
Abstract
Spinal cord injury (SCI) can result in significant neurological impairment and functional and cognitive deficits. It is well established that SCI results in focal neurodegeneration that gradually spreads to other cord areas. On the other hand, traumatic brain injury (TBI) is strongly associated with tau protein pathology and neurodegeneration that can spread in areas throughout the brain. Tau is a microtubule-associated protein abundant in neurons and whose abnormalities result in neuronal cell death. While SCI and TBI have been extensively studied, there is limited research on the relationship between SCI and brain tau pathology. As a result, in this study, we examined tau pathology in spinal cord and brain samples obtained from severe SCI mouse models at various time points. The effects of severe SCI on locomotor function, spatial memory, anxiety/risk-taking behavior were investigated. Immunostaining and immunoblotting confirmed a progressive increase in tau pathology in the spinal cord and brain areas. Moreover, we used electron microscopy to examine brain samples and observed disrupted mitochondria and microtubule structure following SCI. SCI resulted in motor dysfunction, memory impairment, and abnormal risk-taking behavior. Notably, eliminating pathogenic cis P-tau via systemic administration of appropriate monoclonal antibodies restored SCI’s pathological and functional consequences. Thus, our findings suggest that SCI causes severe tauopathy that spreads to brain areas, indicating brain dysfunction. Additionally, tau immunotherapy with an anti-cis P-tau antibody could suppress pathogenic outcomes in SCI mouse models, with significant clinical implications for SCI patients.
Graphical abstract
SCI induces profound pathogenic cis p-tau, which diffuses into the brain through CSF, resulting in brain neurodegeneration and cognitive decline.

Similar content being viewed by others
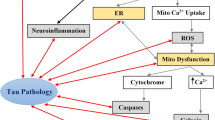


Avoid common mistakes on your manuscript.
Introduction
Each year, approximately 500,000 people worldwide experience a spinal cord injury (SCI) due to motor vehicle collisions, falls, violence, and sporting accidents [1, 2], resulting in significant neurological impairment and significant emotional and psychological distress [3].
SCI can disrupt nerve impulse conduction, resulting in neurological dysfunction [4]. Primary spinal cord injury has been shown to rapidly disrupt cell membranes, myelin, and axons within longitudinal tracts. Additionally, the SCI damages microvessels, causing destructive secondary injury by releasing various harmful factors [5]. Numerous cellular and molecular mechanisms may contribute to extensive neurodegeneration during the secondary injury process [6]. As active biological processes, such cascades offer the possibility of treating SCI with selective inhibitors.
SCI can also alter systemic immune functions, affecting the brain [7]. Moreover, the released agents may enter the brain through the cerebrospinal fluid (CSF). The brain abnormalities caused by SCI result from modified afferent and efferent routes. However, SCI induces distinct neuropathological changes, such as decreasing the number of cortical neurons [4, 8,9,10,11,12]. Additionally, cognitive impairment affects 60% of the SCI population [4, 8, 9, 12].
Despite extensive considerations, the mechanism by which SCI results in brain abnormalities remains unknown. Clearly, abnormal tau protein expression is a significant pathological hallmark of traumatic brain injury (TBI) [13]. Tau is a microtubule-associated protein that promotes the formation and stabilization of microtubules (MTs) [14, 15]. Tau is frequently hyperphosphorylated on Ser/Thr residues in tauopathies, impairing the MT’s function and altering the protein’s integrity, resulting in tau aggregation and tangle formation [16, 17], most notably in chronic traumatic encephalopathy (CTE) [18,19,20,21,22] and Alzheimer’s disease (AD) [16, 17].
It is well established that phosphorylated tau at Thr231 exists in two distinct cis and trans conformations, with the cis pThr231-tau (cis P-tau) conformer being highly neurotoxic and acting as an early initiator of the tauopathy process following TBI. Peptidyl-prolyl cis/trans isomerase (Pin1) inhibits the development of tau pathology and neurodegeneration in AD by converting the neurotoxic cis conformation of phosphorylated tau at the Thr231-Pro motif to the physiological trans conformation [23,24,25,26,27,28,29,30,31]. Additionally, the tauopathy process can be inhibited in vitro and in vivo using a cis P-tau monoclonal antibody (cis mAb) [32,33,34]. Also, pathogenic cis P-tau is prion-like and spreads throughout the brain and CSF in tauopathy mouse models [33,34,35,36,37].
Several studies have examined the concentrations of total tau and P-tau in CSF, serum, and spinal cord tissue from patients and experimental animals with SCI. However, the molecular mechanism underlying tau pathology in SCI is not entirely understood. Furthermore, the causal relationship between SCI and brain dysfunction has remained elusive. Thus, as previously proposed, we examined the tau pathology process in severe SCI (sSCI) mouse models [38]. We induced sSCI and investigated the formation and degeneration of various pathogenic tau species in cord and brain tissues at various time points to determine whether SCI injury can result in brain pathology.
Materials and Methods
Animals and study design
Male Balb/c mice (2–3 months old) weighing 22–26 g were obtained and housed in clear plastic cages with a 12-h light/dark cycle and free access to water and food under controlled temperature and humidity. All animals were given one week to acclimate to their new environment before undergoing experimental procedures. The ethics committee of Tabriz University of Medical Sciences approved all protocols (approval No. IR.TBZMED.VCR.REC.1398.067). Experiments were conducted following the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals (NIH Publications).
A total of 54 adult male mice were randomly assigned to one of six groups (n = 9): Sham (laminectomy surgery without compression injury), sSCI (48 h), sSCI (2 W), sSCI (1 M) (severe compression injury at the 8th thoracic segment ‘T8’ of the spinal cord and sacrificed 48 h, two weeks, and one month after the SCI), sSCI (2 M) + IgG (severe compression injury at T8 and received IgG after the SCI for two months), and sSCI (2 M) + cis mAb group (severe compression injury at T8 and received cis mAb after the SCI for two months).
Laminectomy and spinal cord compression model using calibrated forceps
All procedures were conducted in a sterile environment. Mice were anesthetized with 4% isoflurane, and a laminectomy was performed at T7-9 to expose the T8 segment of the spinal cord without damaging the dura. Pairs of forceps were applied for laterally compressing the spinal cord to the corresponding thickness (0.25 mm) for 15 s [39, 40]. The sham group received laminectomy and forceps placement around the spinal cord without compression. After forceps removal, muscles and skin were stitched, followed by the administration of saline solution (1 ml) for rehydration, buprenorphine (0.05 mg/kg) for pain relief, and ciprofloxacin (5 mg/kg) for bladder infection treatment/prevention, all subcutaneously (SC) twice daily for three days. The animals were monitored in a temperature-controlled room until they recovered, at which point they were transferred to their own cage. SCI mice bladders were manually expressed twice daily until the urinary reflex was established.
Locomotor analysis
Motor function was assessed in mice to ensure that SCI or laminectomy was effective. The Basso mouse scale (BMS) assessed hind-limb function in groups one day after injury in the open field (OF) [41]. In brief, animals were placed individually in the OF chamber (22.5 × 22.5 cm) and allowed to explore freely for 5 min. Two independent evaluators assigned a score of 0 to 9 to each animal, with 0 indicating a complete loss of locomotor function and 9 indicating no locomotor deficits.
To observe the characteristics of cis P-tau, AT8 P-tau, and AT100 P-tau induction following SCI, mice in the sSCI (48 h) and sSCI (2 W) groups were anesthetized with intraperitoneal (IP) injections of ketamine (60 mg/kg) and xylazine (10 mg/kg), 48 h and two weeks after the SCI, and spinal cord tissue samples were collected for immunoblotting and immunofluorescence staining analyses.
Antibody treatment of mice
To assess cis mAb’s efficacy in treating SCI, we examined whether cis mAb could affect intracellular P-tau in sSCI (2 M) + cis mAb and sSCI (2 M) + IgG groups. Antibody-treated sSCI animals were given either mouse cis mAb or IgG at random. Three days prior to the injury, animals received one dose of cis mAb/IgG IP (200 µg/per mouse), a single IP post-injury treatment (20 µg in 5 µl) 15 min after SCI, and then IP (200 µg) every four days for two weeks, followed by 200 µg weekly for the remainder of the two months of treatment [34].
Transmission electron microscopy (TEM)
The ultrastructural examination was performed using the TEM method. Brain and spinal cord specimens were cut into 2 × 2 mm pieces from the sham and SCI mouse models treated with either control IgG or cis mAb. The cells were fixed in glutaraldehyde 2.5%, buffered with 0.1 M phosphate (pH 7.4), post-fixed with 1% osmium tetroxide, and finally embedded in resin. Ultrathin sections measuring 60–90 nm were cut and placed on a copper grid, stained with a solution of uranyl acetate and lead citrate, and examined using a ZEISS electron microscope (EM902A) and a Leo 906 transmission electron microscope (Leo, Germany).
Immunoblotting analysis
Immunoblotting was performed on the spinal cord, and brain samples homogenized in RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP 40, 0.1% SDS, 0.5% Na-deoxycholate, 50 mM NaF) containing proteinase and phosphatase inhibitors and then mixed with SDS sample buffer and loaded onto a gel after boiling. Polyacrylamide gel electrophoresis at 12% resolved the proteins and transferred them to a PVDF membrane. The membranes were then blocked for 1 h with 2% milk in TBST (10 mM Tris–HCl pH 7.6, 150 mM NaCl, 0.1% Tween 20). The membranes were then incubated overnight at 4ºC with the following primary antibodies: cis P-tau mAb (gift from KP. Lu), AT8 P-tau (MN1020), AT100 P-tau (MN1060), and Tau-5 (MAB361). The immunoblots were then incubated with a secondary antibody conjugated to HRP in 2% milk in TBST. Chemiluminescence was used to detect the signals (Perkin Elmer, San Jose, CA). Following each step, the membranes were washed six times with TBST. ImageJ was used to quantify the immunoblotting results.
Subsequently, two-stage normalization was conducted. Initially, the band of interests was normalized against actin. The relative intensity of P-tau markers (cis P-tau, AT8 P-tau, and AT100 P-tau) was normalized to total tau (Tau-5). Afterward, normalized data were visualized as expressions in test samples compared to sham groups.
Immunostaining analysis
Mice were deeply anesthetized, and the left ventricle was perfused with 10% neutral buffered formalin. The spinal cord and brain were immediately removed and post-fixed overnight in 10% neutral buffered formalin. A 1.5-cm segment of spinal cord centered on the injury site as well as mice brain samples were embedded into the paraffin and then cut into 8 µm increments by a microtome.
Dewaxed sections were then dehydrated in a series of ethanol dilutions. A 5% ammonium chloride solution (Merck, 101,145) was used to quench autofluorescence. For antigen enhancement, the sections were placed in a steamer (0.01 M) sodium citrate (Sigma-Aldrich, S4641) for 20 min for antigen enhancement. The slides were then permeabilized for 15 min with 0.5% Triton X-100 (Sigma-Aldrich, T8532) and blocked for one hour with 10% goat serum. Following that, the slides were incubated with cis P-tau mAb (gift from KP. Lu). The samples were incubated with the anti-mouse secondary antibodies (Alexa Fluor 488 or 594) at 37 °C for 1 h. DAPI (Invitrogen, D1306) was used to stain the nuclei, and the images were captured using a fluorescent microscope (BX71; Olympus equipped with DP72 digital camera).
Twenty ROIs (350 × 450 µm2 dimension representative view) were randomly selected and quantified from the cerebral cortices. ImageJ was used to quantify the immunofluorescence intensity (n = 3 per group).
Open-field locomotion
The OF test was used to assess hind-limb functions and spontaneous locomotor activity on days 1, 30, and 60 following injury. We scored locomotor performance using the BMS and a related subscale [42]. We used a computer-based video tracking system (Noldus Ethovision) to record mice’s total traveled distance, speed, and walking pattern for 5 min to determine spontaneous locomotor activity [42,43,44,45,46].
Elevated plus-maze (EPM) test
The EPM task is an experimental model used to examine cortical circuits to assess anxiety/risk-taking behavior [47,48,49,50,51,52]. The test is typically conducted on a plus-shaped apparatus raised 50 cm above the ground and equipped with two opposite open (aversive, no walls) and two opposite closed (safe, 15 cm high walls) arms (30 × 5 cm), as well as a central square. A mouse is placed on the apparatus’s central square, facing an open arm, and is allowed 5 min to explore the maze. We kept track of the number of open arm entries and the amount of time spent exploring open arms (Noldus Ethovision). The maze was thoroughly cleaned following each trial to ensure that no odorant interfered with the test. Mice with normal levels of anxiety/risk-taking behavior enter and stay in the open arms less frequently and for a shorter time. The open arms approach addresses the abnormal risk-taking and anxiety behaviors associated with cognitive decline [34, 53].
Y-maze spontaneous alternation test
The Y-maze test assesses spatial working memory in rodents and quantifies cognitive deficits. The Y-maze is composed of three identical black arms (30 cm long, 6 cm wide, and 15 cm high walls) mounted at a 120° angle to one another in the shape of the letter ‘Y.’ Generally, rodents will explore a new arm rather than revisiting previously visited arms. A random arm (arms A–C) was chosen as the ‘start’ arm, and the mouse was placed at its end and allowed to move through each arm. The number of arm entries (when all four paws enter the arm) was recorded for 10 min. Alternation is defined as non-repeating entries into each arm. The percentage of alternations is calculated as follows: (Number of alternations / Number of arm entries) × 100. A mouse with intact working memory scored significantly ˃ 50% [46, 54, 55].
Mice were anesthetized with ketamine–xylazine IP injections at the end of the second month and following behavioral tests. Brain and spinal cord tissue samples were collected for electron microscopy, immunoblotting, and immunofluorescence staining analyses.
Statistical analysis
To compare the groups’ mean differences, all normally distributed data were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s HSD multiple comparisons post hoc test. Additionally, we used one-way multivariate covariance analysis (MANCOVA) to eliminate the effect of controlling variables or covariates on the relationship between independent and dependent variables. All data were analyzed using SPSS software (v. 22) and reported as mean ± standard deviation (SD), with P < 0.05 considered significant. All graphs were obtained by GraphPad Prism version 8.4.3. F represents the degree of freedom; n denotes the number of different animals.
Results
Compression severe SCI induced tau pathology in cord neurons
We studied tau pathology induced by SCI in SCI mouse models using immunoblotting and immunofluorescence staining of spinal cord tissues. Severe SCI induced pathogenic cis P-tau acutely and persistently 48 h after the injury and maintained it at elevated levels for two weeks. At 48 h and two weeks after injury, robust cis P-tau signals (3.638 ± 0.0442) and (4.975 ± 0.753) were detected in the cords (Fig. 1a–c, P = 0.136, P = 0.0167). Immunoblotting also demonstrated a progressive increase in AT8 P-tau (early tangle) [F (2, 4) = 31.98, P = 0.0229] and AT100 P-tau (late tangle) [F (2, 4) = 57.77, P = 0.0018] in cord tissues following timely trauma (Fig. 1b-e). Evidently, the SCI resulted in the development of tau pathology in the cord tissue.

Severe SCI had a robust and persistent effect on tau pathogenicity induction in the cord. Mice were subjected to SCI via calibrated forceps. (a) The sham and sSCI mouse cords (48 h and 2w after injury) were stained with anti cis P-tau antibody followed by immunofluorescence staining. Cis, green; DNA, blue; scale bar, 100 µm; n = 3. (b–e) Immunoblots were stained with cis P-tau, AT8 P-tau, and AT100 P-tau antibodies, followed by quantification analysis. Protein bands were quantified with ImageJ software and normalized against actin and cis P-tau, AT8 P-tau, and AT100 P-tau relative intensities against total tau (Tau-5). Normalized data were depicted as expressions in test samples versus sham groups, n = 3. One-way ANOVA statistically analyzed data by Tukey’s post hoc test (mean ± SD.). *P < 0.05, **P < 0.01. h, hours; w, weeks
Improved tau pathology effects triggered by sSCI via cis mAb
It is well established that cis P-tau is associated with neurodegenerative outcomes and the progression of TBI-induced tauopathy. As previously stated, we demonstrated that cis P-tau was induced 48 h after sSCI, implying that there may be a link between cis P-tau and pathological changes following injury. To better understand the brain complications caused by SCI, particularly the efficacy of cis mAb in treating neurodegenerative pathologies caused by sSCI, we treated animal models for two months with either cis mAb or a control IgG isotype. Following sSCI, this study demonstrates that pathogenic cis P-tau spreads to the brain and induces tau pathology [34] in various areas, including the cortices (Fig. 2a–d). Moreover, as previously observed, cis mAb treatment effectively suppressed tau pathology (Fig. 2a–d) [34, 56,57,58,59].

Treating sSCI mice with cis mAb blocked cistauosis and tau pathology in the brain. (a, b) Mice with severe SCI were treated with cis mAb or control IgG for two months. The cortices of sham and sSCI mouse brains were stained with cis P-tau followed by immunofluorescence intensity quantification. Cis P-tau, red; DAPI, blue; scale bar, 100 µm; n = 3. (c–f) Immunoblots were stained with cis P-tau, AT8 P-tau, AT100 P-tau, and actin antibodies, followed by quantification analysis. The protein bands were quantified with ImageJ and normalized against actin and cis P-tau, AT8 P-tau, and AT100 P-tau relative intensities against total tau (Tau-5). Normalized data were depicted as expressions in test samples versus sham groups, n = 3. One-way ANOVA statistically analyzed data by Tukey’s post-hoc test (mean ± SD.). *P < 0.05, **P < 0.01. M, month; cis, cis mAb; ns: not significant
One month after injury, immunofluorescence staining of sSCI mouse brains revealed strong cis P-tau signals (11.70 ± 2.078, P = 0.0191) (Fig. 2a, b). Additionally, immunoblotting confirmed the presence of a significant amount of cis P-tau (2.511 ± 0.071, P = 0.0272), AT8 P-tau (5.473 ± 0.432, P = 0.0111), and AT100 P-tau (3.358 ± 0.323, P = 0.0124) in the cortices of sSCI mice, whereas no significant amount of P-tau epitopes was detected in animals in the sham group (Fig. 2c–f). While immunotherapy with control IgG did not affect the formation of cis P-tau in the cortices of sSCI animals (27.94 ± 3.554, P = 0.0234) compared to the sham group, cis mAb administration effectively prevented tau pathology and cistauosis in immunostained sSCI mouse brains (1.611 ± 0.4818, P = 0.7424) (Fig. 2a, b). Similarly, cis mAb administration effectively inhibited the formation of cis P-tau, AT8 P-tau, and AT100 P-tau in sSCI mouse brain immunoblots, when compared to two-month sSCI IgG treated mice (P = 0.0492, P = 0.0219, P = 0.0293, respectively); however, IgG treatment did not affect the number of tau epitopes formed (Fig. 2c–f).
Evidently, sSCI can cause tau pathology in both the spinal cord and brain and immunotherapy with cis mAb can effectively inhibit the formation of pathogenic cis P-tau in the brains of sSCI animals.
Improved SCI-related ultrastructural impairment via cis mAb
According to previous research, SCI causes axonal injury and intracellular Ca2+ and tau hyperphosphorylation. Ca2+ uptake by the mitochondria results in mitochondrial dysfunction and an increase in reactive oxygen species (ROS). Furthermore, hyperphosphorylated tau promotes neuroinflammation, ROS generation, mitochondrial dysfunction (disrupted mitochondrial membrane), microtubule disruption (distributed microtubules), and tangle formation [16, 38, 46]. TBI has previously been shown to cause significant microtubule and mitochondrial disruption [34]. Given that the disruption is caused by pathogenic cis P-tau, cis mAb treatment may restore the impairment [33, 34, 60, 61]. To this end, we examined brain ultrastructural changes employing the respective monoclonal antibody with or without pathogenic cis P-tau elimination. We observed that SCI resulted in pathogenic cis P-tau and structural damage to the spinal cord and brain. Additionally, cis mAb restored the abnormalities effectively (Fig. 3).

Treatment of sSCI mice with cis mAb inhibited the destruction of the spinal cord and brain ultrastructure. Electron micrographs of spinal cord and brain sSCI mouse models; either treated or untreated with cis P-tau mAb. Axonal MTs (blue open arrows) and MIs (red filled arrows). Scale bars, 100 nm. M, month; MT, microtubule; MI, mitochondria
Improved SCI-related motor function impairment via cis mAb
On days 1, 30, and 60 following sSCI, an open field test was used to assess spontaneous locomotor activity and hind-limb functions using BMS scores [41]. On each of the three days of the experiment, mice in the sSCI (1 M) and sSCI (2 M) + IgG groups demonstrated a significant decrease in the total traveled distance (P < 0.001) when compared to mice in the shame group. The decrease in the total traveled distance was mirrored by a decrease in walking speed in sSCI (1 M) and sSCI (2 M) + IgG mice (P < 0.001) compared to the sham group. Notably, on days 30 and 60 following injury, we observed a significant improvement in spontaneous locomotor activity in the sSCI group treated with cis mAb (P < 0.001) compared to the sSCI and IgG-treated sSCI groups (Fig. 4a, b, d).

Cis P-tau elimination in sSCI mice improved motor function. Severe SCI mice were subjected to an open field test to examine spontaneous locomotor activity (a, b), hind-limb locomotor functions, using the BMS score (c), and walking pattern (d). Data expressed as mean ± SD. ***P < 0.001 versus sham, ###P < 0.001 vs sSCI (2 M) + cis mAb. M, month; D, day; BMS, Basso mouse scale
One day after the injury, all mice exhibited a near-complete loss of motor function (BMS score of 1). By day 60, hind-limb functional recovery was significantly improved in cis mAb-treated mice compared to IgG-treated mice (P < 0.001) (Fig. 4c). As a result, cis P-tau elimination improved spontaneous locomotor activity and hind-limb function recovery in sSCI mice.
Restored SCI-related cognitive dysfunction via cis mAb
The Y-maze spontaneous alternation test was performed to measure spatial working memory. The sham group exhibited functional working memory with a spontaneous alteration rate of ~ 65%. Compared to the sham group, severe SCI significantly decreased the percentage of spontaneous alteration (P < 0.001). SCI mice treated with cis mAb had a significantly higher rate of spontaneous alteration than sSCI mice treated with IgG (P = 0.002 or P < 0.01) (Fig. 5a).

Cis P-tau immunotherapy restores cognitive dysfunction in mice with SCI. (a) After two months of treatment with IgG or cis mAb, sSCI mice were subjected to the Y-maze spontaneous alteration test to assess spatial working memory. (b, c) sSCI mice were subjected to the EPM test to assess anxiety/risk-taking behavior, and time spent in the open arms and open arm entries were measured. (d, e) Cognitive functions data (alternation percentage and OAT) were normalized against the locomotor activity (speed and total traveled distance) in IgG/cis mAb treated groups to control locomotor activity as a covariate. The MANCOVA analyses showed that cis mAb independently and significantly restored cognitive deficits (P < 0.01). Data expressed as mean ± SD. ***P < 0.001 vs sham, **P < 0.01 vs sham, ###P < 0.001 vs sSCI (2 M) + cis mAb, ##P < 0.01 vs sSCI (2 M) + cis mAb. M, month; OAT, open arm time; OAE, open arm entry
To assess cis mAb’s anxiolytic efficacy, we used an elevated-plus maze test 60 days after sSCI. In the EPM test, the sham group animals explored the open arms significantly less than IgG-treated sSCI mice (P = 0.009 or P < 0.01). Additionally, similar to the sham group, the cis mAb-treated group spent significantly less time in the open arms (P < 0.001) than the IgG-treated sSCI groups (Fig. 5b). Total open arm entries were increased in SCI (2 M) + IgG mice compared to the sham group and cis mAb-treated sSCI mice (Fig. 5c). As a result, all IgG-treated sSCI mice exhibited anxiety/risk-taking behavior when exploring the two open arms; in comparison, cis mAb-treated mice exhibited minimal anxiety. Our data support the notion that rodents prefer protected environments, such as the nest.
The data for cognitive functions (alternation percentage and OAT) were then normalized against locomotor activity on day 60 (speed and total traveled distance; see Fig. 4a, b) to eliminate the effect of locomotor activity on the relationship between IgG/cis mAb treatment and cognitive functions. The data indicated that cis mAb restores cognitive deficits independently and significantly (P < 0.01) (Fig. 5d, e). As a result, cis mAb abolishes cis P-tau and cistauosis and reverses behavioral deficits triggered by SCI.
Discussion
It is self-evident that SCI impairs brain function and contributes to patients’ cognitive decline. However, the molecular mechanisms by which SCI affects the brain remain unknown. SCI can disrupt the cerebral cortex and neural circuits retrogradely, such as thalamic afferents to the hippocampal compartment [62, 63].
Moreover, injured neurons in the spinal cord produce the chemokine cysteine-cysteine ligand 21, which activates microglia in distant spinal cord sections and the thalamus [64, 65]. Furthermore, SCI impairs systemic immune functions [66], which affects the brain. Neuronal cell injury or death can release intracellular MT binding proteins into the extracellular space, traveling to the brain via the CSF [67, 68]. Several studies have demonstrated an increase in tau levels in the CSF following acute TBI [69] and SCI [70]. Despite extensive consideration, the molecular mechanisms underlying the brain pathology caused by SCI remain unknown [71].
It has been evidenced that single severe TBI (ssTBI) or repetitive mild TBI (rmTBI) induces pathogenic cis P-tau in axons a few hours after injury, resulting in neurodegeneration [34]. Cis P-tau is prion-like and spreads throughout the brain, typically accompanied by neuron pathogenicity [34, 37]. Notably, cis mAb treatment can eliminate pathogenic cis P-tau, restore axonal pathologies such as MT defects, organelle transport, and long-term potentiation (LTP), and prevent the development of numerous short- and long-term pathological and functional consequences following ssTBI or rmTBI [33, 34, 37, 61]. TBI induces oxidative stress and Pin1 oxidation at Cys113 and Ser71, resulting in Pin1 inactivation and cis P-tau accumulation [34]. SCI, on the other hand, results in severe oxidative stress [39, 64, 65]. Similarly, we observed that SCI stress promotes the accumulation of pathogenic cis P-tau, most likely via oxidative stress and Pin1 suppression. We hypothesized that SCI-induced oxidative stress would result in Pin1 inactivation and cis P-tau accumulation.
We investigated tau pathologies induced by SCI in order to identify and treat brain complications such as axonal pathology, functional deficits, and cognitive impairment; as previously proposed [38], we discovered that cis P-tau is the initial and critical mediator of cis P-tau neurodegeneration and functional defects following SCI in the mouse model. Additionally, we assessed the effects of cis mAb therapy on pathological and functional brain complications associated with injury. We observed the following: (1) Severe SCI had a robust and persistent effect on cis P-tau induction and resulted in histological changes in the spinal cord and brain tissues; (2) severe SCI resulted in locomotor impairment, cognitive deficits, and anxiety-like behaviors; (3) treatment with cis mAb effectively prevented the development of extensive tauopathy, improved histopathological consequences, and restored motor and cognitive function in SCI mice. Notably, our histological findings demonstrated that the cis P-tau level was significantly increased at the injury site and spread from the spinal cord to the brain within two months of the injury. These findings suggest that induction of cis P-tau is required for the development of several pathological and functional outcomes following SCI.
Conclusion
We examined the relationship between SCI and tau pathology in the mouse models. We demonstrated that pathogenic tau was induced focally in spinal cord tissue early after a severe compression SCI, spread to brain areas via CSF, and induced prominent tauopathy, resulting in motor dysfunction. Overall, we propose cis P-tau as a reliable biomarker for assessing the pathologic outcomes of SCI.
While we demonstrated that SCI increases cis P-tau accumulation, additional experiments are necessary to determine whether tau pathology is caused by Pin1 inactivation.
Additional research in preclinical SCI models and patients with varying degrees of SCI is required to determine the biomarker value of cis P-tau. The biomarkers may lead to identifying novel SCI therapeutic targets and treatment strategies. In summary, our findings suggest that pathogenic cis P-tau is a tauopathy driver in SCI, making it a potential target for immunotherapy diagnosis and treatment.
Data availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- SCI:
-
Spinal cord injury
- CSF:
-
Cerebrospinal fluid
- TBI:
-
Traumatic brain injury
- MTs:
-
Microtubules
- CTE:
-
Chronic traumatic encephalography
- cis P-tau:
-
cis PThr231-tau
- AD:
-
Alzheimer’s disease
- Pin1:
-
Peptidyl-prolyl cis/trans isomerase
- Cis mAb:
-
cis P-tau monoclonal antibody
- sSCI:
-
Severe SCI
- NIH:
-
National Institute of Health
- SC:
-
Subcutaneously
- BMS:
-
The Basso mouse scale
- OF:
-
Open-field
- IP:
-
Intraperitoneal
- TEM:
-
Transmission electron microscopy
- EPM:
-
Elevated plus-maze
- ROI:
-
Region of interest
- ANOVA:
-
Analysis of variance
- SD:
-
Standard deviation
- ssTBI:
-
Single severe TBI
- rmTBI:
-
Repetitive mild TBI
- LTP:
-
Long-term potentiation
References
Ackery A, Tator C, Krassioukov A (2004) A global perspective on spinal cord injury epidemiology. J Neurotrauma 21(10):1355–1370. https://doi.org/10.1089/neu.2004.21.1355
Van den Berg ME, Castellote JM, Mahillo-Fernandez I, de Pedro-Cuesta J (2010) Incidence of spinal cord injury worldwide: a systematic review. Neuroepidemiology 34(3):184–192. https://doi.org/10.1159/000279335
Singh A, Tetreault L, Kalsi-Ryan S, Nouri A, Fehlings MG (2014) Global prevalence and incidence of traumatic spinal cord injury. Clin Epidemiol 6:309–331. https://doi.org/10.2147/CLEP.S68889
Collins-Praino LE, Corrigan F (2017) Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun 60:369–382. https://doi.org/10.1016/j.bbi.2016.09.027
Bethea JR, Dietrich WD (2002) Targeting the host inflammatory response in traumatic spinal cord injury. Curr Opin Neurol 15(3):355–360. https://doi.org/10.1097/00019052-200206000-00021
Tator CH, Fehlings MG (1991) Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg 75(1):15–26. https://doi.org/10.3171/jns.1991.75.1.0015
Nielson JL, Sears-Kraxberger I, Strong MK, Wong JK, Willenberg R, Steward O (2010) Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J Neurosci 30(34):11516–11528. https://doi.org/10.1523/JNEUROSCI.1433-10.2010
Coulthart MB, Jansen GH, Cashman NR (2016) Evidence for transmissibility of Alzheimer disease pathology: Cause for concern? CMAJ 188(10):E210–E212. https://doi.org/10.1503/cmaj.151257
Craig A, Guest R, Tran Y, Middleton J (2017) Cognitive Impairment and Mood States after Spinal Cord Injury. J Neurotrauma 34(6):1156–1163. https://doi.org/10.1089/neu.2016.4632
Nielson JL, Strong MK, Steward O (2011) A reassessment of whether cortical motor neurons die following spinal cord injury. J Comp Neurol 519(14):2852–2869. https://doi.org/10.1002/cne.22661
Wannier T, Schmidlin E, Bloch J, Rouiller EM (2005) A unilateral section of the corticospinal tract at cervical level in primate does not lead to measurable cell loss in motor cortex. J Neurotrauma 22(6):703–717. https://doi.org/10.1089/neu.2005.22.703
Cohen ML, Tulsky DS, Holdnack JA, Carlozzi NE, Wong A, Magasi S, Heaton RK, Heinemann AW (2017) Cognition among community-dwelling individuals with spinal cord injury. Rehabil Psychol 62(4):425–434. https://doi.org/10.1037/rep0000140
Jeter CB, Hergenroeder GW, Hylin MJ, Redell JB, Moore AN, Dash PK (2013) Biomarkers for the diagnosis and prognosis of mild traumatic brain injury/concussion. J Neurotrauma 30(8):657–670. https://doi.org/10.1089/neu.2012.2439
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A 91(12):5562–5566. https://doi.org/10.1073/pnas.91.12.5562
Gendron TF, Petrucelli L (2009) The role of tau in neurodegeneration. Mol Neurodegener 4:13. https://doi.org/10.1186/1750-1326-4-13
Iqbal K, Liu F, Gong CX (2016) Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12(1):15–27. https://doi.org/10.1038/nrneurol.2015.225
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17(1):5–21. https://doi.org/10.1038/nrn.2015.1
Blennow K, Hardy J, Zetterberg H (2012) The neuropathology and neurobiology of traumatic brain injury. Neuron 76(5):886–899. https://doi.org/10.1016/j.neuron.2012.11.021
DeKosky ST, Blennow K, Ikonomovic MD, Gandy S (2013) Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nat Rev Neurol 9(4):192–200. https://doi.org/10.1038/nrneurol.2013.36
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136(Pt 1):43–64. https://doi.org/10.1093/brain/aws307
Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9(4):211–221. https://doi.org/10.1038/nrneurol.2013.29
Zare-Shahabadi A, Masliah E, Johnson GV, Rezaei N (2015) Autophagy in Alzheimer’s disease. Rev Neurosci 26(4):385–395. https://doi.org/10.1515/revneuro-2014-0076
Chen CH, Li W, Sultana R, You MH, Kondo A, Shahpasand K, Kim BM, Luo ML, Nechama M, Lin YM, Yao Y, Lee TH, Zhou XZ, Swomley AM, Butterfield DA, Zhang Y, Lu KP (2015) Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol Dis 76:13–23. https://doi.org/10.1016/j.nbd.2014.12.027
Lee TH, Chen CH, Suizu F, Huang P, Schiene-Fischer C, Daum S, Zhang YJ, Goate A, Chen RH, Zhou XZ, Lu KP (2011) Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol Cell 42(2):147–159. https://doi.org/10.1016/j.molcel.2011.03.005
Lim J, Balastik M, Lee TH, Nakamura K, Liou YC, Sun A, Finn G, Pastorino L, Lee VM, Lu KP (2008) Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J Clin Invest 118(5):1877–1889. https://doi.org/10.1172/JCI34308
Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP (2003) Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424(6948):556–561. https://doi.org/10.1038/nature01832
Lu KP, Hanes SD, Hunter T (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380(6574):544–547. https://doi.org/10.1038/380544a0
Lu KP, Zhou XZ (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 8(11):904–916. https://doi.org/10.1038/nrm2261
Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP (1999) The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399(6738):784–788. https://doi.org/10.1038/21650
Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP (2006) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 440(7083):528–534. https://doi.org/10.1038/nature04543
Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Markesbery WR, Zhou XZ, Lu KP, Butterfield DA (2006) Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol Aging 27(7):918–925. https://doi.org/10.1016/j.neurobiolaging.2005.05.005
Roqanian S, Ahmadian S, Nabavi SM, Pakdaman H, Shafiezadeh M, Goudarzi G, Shahpasand K (2022) Tau nuclear translocation is a leading step in tau pathology process through P53 stabilization and nucleolar dispersion. J Neurosci Res. https://doi.org/10.1002/jnr.25024
Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, Herbert MK, Qiu J, Monuteaux M, Driver J, Yan S, Gormley W, Puccio AM, Okonkwo DO, Lucke-Wold B, Bailes J, Meehan W, Zeidel M, Lu KP, Zhou XZ (2017) Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 8(1):1000. https://doi.org/10.1038/s41467-017-01068-4
Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y, Wei S, Luo ML, Albayram O, Huang P, Rotenberg A, Ryo A, Goldstein LE, Pascual-Leone A, McKee AC, Meehan W, Zhou XZ, Lu KP (2015) Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523(7561):431–436. https://doi.org/10.1038/nature14658
Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ (2016) Potential of the Antibody Against cis-Phosphorylated Tau in the Early Diagnosis, Treatment, and Prevention of Alzheimer Disease and Brain Injury. JAMA Neurol 73(11):1356–1362. https://doi.org/10.1001/jamaneurol.2016.2027
Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP (2012) Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 149(1):232–244. https://doi.org/10.1016/j.cell.2012.02.016
Albayram O, Herbert MK, Kondo A, Tsai CY, Baxley S, Lian X, Hansen M, Zhou XZ, Lu KP (2016) Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci 6:59. https://doi.org/10.1186/s13578-016-0124-4
Nakhjiri E, Vafaee MS, Hojjati SMM, Shahabi P, Shahpasand K (2020) Tau Pathology Triggered by Spinal Cord Injury Can Play a Critical Role in the Neurotrauma Development. Mol Neurobiol 57(11):4845–4855. https://doi.org/10.1007/s12035-020-02061-7
Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24(9):2143–2155. https://doi.org/10.1523/JNEUROSCI.3547-03.2004
Plemel JR, Duncan G, Chen KW, Shannon C, Park S, Sparling JS, Tetzlaff W (2008) A graded forceps crush spinal cord injury model in mice. J Neurotrauma 25(4):350–370. https://doi.org/10.1089/neu.2007.0426
Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG (2006) Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23(5):635–659. https://doi.org/10.1089/neu.2006.23.635
Wu J, Stoica BA, Luo T, Sabirzhanov B, Zhao Z, Guanciale K, Nayar SK, Foss CA, Pomper MG, Faden AI (2014) Isolated spinal cord contusion in rats induces chronic brain neuroinflammation, neurodegeneration, and cognitive impairment Involvement of cell cycle activation. Cell Cycle 13(15):2446–2458. https://doi.org/10.4161/cc.29420
De Risi M, Tufano M, Alvino FG, Ferraro MG, Torromino G, Gigante Y, Monfregola J, Marrocco E, Pulcrano S, Tunisi L, Lubrano C, Papy-Garcia D, Tuchman Y, Salleo A, Santoro F, Bellenchi GC, Cristino L, Ballabio A, Fraldi A, De Leonibus E (2021) Altered heparan sulfate metabolism during development triggers dopamine-dependent autistic-behaviours in models of lysosomal storage disorders. Nat Commun 12(1):3495. https://doi.org/10.1038/s41467-021-23903-5
Hefner K, Cameron HA, Karlsson RM, Holmes A (2007) Short-term and long-term effects of postnatal exposure to an adult male in C57BL/6J mice. Behav Brain Res 182(2):344–348. https://doi.org/10.1016/j.bbr.2007.03.032
Holmes A, Kinney JW, Wrenn CC, Li Q, Yang RJ, Ma L, Vishwanath J, Saavedra MC, Innerfield CE, Jacoby AS, Shine J, Iismaa TP, Crawley JN (2003) Galanin GAL-R1 receptor null mutant mice display increased anxiety-like behavior specific to the elevated plus-maze. Neuropsychopharmacology 28(6):1031–1044. https://doi.org/10.1038/sj.npp.1300164
Wu J, Zhao Z, Sabirzhanov B, Stoica BA, Kumar A, Luo T, Skovira J, Faden AI (2014) Spinal cord injury causes brain inflammation associated with cognitive and affective changes: role of cell cycle pathways. J Neurosci 34(33):10989–11006. https://doi.org/10.1523/JNEUROSCI.5110-13.2014
Adhikari A, Topiwala MA, Gordon JA (2011) Single units in the medial prefrontal cortex with anxiety-related firing patterns are preferentially influenced by ventral hippocampal activity. Neuron 71(5):898–910. https://doi.org/10.1016/j.neuron.2011.07.027
Gholami M, Saboory E, Khalkhali HR (2014) Chronic morphine and tramadol re-exposure induced an anti-anxiety effect in prepubertal rats exposed neonatally to the same drugs. Clin Exp Pharmacol Physiol 41(10):838–843. https://doi.org/10.1111/1440-1681.12274
Hogg S (1996) A review of the validity and variability of the elevated plus-maze as an animal model of anxiety. Pharmacol Biochem Behav 54(1):21–30. https://doi.org/10.1016/0091-3057(95)02126-4
Korte SM, De Boer SF (2003) A robust animal model of state anxiety: fear-potentiated behaviour in the elevated plus-maze. Eur J Pharmacol 463(1–3):163–175. https://doi.org/10.1016/s0014-2999(03)01279-2
Nakhjiri E, Saboory E, Roshan-Milani S, Rasmi Y, Khalafkhani D (2017) Effect of prenatal restraint stress and morphine co-administration on plasma vasopressin concentration and anxiety behaviors in adult rat offspring. Stress 20(2):205–211. https://doi.org/10.1080/10253890.2017.1306053
Rodgers RJ, Dalvi A (1997) Anxiety, defence and the elevated plus-maze. Neurosci Biobehav Rev 21(6):801–810. https://doi.org/10.1016/s0149-7634(96)00058-9
Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buee L (2006) Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol 169(2):599–616. https://doi.org/10.2353/ajpath.2006.060002
Baratz R, Rubovitch V, Frenk H, Pick CG (2010) The influence of alcohol on behavioral recovery after mTBI in mice. J Neurotrauma 27(3):555–563. https://doi.org/10.1089/neu.2009.0891
Sierksma AS, van den Hove DL, Pfau F, Philippens M, Bruno O, Fedele E, Ricciarelli R, Steinbusch HW, Vanmierlo T, Prickaerts J (2014) Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology 77:120–130. https://doi.org/10.1016/j.neuropharm.2013.09.015
Pedersen JT, Sigurdsson EM (2015) Tau immunotherapy for Alzheimer’s disease. Trends Mol Med 21(6):394–402. https://doi.org/10.1016/j.molmed.2015.03.003
Rosenmann H (2013) Immunotherapy for targeting tau pathology in Alzheimer’s disease and tauopathies. Curr Alzheimer Res 10(3):217–228. https://doi.org/10.2174/1567205011310030001
Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A (2016) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537(7618):50–56. https://doi.org/10.1038/nature19323
Brody DL, Holtzman DM (2008) Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci 31:175–193. https://doi.org/10.1146/annurev.neuro.31.060407.125529
Qiu C, Albayram O, Kondo A, Wang B, Kim N, Arai K, Tsai CY, Bassal MA, Herbert MK, Washida K, Angeli P, Kozono S, Stucky JE, Baxley S, Lin YM, Sun Y, Rotenberg A, Caldarone BJ, Bigio EH, Chen X, Tenen DG, Zeidel M, Lo EH, Zhou XZ, Lu KP (2021) Cis P-tau underlies vascular contribution to cognitive impairment and dementia and can be effectively targeted by immunotherapy in mice. Sci Transl Med 13 (596). https://doi.org/10.1126/scitranslmed.aaz7615
Shahpasand K, Sepehri Shamloo A, Nabavi SM, Ping LuK, Zhen Zhou X (2018) Tau immunotherapy: Hopes and hindrances. Hum Vaccin Immunother 14(2):277–284. https://doi.org/10.1080/21645515.2017.1393594
Cavdar S, Onat FY, Cakmak YO, Yananli HR, Gulcebi M, Aker R (2008) The pathways connecting the hippocampal formation, the thalamic reuniens nucleus and the thalamic reticular nucleus in the rat. J Anat 212(3):249–256. https://doi.org/10.1111/j.1469-7580.2008.00858.x
Nardone R, Holler Y, Brigo F, Seidl M, Christova M, Bergmann J, Golaszewski S, Trinka E (2013) Functional brain reorganization after spinal cord injury: systematic review of animal and human studies. Brain Res 1504:58–73. https://doi.org/10.1016/j.brainres.2012.12.034
Hulsebosch CE, Hains BC, Crown ED, Carlton SM (2009) Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev 60(1):202–213. https://doi.org/10.1016/j.brainresrev.2008.12.010
Zhao P, Waxman SG, Hains BC (2007) Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci 27(33):8893–8902. https://doi.org/10.1523/JNEUROSCI.2209-07.2007
Ankeny DP, Popovich PG (2009) Mechanisms and implications of adaptive immune responses after traumatic spinal cord injury. Neuroscience 158(3):1112–1121. https://doi.org/10.1016/j.neuroscience.2008.07.001
Segal MB (1993) Extracellular and cerebrospinal fluids. J Inherit Metab Dis 16(4):617–638. https://doi.org/10.1007/BF00711896
Zetterberg H (2017) Review: Tau in biofluids - relation to pathology, imaging and clinical features. Neuropathol Appl Neurobiol 43(3):194–199. https://doi.org/10.1111/nan.12378
Shultz SR, Wright DK, Zheng P, Stuchbery R, Liu SJ, Sashindranath M, Medcalf RL, Johnston LA, Hovens CM, Jones NC, O’Brien TJ (2015) Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 138(Pt 5):1297–1313. https://doi.org/10.1093/brain/awv053
Yokobori S, Zhang Z, Moghieb A, Mondello S, Gajavelli S, Dietrich WD, Bramlett H, Hayes RL, Wang M, Wang KK, Bullock MR (2015) Acute diagnostic biomarkers for spinal cord injury: review of the literature and preliminary research report. World Neurosurg 83(5):867–878. https://doi.org/10.1016/j.wneu.2013.03.012
Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35–43. https://doi.org/10.1016/j.expneurol.2012.01.013
Funding
The grant of this study was provided by Neurosciences Research Center of Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran; Council for Stem Cell Sciences and Technologies, Tehran, Iran (Grant number: 11/35721); and grant #1397-A-5443 from Royan Institute for Stem Cell Biology & Technology, Tehran, Iran.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Elnaz Nakhjiri expressed the idea of the article and performed the literature search. Material preparation, data collection and analysis were performed by Selva Zamanzadeh, Ehsan Ehsani, Hamid Soltani Zangbar, Shaqayeq Roqanian, Daryoush Mohammadnejad and Shahin Ahmadian. The first draft of the manuscript was written by Elnaz Nakhjiri, Koorosh Shahpasand and Parviz Shahabi. All authors edited the manuscript and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
This study was conducted following the Declaration of Helsinki’s principles. The Tabriz University of Medical Sciences Ethics Committee approved this study (Code of ethics: IR.TBZMED.VCR.REC.1398.067).
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nakhjiri, E., Roqanian, S., Zangbar, H.S. et al. Spinal Cord Injury Causes Prominent Tau Pathology Associated with Brain Post-Injury Sequela. Mol Neurobiol 59, 4197–4208 (2022). https://doi.org/10.1007/s12035-022-02843-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02843-1