in AD [102, 104].

Unfolded protein response (UPR) in diseased and normal conditions: Under normal conditions, BiP (chaperone) remains associated to the transmembrane proteins (PERK1, IRE1, ATF6). Under diseased conditions, the sensors get activated and cause transcription of genes that regulate transcription, translation, and chaperones. BiP also binds to misfolded proteins and cause refolding
The proteins in the secretory pathway are thermodynamically more stable due to posttranslational modifications (such as glycosylation or disulfide linkage); however, the proteins in the cytosol are less stable due to fewer posttranslational modifications [109]. In the cytosol, misfolded proteins with exposed hydrophobic amino acids are recognized by molecular chaperones (HSP70, HSP90, HSP100, and sHSP) [20] and are subjected to three different fates as described below:
-
A
Degradation of misfolded proteins
Misfolded intracellular proteins can be degraded by the ubiquitin_proteasome pathway (UPP), which involves the covalent binding of ubiquitin with the protein followed by the degradation of the protein. The ubiquitin molecule is released during the process and can be reused [20]. For example, Tau is ubiquitinated at the Lys254, Lys257, Lys311, Lys317, and Lys63 residues and degraded by E3 ubiquitin ligase [101, 110]. Misfolded proteins can also be transferred to acidic compartments, such as lysosomes, where the proteins are exposed to low pH, which ensures their unfolding and cleavage by proteolytic enzymes [75]. The importance of lysosomal enzymes can be predicted, as the inhibition of lysosomal enzymes in rat brains can induce the formation of tangles [111]. Recently, it has been discovered that α-syn can be degraded via selective autophagy [112], which is termed synucleinopathy. According to this mechanism, α-syn in neurons activates microglia, which engulf proteins in autophagosomes. These autophagosomes are then degraded via selective autophagy [112]. Misfolded proteins can also undergo chaperone-mediated autophagy in which HSP70 recognizes the proteins and then degrades them by lysosomes; however, this process is selective for proteins [75]. Notably, α-syn-mediated neurotoxicity was enhanced several folds when chaperone-mediated autophagy was suppressed in neurons [113].
-
B
Refolding
Upon sensing the presence of misfolded proteins, the cell selects several molecular chaperones that cause the refolding of the protein. Chaperones such as BiP are activated as discussed above by dissociating from transmembrane proteins, binding to the target protein and promoting folding. Several other chaperones in the ER are activated by the UPR, which accelerates the folding of the proteins. Heat shock proteins (HSPs) play a major role under stressful conditions, as their transcription is upregulated which promotes protein folding [114]. In the cytoplasm, the unfolded proteins are recognized by Hsp70 and Hsp40 cochaperones. More mature folding intermediates are recognized by Hsp90 or TRiC/CCT (TCP1-ring complex or chaperonin containing TCP1). Protein aggregation is also reduced by sHsps. These chaperones aim to refold misfolded proteins. The activity of each of these molecular chaperones is discussed in detail in a later section. In the ER, the roles of Hsp70, Hsp40, and Hsp90 in damage recognition are conserved, and there is no TRiC/CCT homolog. The role of sHsps is also restricted to the ER [115].
-
C
Protein sequestration
When the concentration of misfolded proteins rises, the cell employs a strategy to avoid chaperone saturation. Other misfolded proteins are sequestered in quality control centers as part of this strategy [20, 28]. The concentrated proteins stored in the centers are later degraded or refolded depending on the cellular condition. Cytosolic proteins that are misfolded or cannot be refolded or degraded accumulate in the juxtanuclear quality control compartment (JUNQ). These proteins accumulate in the JUNQ under unfavorable conditions and can be recycled to the cytoplasm when the conditions are ambient where they can be refolded [20]. When the cell fails to refold, degrade, or sequester the misfolded proteins, the concentration of misfolded protein increases within the cell. Under such conditions, the proteins are likely to form aggregates and cause cytotoxicity.
Role of Molecular Chaperones in Protein Folding/Aggregation During NDs
Under adverse cellular conditions, cells produce proteins that can maintain homeostasis within the cell. Such proteins are termed heat shock proteins (HSPs) or molecular chaperones [15, 114]. They are intricately involved in the prevalence of protein misfolding diseases by (i) preventing aggregation of wrongly folded proteins, (ii) promoting the refolding of proteins, and (iii) facilitating degradation of the misfolded protein [15]. Chaperones interact transiently with protein [116]. However, a change in the interaction between the protein and the chaperone modulates the activity of the chaperone and vice versa [68]. This has been demonstrated in a study where it was observed that a change in the activity of the chaperone modulates the interaction with protein disturbing the balance and leading to PD [68]. Based on the structural and functional similarities of these proteins, they are categorized into different families. Members of these families may be constitutively expressed or may be expressed only when required and possess the ability to recognize misfolded proteins in the cytoplasm and the ER [117, 118]. Table 2 gives an overview of molecular chaperones found within the cell. These chaperones are commonly found in the cytoplasm, but they move to other compartments under stress. It has been recently discovered that HSPs can be secreted by non-neuronal cells into the micro-environment and can be subsequently utilized by neurons. The most studied extracellular chaperone is HSP70 which is known to confer neuroprotective role (by masking toxic extracellular proteins, driving adaptive immune signaling, and stalling expression of proteins playing role in synaptic pruning). Several studies are now being conducted to treat NDs by targeting the extracellular HSPs; however, the complete knowledge of extracellular HSPs and the role played by them is imperative for developing a therapy [119, 120].
One of the largest molecular chaperone families is HSP70. HSP70 in eukaryotes occurs in two different forms: HSC70 (heat shock cognate protein 70), which is constitutively expressed and HSP70 which is expressed under stressful conditions [128]. HSP70 is known to play a major role in diseases involving misfolded proteins such as Alzheimer’s and tauopathies wherein it is responsible for facilitating refolding and disaggregation [129]. HSP70 binds to the hydrophobic region of proteins via its C-terminus in an ATP-dependent manner [114, 130]. HSP70 bound to ATP has a low affinity for the substrate. Hydrolysis of ATP increases the affinity toward the substrate. Thus, ATP hydrolysis regulates the affinity between the chaperone and the target protein. This process necessarily requires the presence of HSP40, which acts as a cochaperone and aids the process. The importance of HSP70 in proteinopathies is demonstrated by the fact that its knockdown makes neurons more vulnerable to cytotoxicity and neurodegeneration [19]. Alternatively, overexpression of HSP70 by pharmacological and genetic approaches promotes neuroprotection in misfolded NDs, such as PD [131, 132], AD [133], and HD [134]. HSP70 binds to the hydrophobic region of these proteins and promotes the recovery of the native conformation, preventing the formation of aggregates.
HSP90 is another important HSP that requires ATP hydrolysis for activation and proper functioning [135]. Several cochaperones influence the ATP cycling process and facilitate bonding with the substrate [136]. HSP90 is a dimeric protein found in the cytosol and ER. It is involved in maintaining protein homeostasis by ensuring the proper folding of proteins and degrading misfolded proteins. The two major HSP90 isoforms HSP90β are expressed constitutively, and HSP90α, whose expression is induced under stress, exists in the cytosol [122]. Selective Hsp90α/β inhibitors have been found to promote clearance of inclusion bodies formed by mutant HTT in rodents [137]. Furthermore, HSP90 binds to HTT via the N-terminus and recruits the deubiquitinating enzyme USP-19 (ubiquitin specific protease 19) to modulate the protein and aggregate of HTT [117]. Hsp90 has several cochaperones that aid in the proper functioning of HSP90 [123]. The most common cochaperones are cdc37 (cell division control protein 37), HSP82, Sti1 (stress-inducible protein 1), and PP5 (protein phosphatase 5) [123, 138]. P23 is another cochaperone of HSP90 that is also a therapeutic target, as gedunin, a p23 inhibitor, prevents neurotoxicity induced by 1-methyl-4-phenyl pyridine (MPP +) [138].
Small heat shock proteins (sHSP) are a group of proteins that weigh less than 40 kDa and are upregulated during stress [126, 139, 140]. These proteins work in an ATP-independent manner and are expressed under unfavorable conditions [139]. Small HSPs play a vital role in refolding proteins, destabilizing, and facilitating the solubilization of aggregates [125, 127, 141]. These proteins carry a conserved sequence called the α-crystallin domain between the N and C terminals. HSP27, a low-molecular weight protein, is known to mitigate cellular damage and neuronal cell death during NDs [75], whereas HSPB5 reduces cellular toxicity and prevents fibril formation by α-syn [127]. It acts as an anti-apoptotic protein and molecular chaperone when it exists in large oligomers [125]; however, it regulates the microfilament dynamics when it exists in small oligomers [141]. Reduction in the level of HSP27 with methylglyoxal results in the aggregation of α-syn, impaired clearance, and neurotoxicity, an effect that was reversed by overexpression of Hsp27 [140]. Studies have demonstrated that the terminal regions of Hsp are important for stable fibril binding and that these regions can play a cytoprotective role in vivo [126].
The Landscape of Therapeutics Targeting Protein Misfolding or Aggregation in NDs
Regardless of the extensive information and advancements in the field of NDs, no effective cure or treatments are available for preventing NDs. This is a consequence of lack of understanding of the tertiary structures of the misfolded proteins, inability of the drug to cross the BBB, recognition of few epitopes by the antibodies, etc. Despite facing these challenges, misfolded protein aggregates remain the primary target for effective therapeutic intervention for NDs. Of note, molecules/drugs are now being formulated that have the potential to diffuse or dilute the aggregates formed in the various brain regions.
Antibodies Targeting Misfolded Proteins
Recently, antibodies have been developed against misfolded proteins that can stimulate both humoral and cell-mediated immunity in the patients [142]. These antibodies are specific to the target proteins and can offer long-term clearance of these proteins. They can dissolve soluble and insoluble aggregates. Both active and passive immunizations have been explored and have shown remarkable potential for the treatment of NDs [143]. However, the major disadvantage of using antibodies is their ability to cross the blood–brain barrier and recognize only a limited number of epitopes. Table 3 shows a list of antibodies that have been generated against α-syn, Aβ, HTT, and tau. The antibodies target different regions of these proteins and aim to reduce oligomerization and prevent cell-to-cell transfer. Several of these antibodies may be supplemented with other drugs to reduce symptoms in patients suffering from NDs.
Monoclonal antibodies are considered a therapeutic strategy for AD. One such monoclonal antibody is bapineuzumab which is currently discontinued due to its lack of efficacy [168]. Crenezumab is another monoclonal IgG4 antibody that binds to monomeric and aggregates of Aβ. It is known to block the aggregation of monomers and has the potential to induce disaggregation. Crenezumab lowers the Aβ concentration in the cerebrospinal fluid in AD patients [161]. Amyloid-beta fibril formation is also inhibited by chronic intranasal treatment of single-chain fragments derived from IgG that can inhibit neurotoxicity and neutralize neurotoxicity related to Aβ in transgenic mice [184]. Another antibody studied is aducanumab which when administered to patients intravenously suffering from AD for a year, caused regression of Aβ plaques in a dose-dependent manner. However, intravenous infusion of the monoclonal antibody has several side effects on patients, including upper respiratory tract infection, urinary tract infection, and headache [162]. Due to a high number of side effects observed, aducanumab is now discontinued. Bosch (2014) showed that immunized dogs show smaller amyloid plaques than nonimmunized dogs [21]. Purified anti-Aβ 40 IgGs were given to dogs, and these exhibited stronger immunoreactivities to disperse plaques. A parallel study carried out with commercial anti-Aβ 40 IgGs demonstrated an increase in the immunoreactivity toward neuritic plaques [21]. Xing et al. (2017) developed a monoclonal antibody (3F5) against 11 amino acid residues of the N-terminal region of Aβ. Treatment with 3F5 has certain disadvantages, as it may result in microhemorrhage lesions and cerebrovascular edema; however, 3F5 also decreased the death rate of neuronal cells in mice and the amount of Aβ deposits [22]. Several other antibodies, such as solaneuzumab, gantenerumab, and ponezumab, offer passive immunity, while AN-1792, ACI-24, CAD-106, ACC-001, and ABvac40 offer active immunity and target Aβ and its aggregates [27, 158, 164, 166, 167, 169, 170, 172].
Several antibodies show the potential to target α-syn protein and cause the reduction of extracellular protein. BIIB054 and prasinezumab are the most advanced projects to date. Administration of BIIB054 has been seen to have several mild side effects, such as headaches. However, no symptoms of cardiovascular disease or stroke were visualized [149]. MEDI1341 is an antibody that targets the C-terminal of α-syn and attenuates cell-to-cell transmission of preformed fibrils in mammalian models such as mice [153]. Administration of α-syn oligomer selective antibodies (mAb47, mAb49G, mAb38E2) can increase the clearance of α-syn added exogenously. Administration of the antibody rescued the astrocyte from mitochondrial damage and decreased the aggregates. Of all the antibodies, mAb47 showed the most promising effect [144]. In addition, AFFITOPE generates anti-α-syn antibodies and reduces neuronal loss. This drug is currently under phase I clinical trial.
Administration of antibodies targeting tau protein has also been shown to reduce the polymerization of tau. Administration of the C10.2 antibody targets tau aggregates and reduces tau seeding [180]. Administration of a single chain variable fragment of MC1 reduced tau pathology in rodents. The delivery of the antibody was achieved by adeno-associated virus (AAV). Administration of the antibody decreased the insoluble and soluble tau forms in the cortex and hippocampus [185]. BIIB092 is a monoclonal antibody that recognizes full-length tau and N-terminal tau fragments. The N-terminal tau fragments are found extracellularly in the cerebrospinal fluid (CSF). Administration of this monoclonal antibody reduced the induction of tau aggregation. Antibodies targeting HTT have also been discovered; however, few antibodies targeting HTT are known. A monoclonal antibody (C6-17) is known to target the exposed region of HTT and inhibit cellular uptake of the protein in vitro [185]. Other antibodies include INT41, sc-Fv-MW1, sc-Fv-MW2, and sc-Fv-C4 target mutant forms of HTT and aggregates [182, 183, 186]. These antibodies reduced the aggregates of the protein and increased cell survival.
Targeting Chaperones
A promising treatment for NDs is the development of neuroprotective therapies that target protein misfolding and aggregation. Chaperones are critical targets in protein folding and degradation. Chaperones are divided into three types: molecular chaperones, chemical chaperones, and pharmacological chaperones. Within the cell, there are molecular chaperones [129]. Targeting the expression of these molecules appears to protect the cell from toxicity caused by protein misfolding. Several natural molecules influence molecular chaperone activity [187]. They may increase or decrease the expression of chaperones that protect the cell from proteinopathies. Furthermore, several synthetic drugs that target molecular chaperones or act as chaperones themselves are being developed to improve the stability of folded proteins [188]. Table 4 gives an overview of several natural and synthetic molecules that cause protein folding, protein degradation, activation of chaperones, and inhibition of chaperones that are responsible for protecting the cell from a toxic gain of function of misfolded proteins.
Geldanamycin and radicicol act as HSP90 inhibitors and impair the interaction of HSP90 and the protein. The interaction between HSP90 and protein was lost after 24 h of treatment with the drug. It was also observed that inhibition of HSP90β leads to the relocalization of α-syn to the mitochondria [68]. Wang et al. (2017) proved that the use of an HSP90 inhibitor (OS47720) can reach the brain and elicit a heat shock-like response by activating heat shock factor (HSF1), which can translocate to the nucleus and activate numerous genes [226]. The HSP90 inhibitor was found to be nontoxic and capable of increasing Synapsin I, which can protect the synapse from damage caused by Aβ. It also improved several cognitive functions of the brain.
It has also been observed that chaperones can indirectly affect the aggregation of misfolded proteins. Silveira et al. (2019) found that a chaperone of glucocerebrosidase (Ambroxol) can raise the level of glucocerebrosidase and increase the clearance rate of aggregate α-syn [24]. Thus, it can be said that Ambroxol acts as a potential treatment for PD.
Antisense Therapy Targets the Expression of Proteins
Recently, the use of antisense oligonucleotide (ASO) has been increased and has shown promising effects to treat NDs [5, 151, 227,228,229]. The ASO is a single-stranded nucleotide that can bind to the target protein and alter its expression. The mechanisms by which oligonucleotides alter the expression of the target protein include (i) mRNA degradation by RNase H recruitment, (ii) inclusion or exclusion of exons by alternate splicing, and (iii) inhibition of the binding of miRNA to the target protein [5]. ASO is resistant to exonuclease and therefore is not targeted by endogenous enzymes that promote the long-lasting effects of ASO. Direct delivery of leucine-rich repeat kinase 2 (LRRK2) ASO prevents α-syn pathology by suppressing of LRRK2 protein expression [229]. Likewise, delivery of amido-bridged nucleic acid ASO (AmNA-ASO) enhanced the targeting potency and decreased the mRNA of α-syn in vivo and in vitro [228]. AmNA-ASO was also reported to decrease motor defects in a mouse model. An advantage of using AmNA-ASO is that no off-target effects were seen in the mouse model. ASO is also a potential target for HTT in HD patients. A study showed that targeting HTT by the ASO technique resulted in the suppression of the protein in the CNS [227]. DeVos et al. (2017) designed an ASO that is specific for tau and inhibited neuronal loss. It also reduced the expression of tau mRNA and protein expression, therefore reversing tau spreading and accumulating activity in a mouse model [5].
Another way of altering the expression of the gene is by synthesizing splice-switching antisense oligonucleotides (SSOs) which are short single-stranded ASOs that can bind to a specific RNA target. The binding of SSO to the target RNA results in the skipping of an exon (alternate splicing), regulating the expression of the protein. The mature RNA formed as a result of alternate splicing lacks the targeted exon. Chang et al. (2018) synthesized an SSO that induced the skipping of exon 17 in APP. The produced mRNA lacked exon 17, which is known to code for γ-secretase. As a result, APP could not produce Aβ, thereby decreasing the concentration of Aβ42. The SSO they synthesized contained a 2-methoxyethyl-ribose and phosphorothioate modified backbone that was resistant to the action of the exonuclease [230]. The effect of SSO was also visualized in a mouse model that had similar results. This proves that SSO can be used to target the expression of the protein in NDs.
Proteolysis-Targeting Chimaera (PROTAC) Targets the Degradation of Undruggable Proteins
An emerging strategy to regulate protein concentration is PROTAC. PROTAC are small molecules that target “undruggable” proteins. These molecules bind to the target protein and E3 ubiquitin ligase and enhance the degradation of the protein (Fig. 5). PROTACs have been shown to have a more promising effect than other therapeutic strategies. Lu et al. (2018) have developed a peptide that can colocalize with Keap1 (substrate for ubiquitin E3 ligase) and downregulate intracellular tau dependent on the concentration [231]. Administration of the proteasome inhibitor MG132 proved that the developed peptide can cause proteasome-dependent degradation of Tau. Similar results were obtained when Chu et al. (2016) synthesized TH006, which has tau and E3 ubiquitin ligase binding moieties [232]. TH006 has the potential to interact with Tau and cause its degradation by ubiquitination. QC-01–175 is a heterobifunctional molecule that can bind to Tau and carbon, which forms a part of the E3 ubiquitin ligase and triggers proteasomal degradation of Tau [233]. The direct introduction of a PROTAC targeting α-syn into the cell lines demonstrates its ability to regulate the expression of the protein. The designed peptide-induced degradation of α-syn in a proteasome-dependent pathway attenuates neurotoxicity [25].

Mechanism of PROTAC: Proteolysis-targeting chimaeras (PROTAC) have protein binding and E3 ubiquitin ligase binding domain. On binding of the target protein and E3 ligase, E2 bound ubiquitin is recruited that adds ubiquitin to the target protein. Thus, the protein is directed toward the proteasome where it is degraded
Other Therapies
Neuronal survival can also be achieved by brain-derived neurotrophic factor (BDNF), which is present naturally throughout the CNS. Increased or decreased production of BDNF plays a vital role in the pathogenesis of NDs [58]. Overexpression of BDNF results in cognitive decline, while underexpression is linked to the progression of AD. Arancibia (2008) demonstrated its protective function against the amyloid-beta protein in rodents suffering from Alzheimer’s disease [234]. Rats given BDNF therapy were shown to have improved learning skills and reversed loss caused by APP mutation [58, 235]. BDNF can also act on tau protein and interfere with its toxicity. Scientists have also been working on antibodies that can pass through the barrier, bind to misfolded proteins, and enhance their clearance; however, none of the antibodies produced to date is highly efficient in the process. Inhibitors of the enzyme acetylcholinesterase have been shown to improve performance in PDD (Parkinson’s disease dementia) patients [236]. CRISPR/Cas9-mediated inactivation of the target protein is also a therapeutic strategy to treat NDs. CRISPR/Cas9-mediated strategy results in the elimination of the pathogenic protein and is highly selective toward the target protein. Using this strategy, HTT aggregates have been attenuated in a mouse model of HD [237]. This technique has also been studied in AD [238]. However, this technique also results in off-target effects that make this strategy difficult to adopt.
Conclusion
Proteins are the structural unit of a cell. The secretory proteins are translated into the cytoplasm and then modified in the endoplasmic reticulum and Golgi body. These modifications fold the primary structure of proteins into tertiary or quaternary structures by the formation of chemical bonds. Proteins translated in the cytoplasm are modified and folded in the cytosol itself and can be folded into their native states with the help of chaperones in the cytosol. In a normal healthy cell, if the protein formed is not modified properly, the cell is likely to degrade the protein or activate proteins that in turn can remodify the protein [28, 75, 100]. In NDs, these proteins are not destroyed and thus form aggregates. A lack of understanding of the tertiary structure of these proteins is a major disadvantage. Under abnormal conditions, proteins can occur in multiple conformations; thus, targeting them is a challenge. The transcription of molecular chaperones under stress conditions is therefore increased in an attempt to restore the cellular condition [138, 140, 239]. Several different proteins form aggregates, such as Aβ, tau, α-Syn, SOD, and prion-like proteins [10, 15, 31, 40, 80]. It is believed that the polymerization of HTT is responsible for the aggregates formed in Huntington patients. These misfolded proteins are known to cause misfolding of other proteins, which results in a neuronal cell that contains a very high number of misfolded proteins. These proteins form aggregates that can be transported from one neuron to another via synaptic junctions and nanotube formation. Several drugs have been synthesized that target chaperones, the degradation of proteins, and the extracellular clearance of aggregates. However, these drugs lack efficacy, and none of them can remove or clear all the aggregates formed. Efforts are being made to produce a compound that has high penetration through the BBB and that can specifically bind to the target protein and dissolve the neuronal aggregates formed.
Future Perspective
With an advanced understanding of the underlying mechanisms of NDs, targeting these diseases is now possible. Since the salient feature of these diseases is the aggregation of misfolded proteins, scientists are synthesizing drugs that can target these proteins and cause their clearance. However, the dynamic nature of these misfolded proteins poses a challenge. These proteins occupy multiple conformations, making the understanding of the tertiary structure of these proteins difficult [28]. Several molecular chaperones are known to cause refolding, thus preventing aggregation. HSP70 is the most effective chaperone, which can start its process within 2 h following the minutest assault via the environment, as shown by our previous work [200, 240, 241]. Thus, targeting the genes of chaperones, causing their overexpression or supplying inhibitors of chaperones that promote cytotoxicity, such as HSP90, can be used as a therapy. Treatment by targeting chaperones, however, lacks effectiveness. Thus, focusing on developing a chaperone-targeted drug may be achieved by the application of the mathematical model. Additionally, working on increasing the short life of these drugs might be effective [226]. Another major challenge in the treatment of NDs is delivering the molecules across the BBB. Many of the antibodies that are under trial have low penetrability. However, intranasal delivery of drugs offers the shortest path to reaching the CNS and is considered safe. Thus, targeted delivery to the brain via the nasal pathway should be studied more [184]. Additionally, these proteins can be delivered in liposomes. Since liposomes are hydrophobic, they can cross the BBB and can carry such proteins inside them. Targeting the brain through liposomes has shown successful transfers in various experimental procedures, and thus, the aggregates present in the body can be cleared, deformed, or refolded into proper conformations. The use of ASO therapy has also shown promising effects and can be used to modulate the expression of misfolded proteins [230]. Thus, more research should be conducted targeting the expression of misfolded proteins. Furthermore, treating patients with a combination of multiple therapeutic strategies might prove to be an effective plan of action since use of single strategy fails to cure NDs.
Data Availability
Not applicable.
References
Himalian R, Singh SK, Singh MP (2022) Ameliorative role of nutraceuticals on neurodegenerative diseases using the Drosophila melanogaster as a discovery model to define bioefficacy. J Am Nutr Assoc 41(5):511–539. https://doi.org/10.1080/07315724.2021.1904305
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42(3 Pt 1):631–639. https://doi.org/10.1212/wnl.42.3.631
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Coyne AN, Zaepfel BL, Zarnescu DC (2017) Failure to deliver and translate-new insights into RNA dysregulation in ALS. Front Cell Neurosci 11:243. https://doi.org/10.3389/fncel.2017.00243
DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T et al. (2017) Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 9 (374). https://doi.org/10.1126/scitranslmed.aag0481
Kritsilis M, S VR, Koutsoudaki PN, Evangelou K, Gorgoulis VG, Papadopoulos D (2018) Ageing, cellular senescence and neurodegenerative disease. Int J Mol Sci 19 (10). https://doi.org/10.3390/ijms19102937
Aleksis R, Oleskovs F, Jaudzems K, Pahnke J, Biverstal H (2017) Structural studies of amyloid-beta peptides: unlocking the mechanism of aggregation and the associated toxicity. Biochimie 140:176–192. https://doi.org/10.1016/j.biochi.2017.07.011
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3(4):519–526. https://doi.org/10.1016/0896-6273(89)90210-9
Guo Q, Bin H, Cheng J, Seefelder M, Engler T, Pfeifer G, Oeckl P, Otto M et al. (2018) The cryo-electron microscopy structure of huntingtin. Nature 555(7694):117–120. https://doi.org/10.1038/nature25502
Pearce MMP, Spartz EJ, Hong W, Luo L, Kopito RR (2015) Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun 6:6768. https://doi.org/10.1038/ncomms7768
Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK (2019) Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 12:25. https://doi.org/10.3389/fnmol.2019.00025
Falcon B, Zhang W, Schweighauser M, Murzin AG, Vidal R, Garringer HJ, Ghetti B, Scheres SHW et al. (2018) Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol 136(5):699–708. https://doi.org/10.1007/s00401-018-1914-z
Muntane G, Dalfo E, Martinez A, Ferrer I (2008) Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related alpha-synucleinopathies. Neuroscience 152(4):913–923. https://doi.org/10.1016/j.neuroscience.2008.01.030
Moore DJ, West AB, Dawson VL, Dawson TM (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87. https://doi.org/10.1146/annurev.neuro.28.061604.135718
Bourdenx M, Koulakiotis NS, Sanoudou D, Bezard E, Dehay B, Tsarbopoulos A (2017) Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: examples of amyloidopathies, tauopathies and synucleinopathies. Prog Neurobiol 155:171–193. https://doi.org/10.1016/j.pneurobio.2015.07.003
Benilova I, Karran E, De Strooper B (2012) The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 15(3):349–357. https://doi.org/10.1038/nn.3028
Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Kayed R (2014) Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis 40(Suppl 1):S97–S111. https://doi.org/10.3233/JAD-132477
Cohen TJ, Lee VM, Trojanowski JQ (2011) TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med 17(11):659–667. https://doi.org/10.1016/j.molmed.2011.06.004
Ekimova IV, Plaksina DV, Pastukhov YF, Lapshina KV, Lazarev VF, Mikhaylova ER, Polonik SG, Pani B et al. (2018) New HSF1 inducer as a therapeutic agent in a rodent model of Parkinson’s disease. Exp Neurol 306:199–208. https://doi.org/10.1016/j.expneurol.2018.04.012
Buchberger A, Bukau B, Sommer T (2010) Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 40(2):238–252. https://doi.org/10.1016/j.molcel.2010.10.001
Neus Bosch M, Pugliese M, Andrade C, Gimeno-Bayon J, Mahy N, Rodriguez MJ (2015) Amyloid-beta immunotherapy reduces amyloid plaques and astroglial reaction in aged domestic dogs. Neurodegener Dis 15(1):24–37. https://doi.org/10.1159/000368672
Xing HY, Li B, Peng D, Wang CY, Wang GY, Li P, Le YY, Wang JM et al. (2017) A novel monoclonal antibody against the N-terminus of Abeta1-42 reduces plaques and improves cognition in a mouse model of Alzheimer’s disease. PLoS ONE 12(6):e0180076. https://doi.org/10.1371/journal.pone.0180076
Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W, Soto J, Atiee G, Ostrowitzki S, Kinney GG (2017) First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 32(2):211–218. https://doi.org/10.1002/mds.26878
Silveira CRA, MacKinley J, Coleman K, Li Z, Finger E, Bartha R, Morrow SA, Wells J et al. (2019) Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol 19(1):20. https://doi.org/10.1186/s12883-019-1252-3
Qu J, Ren X, Xue F, He Y, Zhang R, Zheng Y, Huang H, Wang W et al. (2020) Specific knockdown of alpha-synuclein by peptide-directed proteasome degradation rescued its associated neurotoxicity. Cell Chem Biol 27(6):751-762 e754. https://doi.org/10.1016/j.chembiol.2020.03.010
McFarthing K, Simuni T (2019) Clinical trial highlights: targetting alpha-synuclein. J Parkinsons Dis 9(1):5–16. https://doi.org/10.3233/JPD-189004
Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N et al. (2018) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378(4):321–330. https://doi.org/10.1056/NEJMoa1705971
Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, Kopito R, McCampbell A, Leblanc G et al. (2017) Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener 6:6. https://doi.org/10.1186/s40035-017-0077-5
Gurry T, Stultz CM (2014) Mechanism of amyloid-beta fibril elongation. Biochemistry 53(44):6981–6991. https://doi.org/10.1021/bi500695g
Yang PH, Zhu JX, Huang YD, Zhang XY, Lei P, Bush AI, Xiang Q, Su ZJ et al. (2016) Human basic fibroblast growth factor inhibits tau phosphorylation via the PI3K/Akt-GSK3beta signaling pathway in a 6-hydroxydopamine-induced model of Parkinson’s disease. Neurodegener Dis 16(5–6):357–369. https://doi.org/10.1159/000445871
Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2(7):492–501. https://doi.org/10.1038/35081564
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33(1):95–130. https://doi.org/10.1016/s0165-0173(00)00019-9
Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol 12(6):609–622. https://doi.org/10.1016/S1474-4422(13)70090-5
Sanghai N, Tranmer GK (2021) Hydrogen peroxide and amyotrophic lateral sclerosis: from biochemistry to pathophysiology. Antioxidants (Basel) 11 (1). https://doi.org/10.3390/antiox11010052
Koyuncu S, Fatima A, Gutierrez-Garcia R, Vilchez D (2017) Proteostasis of huntingtin in health and disease. Int J Mol Sci 18 (7). https://doi.org/10.3390/ijms18071568
Baldwin KJ, Correll CM (2019) Prion disease. Semin Neurol 39(4):428–439. https://doi.org/10.1055/s-0039-1687841
DeTure MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14(1):32. https://doi.org/10.1186/s13024-019-0333-5
Rajasekhar K, Chakrabarti M, Govindaraju T (2015) Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem Commun (Camb) 51(70):13434–13450. https://doi.org/10.1039/c5cc05264e
Pampuscenko K, Morkuniene R, Sneideris T, Smirnovas V, Budvytyte R, Valincius G, Brown GC, Borutaite V (2020) Extracellular tau induces microglial phagocytosis of living neurons in cell cultures. J Neurochem 154(3):316–329. https://doi.org/10.1111/jnc.14940
Butterfield DA, Swomley AM, Sultana R (2013) Amyloid beta-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal 19(8):823–835. https://doi.org/10.1089/ars.2012.5027
Oliveira LF, Rodrigues LD, Cardillo GM, Nejm MB, Guimaraes-Marques M, Reyes-Garcia SZ, Zuqui K, Vassallo DV et al. (2020) Deleterious effects of chronic mercury exposure on in vitro LTP, memory process, and oxidative stress. Environ Sci Pollut Res Int 27(7):7559–7569. https://doi.org/10.1007/s11356-019-06625-6
He Y, Yao PF, Chen SB, Huang ZH, Huang SL, Tan JH, Li D, Gu LQ et al. (2013) Synthesis and evaluation of 7,8-dehydrorutaecarpine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem 63:299–312. https://doi.org/10.1016/j.ejmech.2013.02.014
Nimmrich V, Ebert U (2009) Is Alzheimer’s disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid. Rev Neurosci 20(1):1–12. https://doi.org/10.1515/revneuro.2009.20.1.1
Nesi G, Sestito S, Digiacomo M, Rapposelli S (2017) Oxidative stress, mitochondrial abnormalities and proteins deposition: multitarget approaches in Alzheimer’s disease. Curr Top Med Chem 17(27):3062–3079. https://doi.org/10.2174/1568026617666170607114232
Shi C, Zhu X, Wang J, Long D (2014) Intromitochondrial IkappaB/NF-kappaB signaling pathway is involved in amyloid beta peptide-induced mitochondrial dysfunction. J Bioenerg Biomembr 46(5):371–376. https://doi.org/10.1007/s10863-014-9567-7
Sivandzade F, Prasad S, Bhalerao A, Cucullo L (2019) NRF2 and NF-B interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol 21:101059. https://doi.org/10.1016/j.redox.2018.11.017
Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL (1993) Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci 13(7):3136–3142
Estus S, Golde TE, Kunishita T, Blades D, Lowery D, Eisen M, Usiak M, Qu XM et al. (1992) Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science 255(5045):726–728. https://doi.org/10.1126/science.1738846
Naslund J, Jensen M, Tjernberg LO, Thyberg J, Terenius L, Nordstedt C (1994) The metabolic pathway generating p3, an A beta-peptide fragment, is probably non-amyloidogenic. Biochem Biophys Res Commun 204(2):780–787. https://doi.org/10.1006/bbrc.1994.2527
Giuffrida ML, Caraci F, De Bona P, Pappalardo G, Nicoletti F, Rizzarelli E, Copani A (2010) The monomer state of beta-amyloid: where the Alzheimer’s disease protein meets physiology. Rev Neurosci 21(2):83–93. https://doi.org/10.1515/revneuro.2010.21.2.83
Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, Schmidt M, Sigurdson CJ et al. (2019) Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nat Commun 10(1):4760. https://doi.org/10.1038/s41467-019-12683-8
Fitzpatrick AW, Saibil HR (2019) Cryo-EM of amyloid fibrils and cellular aggregates. Curr Opin Struct Biol 58:34–42. https://doi.org/10.1016/j.sbi.2019.05.003
Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M et al. (2013) Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110(24):9758–9763. https://doi.org/10.1073/pnas.1218402110
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18(21):4153–4170. https://doi.org/10.1093/hmg/ddp367
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC et al. (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142(3):387–397. https://doi.org/10.1016/j.cell.2010.06.036
Iwata M, Watanabe S, Yamane A, Miyasaka T, Misonou H (2019) Regulatory mechanisms for the axonal localization of tau protein in neurons. Mol Biol Cell 30(19):2441–2457. https://doi.org/10.1091/mbc.E19-03-0183
Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M et al. (2005) Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25(46):10637–10647. https://doi.org/10.1523/JNEUROSCI.3279-05.2005
Jiao SS, Shen LL, Zhu C, Bu XL, Liu YH, Liu CH, Yao XQ, Zhang LL et al. (2016) Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl Psychiatry 6(10):e907. https://doi.org/10.1038/tp.2016.186
Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E (1994) Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem 269(39):24290–24297
Goedert M, Falcon B, Zhang W, Ghetti B, Scheres SHW (2018) Distinct conformers of assembled tau in Alzheimer’s and Pick’s diseases. Cold Spring Harb Symp Quant Biol 83:163–171. https://doi.org/10.1101/sqb.2018.83.037580
Hisanaga SI, Krishnankutty A, Kimura T (2022) In vivo analysis of the phosphorylation of tau and the tau protein kinases Cdk5-p35 and GSK3beta by using Phos-tag SDS-PAGE. J Proteomics 262:104591. https://doi.org/10.1016/j.jprot.2022.104591
Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K (2013) Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis 33(Suppl 1):S123-139. https://doi.org/10.3233/JAD-2012-129031
Lei P, Ayton S, Finkelstein DI, Adlard PA, Masters CL, Bush AI (2010) Tau protein: relevance to Parkinson’s disease. Int J Biochem Cell Biol 42(11):1775–1778. https://doi.org/10.1016/j.biocel.2010.07.016
Fernandez-Nogales M, Cabrera JR, Santos-Galindo M, Hoozemans JJ, Ferrer I, Rozemuller AJ, Hernandez F, Avila J et al. (2014) Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat Med 20(8):881–885. https://doi.org/10.1038/nm.3617
Cisbani G, Maxan A, Kordower JH, Planel E, Freeman TB, Cicchetti F (2017) Presence of tau pathology within foetal neural allografts in patients with Huntington’s and Parkinson’s disease. Brain 140(11):2982–2992. https://doi.org/10.1093/brain/awx255
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM (2019) Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 216(11):2546–2561. https://doi.org/10.1084/jem.20190980
Pascoal TA, Benedet AL, Ashton NJ, Kang MS, Therriault J, Chamoun M, Savard M, Lussier FZ et al. (2021) Microglial activation and tau propagate jointly across Braak stages. Nat Med 27(9):1592–1599. https://doi.org/10.1038/s41591-021-01456-w
Burmann BM, Gerez JA, Matecko-Burmann I, Campioni S, Kumari P, Ghosh D, Mazur A, Aspholm EE et al. (2020) Regulation of alpha-synuclein by chaperones in mammalian cells. Nature 577(7788):127–132. https://doi.org/10.1038/s41586-019-1808-9
Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T et al. (2020) Structures of alpha-synuclein filaments from multiple system atrophy. Nature 585(7825):464–469. https://doi.org/10.1038/s41586-020-2317-6
Flynn JD, McGlinchey RP, Walker RL 3rd, Lee JC (2018) Structural features of alpha-synuclein amyloid fibrils revealed by Raman spectroscopy. J Biol Chem 293(3):767–776. https://doi.org/10.1074/jbc.M117.812388
Greenbau EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI (2005) The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem 280(9):7800–7807. https://doi.org/10.1074/jbc.M411638200
Conway KA, Harper JD, Lansbury PT Jr (2000) Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 39(10):2552–2563. https://doi.org/10.1021/bi991447r
Fares MB, Jagannath S, Lashuel HA (2021) Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat Rev Neurosci 22(2):111–131. https://doi.org/10.1038/s41583-020-00416-6
Uversky VN, Li J, Fink AL (2001) Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem 276(14):10737–10744. https://doi.org/10.1074/jbc.M010907200
Bandopadhyay R, de Belleroche J (2010) Pathogenesis of Parkinson’s disease: emerging role of molecular chaperones. Trends Mol Med 16(1):27–36. https://doi.org/10.1016/j.molmed.2009.11.004
Bartels T, Choi JG, Selkoe DJ (2011) alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477(7362):107–110. https://doi.org/10.1038/nature10324
Burre J, Vivona S, Diao J, Sharma M, Brunger AT, Sudhof TC (2013) Properties of native brain alpha-synuclein. Nature 498 (7453):E4–6; discussion E6–7. https://doi.org/10.1038/nature12125
Pfefferkorn CM, Lee JC (2010) Tryptophan probes at the alpha-synuclein and membrane interface. J Phys Chem B 114(13):4615–4622. https://doi.org/10.1021/jp908092e
Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P, Zurzolo C (2013) Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci 126(Pt 16):3678–3685. https://doi.org/10.1242/jcs.126086
Victoria GS, Zurzolo C (2017) The spread of prion-like proteins by lysosomes and tunneling nanotubes: implications for neurodegenerative diseases. J Cell Biol 216(9):2633–2644. https://doi.org/10.1083/jcb.201701047
Aiken CT, Steffan JS, Guerrero CM, Khashwji H, Lukacsovich T, Simmons D, Purcell JM, Menhaji K et al. (2009) Phosphorylation of threonine 3: implications for huntingtin aggregation and neurotoxicity. J Biol Chem 284(43):29427–29436. https://doi.org/10.1074/jbc.M109.013193
Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM et al. (2009) Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64(6):828–840. https://doi.org/10.1016/j.neuron.2009.11.020
Chaibva M, Jawahery S, Pilkington AWT, Arndt JR, Sarver O, Valentine S, Matysiak S, Legleiter J (2016) Acetylation within the first 17 residues of huntingtin exon 1 alters aggregation and lipid binding. Biophys J 111(2):349–362. https://doi.org/10.1016/j.bpj.2016.06.018
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K et al. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351(3):602–611. https://doi.org/10.1016/j.bbrc.2006.10.093
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133. https://doi.org/10.1126/science.1134108
Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121(Pt 22):3778–3785. https://doi.org/10.1242/jcs.038950
Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF et al. (2013) Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat Struct Mol Biol 20(12):1443–1449. https://doi.org/10.1038/nsmb.2698
Mompean M, Romano V, Pantoja-Uceda D, Stuani C, Baralle FE, Buratti E, Laurents DV (2016) The TDP-43 N-terminal domain structure at high resolution. FEBS J 283(7):1242–1260. https://doi.org/10.1111/febs.13651
Kuo PH, Chiang CH, Wang YT, Doudeva LG, Yuan HS (2014) The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res 42(7):4712–4722. https://doi.org/10.1093/nar/gkt1407
Prusiner SB (2013) Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47:601–623. https://doi.org/10.1146/annurev-genet-110711-155524
Pignataro A, Middei S (2017) Trans-synaptic spread of amyloid-beta in Alzheimer’s disease: paths to beta-amyloidosis. Neural Plast 2017:5281829. https://doi.org/10.1155/2017/5281829
Ariazi J, Benowitz A, De Biasi V, Den Boer ML, Cherqui S, Cui H, Douillet N, Eugenin EA et al. (2017) Tunneling nanotubes and gap junctions-their role in long-range intercellular communication during development, health, and disease conditions. Front Mol Neurosci 10:333. https://doi.org/10.3389/fnmol.2017.00333
Kelly JW (1998) The environmental dependency of protein folding best explains prion and amyloid diseases. Proc Natl Acad Sci U S A 95(3):930–932. https://doi.org/10.1073/pnas.95.3.930
Marrero-Winkens C, Sankaran C, Schatzl HM (2020) From seeds to fibrils and back: fragmentation as an overlooked step in the propagation of prions and prion-like proteins. Biomolecules 10 (9). https://doi.org/10.3390/biom10091305
Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M et al. (2013) Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288(3):1856–1870. https://doi.org/10.1074/jbc.M112.394528
Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E (2014) Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62(3):387–398. https://doi.org/10.1002/glia.22611
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC et al. (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82(6):1271–1288. https://doi.org/10.1016/j.neuron.2014.04.047
Nath S, Agholme L, Kurudenkandy FR, Granseth B, Marcusson J, Hallbeck M (2012) Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J Neurosci 32(26):8767–8777. https://doi.org/10.1523/JNEUROSCI.0615-12.2012
Jackson RJ, Rudinskiy N, Herrmann AG, Croft S, Kim JM, Petrova V, Ramos-Rodriguez JJ, Pitstick R et al. (2016) Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer’s disease. Eur J Neurosci 44(12):3056–3066. https://doi.org/10.1111/ejn.13442
Smith HL, Mallucci GR (2016) The unfolded protein response: mechanisms and therapy of neurodegeneration. Brain 139(Pt 8):2113–2121. https://doi.org/10.1093/brain/aww101
Shimura H, Schwartz D, Gygi SP, Kosik KS (2004) CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 279(6):4869–4876. https://doi.org/10.1074/jbc.M305838200
Kohler C, Dinekov M, Gotz J (2014) Granulovacuolar degeneration and unfolded protein response in mouse models of tauopathy and Abeta amyloidosis. Neurobiol Dis 71:169–179. https://doi.org/10.1016/j.nbd.2014.07.006
Mayer M, Kies U, Kammermeier R, Buchner J (2000) BiP and PDI cooperate in the oxidative folding of antibodies in vitro. J Biol Chem 275(38):29421–29425. https://doi.org/10.1074/jbc.M002655200
Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W (2005) The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110(2):165–172. https://doi.org/10.1007/s00401-005-1038-0
Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I (2016) The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med 16(6):533–544. https://doi.org/10.2174/1566524016666160523143937
Thorpe JA, Schwarze SR (2010) IRE1alpha controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress Chaperones 15(5):497–508. https://doi.org/10.1007/s12192-009-0163-4
Duran-Aniotz C, Cornejo VH, Espinoza S, Ardiles AO, Medinas DB, Salazar C, Foley A, Gajardo I et al. (2017) IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol 134(3):489–506. https://doi.org/10.1007/s00401-017-1694-x
Tam AB, Roberts LS, Chandra V, Rivera IG, Nomura DK, Forbes DJ, Niwa M (2018) The UPR activator ATF6 responds to proteotoxic and lipotoxic stress by distinct mechanisms. Dev Cell 46(3):327-343 e327. https://doi.org/10.1016/j.devcel.2018.04.023
Patwardhan A, Cheng N, Trejo J (2021) Post-translational modifications of G protein-coupled receptors control cellular signaling dynamics in space and time. Pharmacol Rev 73(1):120–151. https://doi.org/10.1124/pharmrev.120.000082
Babu JR, Geetha T, Wooten MW (2005) Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem 94(1):192–203. https://doi.org/10.1111/j.1471-4159.2005.03181.x
Ikeda K, Akiyama H, Arai T, Kondo H, Haga C, Iritani S, Tsuchiya K (1998) Alz-50/Gallyas-positive lysosome-like intraneuronal granules in Alzheimer’s disease and control brains. Neurosci Lett 258(2):113–116. https://doi.org/10.1016/s0304-3940(98)00867-2
Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, Zhang B, Yue Z (2020) Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun 11(1):1386. https://doi.org/10.1038/s41467-020-15119-w
Sun L, Lian Y, Ding J, Meng Y, Li C, Chen L, Qiu P (2019) The role of chaperone-mediated autophagy in neurotoxicity induced by alpha-synuclein after methamphetamine exposure. Brain Behav 9(8):e01352. https://doi.org/10.1002/brb3.1352
Behnke J, Mann MJ, Scruggs FL, Feige MJ, Hendershot LM (2016) Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol Cell 63(5):739–752. https://doi.org/10.1016/j.molcel.2016.07.012
Agashe VR, Hartl FU (2000) Roles of molecular chaperones in cytoplasmic protein folding. Semin Cell Dev Biol 11(1):15–25. https://doi.org/10.1006/scdb.1999.0347
Arhar T, Shkedi A, Nadel CM, Gestwicki JE (2021) The interactions of molecular chaperones with client proteins: why are they so weak? J Biol Chem 297(5):101282. https://doi.org/10.1016/j.jbc.2021.101282
He WT, Xue W, Gao YG, Hong JY, Yue HW, Jiang LL, Hu HY (2017) HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci Rep 7(1):14797. https://doi.org/10.1038/s41598-017-13711-7
Zhang M, Qian C, Zheng ZG, Qian F, Wang Y, Thu PM, Zhang X, Zhou Y et al. (2018) Jujuboside A promotes Abeta clearance and ameliorates cognitive deficiency in Alzheimer’s disease through activating Axl/HSP90/PPARgamma pathway. Theranostics 8(15):4262–4278. https://doi.org/10.7150/thno.26164
Robinson MB, Tidwell JL, Gould T, Taylor AR, Newbern JM, Graves J, Tytell M, Milligan CE (2005) Extracellular heat shock protein 70: a critical component for motoneuron survival. J Neurosci 25(42):9735–9745. https://doi.org/10.1523/JNEUROSCI.1912-05.2005
Lyon MS, Milligan C (2019) Extracellular heat shock proteins in neurodegenerative diseases: new perspectives. Neurosci Lett 711:134462. https://doi.org/10.1016/j.neulet.2019.134462
Magen D, Georgopoulos C, Bross P, Ang D, Segev Y, Goldsher D, Nemirovski A, Shahar E et al. (2008) Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 83(1):30–42. https://doi.org/10.1016/j.ajhg.2008.05.016
Biebl MM, Buchner J (2019) Structure, function, and regulation of the Hsp90 machinery. Cold Spring Harb Perspect Biol 11 (9). https://doi.org/10.1101/cshperspect.a034017
Bohush A, Niewiadomska G, Weis S, Filipek A (2019) HSP90 and its novel co-chaperones, SGT1 and CHP-1, in brain of patients with Parkinson’s disease and dementia with Lewy bodies. J Parkinsons Dis 9(1):97–107. https://doi.org/10.3233/JPD-181443
Soti C, Csermely P (2002) Chaperones and aging: role in neurodegeneration and in other civilizational diseases. Neurochem Int 41(6):383–389. https://doi.org/10.1016/s0197-0186(02)00043-8
Abisambra JF, Blair LJ, Hill SE, Jones JR, Kraft C, Rogers J, Koren J 3rd, Jinwal UK et al. (2010) Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci 30(46):15374–15382. https://doi.org/10.1523/JNEUROSCI.3155-10.2010
Selig EE, Zlatic CO, Cox D, Mok YF, Gooley PR, Ecroyd H, Griffin MDW (2020) N- and C-terminal regions of alphaB-crystallin and Hsp27 mediate inhibition of amyloid nucleation, fibril binding, and fibril disaggregation. J Biol Chem 295(29):9838–9854. https://doi.org/10.1074/jbc.RA120.012748
Outeiro TF, Klucken J, Strathearn KE, Liu F, Nguyen P, Rochet JC, Hyman BT, McLean PJ (2006) Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem Biophys Res Commun 351(3):631–638. https://doi.org/10.1016/j.bbrc.2006.10.085
Goldfarb SB, Kashlan OB, Watkins JN, Suaud L, Yan W, Kleyman TR, Rubenstein RC (2006) Differential effects of Hsc70 and Hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc Natl Acad Sci U S A 103(15):5817–5822. https://doi.org/10.1073/pnas.0507903103
Brandvold KR, Morimoto RI (2015) The chemical biology of molecular chaperones–implications for modulation of proteostasis. J Mol Biol 427(18):2931–2947. https://doi.org/10.1016/j.jmb.2015.05.010
Xu H (2018) Cochaperones enable Hsp70 to use ATP energy to stabilize native proteins out of the folding equilibrium. Sci Rep 8(1):13213. https://doi.org/10.1038/s41598-018-31641-w
Li H, Yang J, Wang Y, Liu Q, Cheng J, Wang F (2019) Neuroprotective effects of increasing levels of HSP70 against neuroinflammation in Parkinson’s disease model by inhibition of NF-kappaB and STAT3. Life Sci 234:116747. https://doi.org/10.1016/j.lfs.2019.116747
Zheng Q, Huang C, Guo J, Tan J, Wang C, Tang B, Zhang H (2018) Hsp70 participates in PINK1-mediated mitophagy by regulating the stability of PINK1. Neurosci Lett 662:264–270. https://doi.org/10.1016/j.neulet.2017.10.051
Evgen’ev M, Bobkova N, Krasnov G, Garbuz D, Funikov S, Kudryavtseva A, Kulikov A, Samokhin A et al. (2019) The effect of human HSP70 administration on a mouse model of Alzheimer’s disease strongly depends on transgenicity and age. J Alzheimers Dis 67(4):1391–1404. https://doi.org/10.3233/JAD-180987
Chen JY, Parekh M, Seliman H, Bakshinskaya D, Dai W, Kwan K, Chen KY, Liu AYC (2018) Heat shock promotes inclusion body formation of mutant huntingtin (mHtt) and alleviates mHtt-induced transcription factor dysfunction. J Biol Chem 293(40):15581–15593. https://doi.org/10.1074/jbc.RA118.002933
Csermely P, Kajtar J, Hollosi M, Jalsovszky G, Holly S, Kahn CR, Gergely P Jr, Soti C et al. (1993) ATP induces a conformational change of the 90-kDa heat shock protein (hsp90). J Biol Chem 268(3):1901–1907
Obermann WM, Sondermann H, Russo AA, Pavletich NP, Hartl FU (1998) In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J Cell Biol 143(4):901–910. https://doi.org/10.1083/jcb.143.4.901
Ernst JT, Neubert T, Liu M, Sperry S, Zuccola H, Turnbull A, Fleck B, Kargo W et al. (2014) Identification of novel HSP90alpha/beta isoform selective inhibitors using structure-based drug design. Demonstration of potential utility in treating CNS disorders such as Huntington’s disease. J Med Chem 57(8):3382–3400. https://doi.org/10.1021/jm500042s
Rane A, Rajagopalan S, Ahuja M, Thomas B, Chinta SJ, Andersen JK (2018) Hsp90 co-chaperone p23 contributes to dopaminergic mitochondrial stress via stabilization of PHD2: implications for Parkinson’s disease. Neurotoxicology 65:166–173. https://doi.org/10.1016/j.neuro.2018.02.012
Janowska MK, Baughman HER, Woods CN, Klevit RE (2019) Mechanisms of small heat shock proteins. Cold Spring Harb Perspect Biol 11 (10). https://doi.org/10.1101/cshperspect.a034025
Vicente Miranda H, Chegao A, Oliveira MS, Fernandes Gomes B, Enguita FJ, Outeiro TF (2020) Hsp27 reduces glycation-induced toxicity and aggregation of alpha-synuclein. FASEB J 34(5):6718–6728. https://doi.org/10.1096/fj.201902936R
Lavoie JN, Hickey E, Weber LA, Landry J (1993) Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J Biol Chem 268(32):24210–24214
Gonzales SJ, Reyes RA, Braddom AE, Batugedara G, Bol S, Bunnik EM (2020) Naturally acquired humoral immunity against Plasmodium falciparum malaria. Front Immunol 11:594653. https://doi.org/10.3389/fimmu.2020.594653
Brody DL, Holtzman DM (2008) Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci 31:175–193. https://doi.org/10.1146/annurev.neuro.31.060407.125529
Gustafsson G, Eriksson F, Moller C, da Fonseca TL, Outeiro TF, Lannfelt L, Bergstrom J, Ingelsson M (2017) Cellular uptake of alpha-synuclein oligomer-selective antibodies is enhanced by the extracellular presence of alpha-synuclein and mediated via Fcgamma receptors. Cell Mol Neurobiol 37(1):121–131. https://doi.org/10.1007/s10571-016-0352-5
Nordstrom E, Eriksson F, Sigvardson J, Johannesson M, Kasrayan A, Jones-Kostalla M, Appelkvist P, Soderberg L et al. (2021) ABBV-0805, a novel antibody selective for soluble aggregated alpha-synuclein, prolongs lifespan and prevents buildup of alpha-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol Dis 161:105543. https://doi.org/10.1016/j.nbd.2021.105543
Huang YR, Xie XX, Ji M, Yu XL, Zhu J, Zhang LX, Liu XG, Wei C et al. (2019) Naturally occurring autoantibodies against alpha-synuclein rescues memory and motor deficits and attenuates alpha-synuclein pathology in mouse model of Parkinson’s disease. Neurobiol Dis 124:202–217. https://doi.org/10.1016/j.nbd.2018.11.024
Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, Zago W, Ness DK et al. (2018) Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol 75(10):1206–1214. https://doi.org/10.1001/jamaneurol.2018.1487
Kuchimanchi M, Monine M, Kandadi Muralidharan K, Woodward C, Penner N (2020) Phase II dose selection for alpha synuclein-targeting antibody cinpanemab (BIIB054) based on target protein binding levels in the brain. CPT Pharmacometrics Syst Pharmacol 9(9):515–522. https://doi.org/10.1002/psp4.12538
Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, Welch M, Koenig E et al. (2019) Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov Disord 34(8):1154–1163. https://doi.org/10.1002/mds.27738
Valera E, Masliah E (2013) Immunotherapy for neurodegenerative diseases: focus on alpha-synucleinopathies. Pharmacol Ther 138(3):311–322. https://doi.org/10.1016/j.pharmthera.2013.01.013
Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, Adame A, Schmidhuber S et al. (2015) Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener 10:10. https://doi.org/10.1186/s13024-015-0008-9
Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, Santic R, Meindl S et al. (2014) Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol 127(6):861–879. https://doi.org/10.1007/s00401-014-1256-4
Schofield DJ, Irving L, Calo L, Bogstedt A, Rees G, Nuccitelli A, Narwal R, Petrone M et al. (2019) Preclinical development of a high affinity alpha-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular alpha-synuclein and attenuate alpha-synuclein spreading in vivo. Neurobiol Dis 132:104582. https://doi.org/10.1016/j.nbd.2019.104582
Cheng L, Zhang J, Li XY, Yuan L, Pan YF, Chen XR, Gao TM, Qiao JT et al. (2017) A novel antibody targeting sequence 31–35 in amyloid beta protein attenuates Alzheimer’s disease-related neuronal damage. Hippocampus 27(2):122–133. https://doi.org/10.1002/hipo.22676
Abushouk AI, Elmaraezy A, Aglan A, Salama R, Fouda S, Fouda R, AlSafadi AM (2017) Bapineuzumab for mild to moderate Alzheimer’s disease: a meta-analysis of randomized controlled trials. BMC Neurol 17(1):66. https://doi.org/10.1186/s12883-017-0850-1
Lu M, Brashear HR (2019) Pharmacokinetics, pharmacodynamics, and safety of subcutaneous bapineuzumab: a single-ascending-dose study in patients with mild to moderate Alzheimer disease. Clin Pharmacol Drug Dev 8(3):326–335. https://doi.org/10.1002/cpdd.584
Doggrell SA (2018) Grasping at straws: the failure of solanezumab to modify mild Alzheimer’s disease. Expert Opin Biol Ther 18(12):1189–1192. https://doi.org/10.1080/14712598.2018.1543397
Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, Andjelkovic M, Ristic S et al. (2019) Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther 11(1):101. https://doi.org/10.1186/s13195-019-0559-z
Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, Ashford E, Retout S et al. (2017) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9(1):95. https://doi.org/10.1186/s13195-017-0318-y
Ultsch M, Li B, Maurer T, Mathieu M, Adolfsson O, Muhs A, Pfeifer A, Pihlgren M et al. (2016) Structure of crenezumab complex with Abeta shows loss of beta-hairpin. Sci Rep 6:39374. https://doi.org/10.1038/srep39374
Yang T, Dang Y, Ostaszewski B, Mengel D, Steffen V, Rabe C, Bittner T, Walsh DM et al. (2019) Target engagement in an alzheimer trial: crenezumab lowers amyloid beta oligomers in cerebrospinal fluid. Ann Neurol 86(2):215–224. https://doi.org/10.1002/ana.25513
Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S et al. (2016) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537(7618):50–56. https://doi.org/10.1038/nature19323
Arndt JW, Qian F, Smith BA, Quan C, Kilambi KP, Bush MW, Walz T, Pepinsky RB et al. (2018) Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-beta. Sci Rep 8(1):6412. https://doi.org/10.1038/s41598-018-24501-0
Landen JW, Andreasen N, Cronenberger CL, Schwartz PF, Borjesson-Hanson A, Ostlund H, Sattler CA, Binneman B et al. (2017) Ponezumab in mild-to-moderate Alzheimer’s disease: randomized phase II PET-PIB study. Alzheimers Dement (N Y) 3(3):393–401. https://doi.org/10.1016/j.trci.2017.05.003
Landen JW, Cohen S, Billing CB Jr, Cronenberger C, Styren S, Burstein AH, Sattler C, Lee JH et al. (2017) Multiple-dose ponezumab for mild-to-moderate Alzheimer’s disease: safety and efficacy. Alzheimers Dement (N Y) 3(3):339–347. https://doi.org/10.1016/j.trci.2017.04.003
Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S (2005) Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 64(1):94–101. https://doi.org/10.1212/01.WNL.0000148604.77591.67
Vukicevic M, Fiorini E, Siegert S, Carpintero R, Rincon-Restrepo M, Lopez-Deber P, Piot N, Ayer M et al. (2022) An amyloid beta vaccine that safely drives immunity to a key pathological species in Alzheimer’s disease: pyroglutamate amyloid beta. Brain Commun 4(1):fcac022. https://doi.org/10.1093/braincomms/fcac022
Vandenberghe R, Riviere ME, Caputo A, Sovago J, Maguire RP, Farlow M, Marotta G, Sanchez-Valle R et al. (2017) Active Abeta immunotherapy CAD106 in Alzheimer’s disease: a phase 2b study. Alzheimers Dement (N Y) 3(1):10–22. https://doi.org/10.1016/j.trci.2016.12.003
Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, Maguire RP, Blennow K et al. (2012) Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol 11(7):597–604. https://doi.org/10.1016/S1474-4422(12)70140-0
Hull M, Sadowsky C, Arai H, Le Prince LG, Holstein A, Booth K, Peng Y, Yoshiyama T et al. (2017) Long-term extensions of randomized vaccination trials of ACC-001 and QS-21 in mild to moderate Alzheimer’s disease. Curr Alzheimer Res 14(7):696–708. https://doi.org/10.2174/1567205014666170117101537
Ketter N, Liu E, Di J, Honig LS, Lu M, Novak G, Werth J, LePrinceLeterme G et al. (2016) A randomized double-blind, phase 2 study of the effects of the vaccine Vanutide Cridificar with QS-21 adjuvant on immunogenicity safety and amyloid imaging in patients with mild to moderate Alzheimer’s disease. J Prev Alzheimers Dis 3(4):192–201. https://doi.org/10.14283/jpad.2016.118
Lacosta AM, Pascual-Lucas M, Pesini P, Casabona D, Perez-Grijalba V, Marcos-Campos I, Sarasa L, Canudas J et al. (2018) Safety, tolerability and immunogenicity of an active anti-Abeta40 vaccine (ABvac40) in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res Ther 10(1):12. https://doi.org/10.1186/s13195-018-0340-8
Rajamohamedsait H, Rasool S, Rajamohamedsait W, Lin Y, Sigurdsson EM (2017) Prophylactic active tau immunization leads to sustained reduction in both tau and amyloid-beta pathologies in 3xTg mice. Sci Rep 7(1):17034. https://doi.org/10.1038/s41598-017-17313-1
Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M (2014) First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res Ther 6(4):44. https://doi.org/10.1186/alzrt278
Ayalon G, Lee SH, Adolfsson O, Foo-Atkins C, Atwal JK, Blendstrup M, Booler H, Bravo J et al. (2021) Antibody semorinemab reduces tau pathology in a transgenic mouse model and engages tau in patients with Alzheimer’s disease. Sci Transl Med 13 (593). https://doi.org/10.1126/scitranslmed.abb2639
van Ameijde J, Crespo R, Janson R, Juraszek J, Siregar B, Verveen H, Sprengers I, Nahar T et al. (2018) Enhancement of therapeutic potential of a naturally occurring human antibody targeting a phosphorylated Ser(422) containing epitope on pathological tau. Acta Neuropathol Commun 6(1):59. https://doi.org/10.1186/s40478-018-0562-9
Dai CL, Hu W, Tung YC, Liu F, Gong CX, Iqbal K (2018) Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 x Tg-AD mice. Alzheimers Res Ther 10(1):13. https://doi.org/10.1186/s13195-018-0341-7
Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, Estes DM, Barrett AD et al. (2014) Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34(12):4260–4272. https://doi.org/10.1523/JNEUROSCI.3192-13.2014
Breijyeh Z, Karaman R (2020) Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25 (24). https://doi.org/10.3390/molecules25245789
Rosenqvist N, Asuni AA, Andersson CR, Christensen S, Daechsel JA, Egebjerg J, Falsig J, Helboe L et al. (2018) Highly specific and selective anti-pS396-tau antibody C10.2 targets seeding-competent tau. Alzheimers Dement (N Y) 4:521–534. https://doi.org/10.1016/j.trci.2018.09.005
West T, Hu Y, Verghese PB, Bateman RJ, Braunstein JB, Fogelman I, Budur K, Florian H et al. (2017) Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis 4(4):236–241. https://doi.org/10.14283/jpad.2017.36
Amaro IA, Henderson LA (2016) An intrabody drug (rAAV6-INT41) reduces the binding of N-terminal huntingtin fragment(s) to DNA to basal levels in PC12 cells and delays cognitive loss in the R6/2 animal model. J Neurodegener Dis 2016:7120753. https://doi.org/10.1155/2016/7120753
Lecerf JM, Shirley TL, Zhu Q, Kazantsev A, Amersdorfer P, Housman DE, Messer A, Huston JS (2001) Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc Natl Acad Sci U S A 98(8):4764–4769. https://doi.org/10.1073/pnas.071058398
Cattepoel S, Hanenberg M, Kulic L, Nitsch RM (2011) Chronic intranasal treatment with an anti-Abeta(30–42) scFv antibody ameliorates amyloid pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 6(4):e18296. https://doi.org/10.1371/journal.pone.0018296
Vitale F, Giliberto L, Ruiz S, Steslow K, Marambaud P, d’Abramo C (2018) Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol Commun 6(1):82. https://doi.org/10.1186/s40478-018-0585-2
Khoshnan A, Ko J, Patterson PH (2002) Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc Natl Acad Sci U S A 99(2):1002–1007. https://doi.org/10.1073/pnas.022631799
Lorenzen N, Nielsen SB, Yoshimura Y, Vad BS, Andersen CB, Betzer C, Kaspersen JD, Christiansen G et al. (2014) How epigallocatechin gallate can inhibit alpha-synuclein oligomer toxicity in vitro. J Biol Chem 289(31):21299–21310. https://doi.org/10.1074/jbc.M114.554667
Zhao D, Xu YM, Cao LQ, Yu F, Zhou H, Qin W, Li HJ, He CX et al. (2021) Complex crystal structure determination and in vitro anti-non-small cell lung cancer activity of Hsp90 (N) inhibitor SNX-2112. Front Cell Dev Biol 9:650106. https://doi.org/10.3389/fcell.2021.650106
Opattova A, Filipcik P, Cente M, Novak M (2013) Intracellular degradation of misfolded tau protein induced by geldanamycin is associated with activation of proteasome. J Alzheimers Dis 33(2):339–348. https://doi.org/10.3233/JAD-2012-121072
Zare N, Motamedi F, Digaleh H, Khodagholi F, Maghsoudi N (2015) Collaboration of geldanamycin-activated P70S6K and Hsp70 against beta-amyloid-induced hippocampal apoptosis: an approach to long-term memory and learning. Cell Stress Chaperones 20(2):309–319. https://doi.org/10.1007/s12192-014-0550-3
Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE (2001) Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet 10(12):1307–1315. https://doi.org/10.1093/hmg/10.12.1307
Chen Y, Wang B, Liu D, Li JJ, Xue Y, Sakata K, Zhu LQ, Heldt SA et al. (2014) Hsp90 chaperone inhibitor 17-AAG attenuates Abeta-induced synaptic toxicity and memory impairment. J Neurosci 34(7):2464–2470. https://doi.org/10.1523/JNEUROSCI.0151-13.2014
Sharma S, Sharma N, Saini A, Nehru B (2019) Carbenoxolone reverses the amyloid beta 1–42 oligomer-induced oxidative damage and anxiety-related behavior in rats. Neurotox Res 35(3):654–667. https://doi.org/10.1007/s12640-018-9975-2
Cao F, Wang Y, Peng B, Zhang X, Zhang D, Xu L (2018) Effects of celastrol on Tau hyperphosphorylation and expression of HSF-1 and HSP70 in SH-SY5Y neuroblastoma cells induced by amyloid-beta peptides. Biotechnol Appl Biochem 65(3):390–396. https://doi.org/10.1002/bab.1633
Paris D, Ganey NJ, Laporte V, Patel NS, Beaulieu-Abdelahad D, Bachmeier C, March A, Ait-Ghezala G et al. (2010) Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation 7:17. https://doi.org/10.1186/1742-2094-7-17
Wang J, Gines S, MacDonald ME, Gusella JF (2005) Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci 6:1. https://doi.org/10.1186/1471-2202-6-1
Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M et al. (2004) Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10(2):148–154. https://doi.org/10.1038/nm985
Shaltiel-Karyo R, Frenkel-Pinter M, Rockenstein E, Patrick C, Levy-Sakin M, Schiller A, Egoz-Matia N, Masliah E et al. (2013) A blood-brain barrier (BBB) disrupter is also a potent alpha-synuclein (alpha-syn) aggregation inhibitor: a novel dual mechanism of mannitol for the treatment of Parkinson disease (PD). J Biol Chem 288(24):17579–17588. https://doi.org/10.1074/jbc.M112.434787
Huang F, Wang J, Qu A, Shen L, Liu J, Liu J, Zhang Z, An Y et al. (2014) Maintenance of amyloid beta peptide homeostasis by artificial chaperones based on mixed-shell polymeric micelles. Angew Chem Int Ed Engl 53(34):8985–8990. https://doi.org/10.1002/anie.201400735
Singh MP, Mishra M, Sharma A, Shukla AK, Mudiam MK, Patel DK, Ram KR, Chowdhuri DK (2011) Genotoxicity and apoptosis in Drosophila melanogaster exposed to benzene, toluene and xylene: attenuation by quercetin and curcumin. Toxicol Appl Pharmacol 253(1):14–30. https://doi.org/10.1016/j.taap.2011.03.006
Choi JH, Choi AY, Yoon H, Choe W, Yoon KS, Ha J, Yeo EJ, Kang I (2010) Baicalein protects HT22 murine hippocampal neuronal cells against endoplasmic reticulum stress-induced apoptosis through inhibition of reactive oxygen species production and CHOP induction. Exp Mol Med 42(12):811–822. https://doi.org/10.3858/emm.2010.42.12.084
Tatenhorst L, Eckermann K, Dambeck V, Fonseca-Ornelas L, Walle H, Lopes da Fonseca T, Koch JC, Becker S et al. (2016) Fasudil attenuates aggregation of alpha-synuclein in models of Parkinson’s disease. Acta Neuropathol Commun 4:39. https://doi.org/10.1186/s40478-016-0310-y
Limbocker R, Staats R, Chia S, Ruggeri FS, Mannini B, Xu CK, Perni M, Cascella R et al. (2021) Squalamine and its derivatives modulate the aggregation of amyloid-beta and alpha-synuclein and suppress the toxicity of their oligomers. Front Neurosci 15:680026. https://doi.org/10.3389/fnins.2021.680026
Perni M, Flagmeier P, Limbocker R, Cascella R, Aprile FA, Galvagnion C, Heller GT, Meisl G et al. (2018) Multistep inhibition of alpha-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem Biol 13(8):2308–2319. https://doi.org/10.1021/acschembio.8b00466
Schwab K, Frahm S, Horsley D, Rickard JE, Melis V, Goatman EA, Magbagbeolu M, Douglas M et al. (2017) A protein aggregation inhibitor, leuco-methylthioninium bis(hydromethanesulfonate), decreases alpha-synuclein inclusions in a transgenic mouse model of synucleinopathy. Front Mol Neurosci 10:447. https://doi.org/10.3389/fnmol.2017.00447
Attar A, Chan WT, Klarner FG, Schrader T, Bitan G (2014) Safety and pharmacological characterization of the molecular tweezer CLR01 - a broad-spectrum inhibitor of amyloid proteins’ toxicity. BMC Pharmacol Toxicol 15:23. https://doi.org/10.1186/2050-6511-15-23
Deeg AA, Reiner AM, Schmidt F, Schueder F, Ryazanov S, Ruf VC, Giller K, Becker S et al. (1850) Zinth W (2015) Anle138b and related compounds are aggregation specific fluorescence markers and reveal high affinity binding to alpha-synuclein aggregates. Biochim Biophys Acta 9:1884–1890. https://doi.org/10.1016/j.bbagen.2015.05.021
Mahia A, Pena-Diaz S, Navarro S, Jose Galano-Frutos J, Pallares I, Pujols J, Diaz-de-Villegas MD, Galvez JA et al. (2021) Design, synthesis and structure-activity evaluation of novel 2-pyridone-based inhibitors of alpha-synuclein aggregation with potentially improved BBB permeability. Bioorg Chem 117:105472. https://doi.org/10.1016/j.bioorg.2021.105472
Castro-Caldas M, Carvalho AN, Rodrigues E, Henderson CJ, Wolf CR, Rodrigues CM, Gama MJ (2012) Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s disease. Mol Neurobiol 46(2):475–486. https://doi.org/10.1007/s12035-012-8295-4
Nunes AF, Amaral JD, Lo AC, Fonseca MB, Viana RJ, Callaerts-Vegh Z, D’Hooge R, Rodrigues CM (2012) TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-beta deposition in APP/PS1 mice. Mol Neurobiol 45(3):440–454. https://doi.org/10.1007/s12035-012-8256-y
Qi H, Shen D, Jiang C, Wang H, Chang M (2021) Ursodeoxycholic acid protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis in MPTP/MPP(+)-induced Parkinson’s disease. Neurosci Lett 741:135493. https://doi.org/10.1016/j.neulet.2020.135493
Batarseh YS, Mohamed LA, Al Rihani SB, Mousa YM, Siddique AB, El Sayed KA, Kaddoumi A (2017) Oleocanthal ameliorates amyloid-beta oligomers’ toxicity on astrocytes and neuronal cells: in vitro studies. Neuroscience 352:204–215. https://doi.org/10.1016/j.neuroscience.2017.03.059
Schneider SA, Alcalay RN (2020) Precision medicine for Parkinson’s disease: Ambroxol for glucocerebrosidase-associated Parkinson’s disease, first trial completed. Mov Disord 35(7):1134–1135. https://doi.org/10.1002/mds.28072
Friesen EL, De Snoo ML, Rajendran L, Kalia LV, Kalia SK (2017) Chaperone-based therapies for disease modification in Parkinson’s disease. Parkinsons Dis 2017:5015307. https://doi.org/10.1155/2017/5015307
Vidoni C, Secomandi E, Castiglioni A, Melone MAB, Isidoro C (2018) Resveratrol protects neuronal-like cells expressing mutant huntingtin from dopamine toxicity by rescuing ATG4-mediated autophagosome formation. Neurochem Int 117:174–187. https://doi.org/10.1016/j.neuint.2017.05.013
Choi JG, Kim N, Huh E, Lee H, Oh MH, Park JD, Pyo MK, Oh MS (2017) White ginseng protects mouse hippocampal cells against amyloid-beta oligomer toxicity. Phytother Res 31(3):497–506. https://doi.org/10.1002/ptr.5776
Bukhari SN, Jantan I, Masand VH, Mahajan DT, Sher M, Naeem-ul-Hassan M, Amjad MW (2014) Synthesis of alpha, beta-unsaturated carbonyl based compounds as acetylcholinesterase and butyrylcholinesterase inhibitors: characterization, molecular modeling, QSAR studies and effect against amyloid beta-induced cytotoxicity. Eur J Med Chem 83:355–365. https://doi.org/10.1016/j.ejmech.2014.06.034
Nguyen P, Oumata N, Soubigou F, Evrard J, Desban N, Lemoine P, Bouaziz S, Blondel M et al. (2014) Evaluation of the antiprion activity of 6-aminophenanthridines and related heterocycles. Eur J Med Chem 82:363–371. https://doi.org/10.1016/j.ejmech.2014.05.083
Guzior N, Bajda M, Skrok M, Kurpiewska K, Lewinski K, Brus B, Pislar A, Kos J et al. (2015) Development of multifunctional, heterodimeric isoindoline-1,3-dione derivatives as cholinesterase and beta-amyloid aggregation inhibitors with neuroprotective properties. Eur J Med Chem 92:738–749. https://doi.org/10.1016/j.ejmech.2015.01.027
Ahmad S, Jo MH, Ikram M, Khan A, Kim MO (2021) Deciphering the potential neuroprotective effects of luteolin against Abeta1–42-induced Alzheimer’s disease. Int J Mol Sci 22 (17). https://doi.org/10.3390/ijms22179583
Oliveira AM, Cardoso SM, Ribeiro M, Seixas RS, Silva AM, Rego AC (2015) Protective effects of 3-alkyl luteolin derivatives are mediated by Nrf2 transcriptional activity and decreased oxidative stress in Huntington’s disease mouse striatal cells. Neurochem Int 91:1–12. https://doi.org/10.1016/j.neuint.2015.10.004
Zhou Y, Vu K, Chen Y, Pham J, Brady T, Liu G, Chen J, Nam J et al. (2009) Chloro-oxime derivatives as novel small molecule chaperone amplifiers. Bioorg Med Chem Lett 19(11):3128–3135. https://doi.org/10.1016/j.bmcl.2009.03.011
Okamoto M, Gray JD, Larson CS, Kazim SF, Soya H, McEwen BS, Pereira AC (2018) Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early-onset Alzheimer’s disease. Transl Psychiatry 8(1):153. https://doi.org/10.1038/s41398-018-0201-z
Schiefer J, Landwehrmeyer GB, Luesse HG, Sprunken A, Puls C, Milkereit A, Milkereit E, Kosinski CM (2002) Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington’s disease. Mov Disord 17(4):748–757. https://doi.org/10.1002/mds.10229
Zhang B, Au Q, Yoon IS, Tremblay MH, Yip G, Zhou Y, Barber JR, Ng SC (2009) Identification of small-molecule HSF1 amplifiers by high content screening in protection of cells from stress induced injury. Biochem Biophys Res Commun 390(3):925–930. https://doi.org/10.1016/j.bbrc.2009.10.079
Wang B, Liu Y, Huang L, Chen J, Li JJ, Wang R, Kim E, Chen Y et al. (2017) A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol Psychiatry 22(7):990–1001. https://doi.org/10.1038/mp.2016.104
Smith AV, Tabrizi SJ (2020) Therapeutic antisense targeting of huntingtin. DNA Cell Biol 39(2):154–158. https://doi.org/10.1089/dna.2019.5188
Uehara T, Choong CJ, Nakamori M, Hayakawa H, Nishiyama K, Kasahara Y, Baba K, Nagata T et al. (2019) Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting alpha-synuclein as a novel therapy for Parkinson’s disease. Sci Rep 9(1):7567. https://doi.org/10.1038/s41598-019-43772-9
Zhao HT, John N, Delic V, Ikeda-Lee K, Kim A, Weihofen A, Swayze EE, Kordasiewicz HB et al. (2017) LRRK2 antisense oligonucleotides ameliorate alpha-synuclein inclusion formation in a Parkinson’s disease mouse model. Mol Ther Nucleic Acids 8:508–519. https://doi.org/10.1016/j.omtn.2017.08.002
Chang JL, Hinrich AJ, Roman B, Norrbom M, Rigo F, Marr RA, Norstrom EM, Hastings ML (2018) Targeting amyloid-beta precursor protein, APP, splicing with antisense oligonucleotides reduces toxic amyloid-beta production. Mol Ther 26(6):1539–1551. https://doi.org/10.1016/j.ymthe.2018.02.029
Lu M, Liu T, Jiao Q, Ji J, Tao M, Liu Y, You Q, Jiang Z (2018) Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem 146:251–259. https://doi.org/10.1016/j.ejmech.2018.01.063
Chu Y, Muller S, Tavares A, Barret O, Alagille D, Seibyl J, Tamagnan G, Marek K et al. (2019) Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain 142(11):3565–3579. https://doi.org/10.1093/brain/awz296
Silva MC, Ferguson FM, Cai Q, Donovan KA, Nandi G, Patnaik D, Zhang T, Huang HT et al. (2019) Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 8. https://doi.org/10.7554/eLife.45457
Arancibia S, Silhol M, Mouliere F, Meffre J, Hollinger I, Maurice T, Tapia-Arancibia L (2008) Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis 31(3):316–326. https://doi.org/10.1016/j.nbd.2008.05.012
Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A et al. (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15(3):331–337. https://doi.org/10.1038/nm.1912
Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14(9):626–636. https://doi.org/10.1038/nrn3549
Yang S, Chang R, Yang H, Zhao T, Hong Y, Kong HE, Sun X, Qin Z et al. (2017) CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest 127(7):2719–2724. https://doi.org/10.1172/JCI92087
Gyorgy B, Loov C, Zaborowski MP, Takeda S, Kleinstiver BP, Commins C, Kastanenka K, Mu D et al. (2018) CRISPR/Cas9 mediated disruption of the Swedish APP allele as a therapeutic approach for early-onset Alzheimer’s disease. Mol Ther Nucleic Acids 11:429–440. https://doi.org/10.1016/j.omtn.2018.03.007
Taguchi YV, Gorenberg EL, Nagy M, Thrasher D, Fenton WA, Volpicelli-Daley L, Horwich AL, Chandra SS (2019) Hsp110 mitigates alpha-synuclein pathology in vivo. Proc Natl Acad Sci U S A 116(48):24310–24316. https://doi.org/10.1073/pnas.1903268116
Singh MP, Ram KR, Mishra M, Shrivastava M, Saxena DK, Chowdhuri DK (2010) Effects of co-exposure of benzene, toluene and xylene to Drosophila melanogaster: alteration in hsp70, hsp60, hsp83, hsp26, ROS generation and oxidative stress markers. Chemosphere 79(5):577–587. https://doi.org/10.1016/j.chemosphere.2010.01.054
Singh MP, Reddy MM, Mathur N, Saxena DK, Chowdhuri DK (2009) Induction of hsp70, hsp60, hsp83 and hsp26 and oxidative stress markers in benzene, toluene and xylene exposed Drosophila melanogaster: role of ROS generation. Toxicol Appl Pharmacol 235(2):226–243. https://doi.org/10.1016/j.taap.2008.12.002
Acknowledgements
The authors are grateful to Lovely Professional University for providing infrastructure and library facilities.
Author information
Authors and Affiliations
Contributions
Conceptualization, GC, and MPS; writing—original draft preparation, GC, SS, SY, S, AHM, MPS; writing—review and editing, MPS, SAB. “All authors have read and agreed to the published version of the manuscript.”
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chopra, G., Shabir, S., Yousuf, S. et al. Proteinopathies: Deciphering Physiology and Mechanisms to Develop Effective Therapies for Neurodegenerative Diseases. Mol Neurobiol 59, 7513–7540 (2022). https://doi.org/10.1007/s12035-022-03042-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-03042-8