
Abstract
Traumatic spinal cord injury (SCI) can result in substantial neurological impairment along with significant emotional and psychological distress. It is clear that there is profound neurodegeneration upon SCI, gradually spread to other spinal cord regions and brain areas. Despite extensive considerations, it remains uncertain how pathogenicity diffuses in the cord. It has been reported that tau protein abnormal hyperphosphorylation plays a central role in neurodegeneration triggered by traumatic brain injury (TBI). Tau is a microtubule-associated protein, heavily implicated in neurodegenerative diseases. Importantly, tau pathology spreads in a traumatic brain in a timely manner. In particular, we have recently demonstrated that phosphorylated tau at Thr231 exists in two distinct cis and trans conformations, in which that cis P-tau is extremely neurotoxic, has a prion nature, and spreads to various brain areas and cerebrospinal fluid (CSF) upon trauma. On the other hand, tau pathology, in particular hyperphosphorylation at Thr231, has been observed upon SCI. Taken these together, we conclude that cis pT231-tau may accumulate and spread in the spinal cord as well as CSF and diffuse tau pathology in the central nervous system (CNS). Moreover, antibody against cis P-tau can target intracellular cis P-tau and protect pathology spreading. Thus, considering cis P-tau as a driver of tau pathology and neurodegeneration upon SCI would open new windows toward understanding the disease development and early biomarkers. Furthermore, it would help us develop effective therapies for SCI patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Spinal cord injury (SCI) is a global disability, affecting several thousands of people [1,2,3,4]. The initial mechanical injury upon SCI can drive various pathogenic cascades, including inflammatory cell infiltration, microglia activation, and oxidative stress. In brief, the blood-brain barrier (BBB) leakage reflects an influx of inflammatory cells into the spinal cord. Lymphocytes release oxidative and proteolytic enzymes, reactive oxygen species (ROS), and pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) [5,6,7]. This acute pro-inflammatory response to damage can induce secondary damages in the neighboring tissues, resulting in neuronal dysfunction [5]. A reactive process of secondary damage was induced by the acute pro-inflammatory response to injury that surrounds the original injury site [5]. These secondary damage cascades may prolong for several weeks, reflecting an expanding matrix of proteins associated with neural cell apoptosis, such as soluble cluster of differentiation 95 (CD95) ligand (an initiator of the Fas apoptotic pathway) [8]. In addition, reactive astrocytes, responding to the damage at the traumatic region, secret several pro-inflammatory cytokines and neurotoxins that results in more neural cell death and tissue degeneration [9,10,11,12]. It mediates further glial scarring, mostly consist of reactive astrocytes and proteoglycans, resulting in a physical and chemical barrier against axonal regenerations [13,14,15].
The secondary responses are almost identical with traumatic brain injury (TBI) outcomes, such as cognitive decline and depression. However, the underlying mechanisms mediating SCI effects on the brain have remained elusive thus far. SCI can disorganize the cerebral cortex and thalamus circuits, likely through disrupted anterograde, and retrograde transports [16]. Previous research has reported that the thalamic nucleus sends some of its major cortical efferents to the hippocampus compartments [17]. Degenerated neurons upon SCI generate cysteine-cysteine chemokine ligand 21, released in distal spinal segments and the thalamus area, which trigger microglial activation [18, 19]. On the other hand, supratentorial alterations in particular brain areas are addressed to pain modulation upon SCI-related hyperesthesia [19,20,21,22,23]. It is clear that SCI-related hyperpathia results in sustained neuroinflammation in some brain areas regulating pain sensation, such as cortex, thalamus, and hippocampus [24]. Strikingly, there are reports demonstrating that there is no motor cortex dysfunction and neurodegeneration upon SCI [25,26,27]. However, there are remarkable neuropathological hallmarks triggered by SCI such as decreased number of cortical neurons [28,29,30,31,32,33].
Moreover, there is cognitive impairment in 60% of the SCI population. Also, individuals with SCI are 13 times more likely to have cognitive impairment than healthy subjects [34,35,36,37]. While this could be addressed to concomitant head injury [38], another possibility is encephalitis upon isolated SCI, observed in recent animal studies [39,40,41]. In addition, the BBB is disrupted for approximately 14 days upon either TBI or SCI [42,43,44], releasing the central nervous system (CNS) antigens into the systemic circulation.
Traumatic Brain Injury
Acute and chronic injuries are the two main categories of TBI. Acute brain injury contains mild TBI or concussion that may result in instant death. Chronic brain injury, named chronic traumatic encephalopathy (CTE), is considered a progressive degenerative disease results from repeated traumas on the brain and symptoms start years to decades after head trauma such as behavioral problems, and finally, it can lead to dementia [45]. TBI can be focal (a specific area of the brain) or diffuse (a more widespread area of the brain), in which focal injury is due to a severe direct impact on the brain upon severe TBI, and diffuse injury is due to tearing of the brain tissue upon mild TBI [45]. TBI drives a multifaceted cascade of neurochemical changes affecting brain function. The initiating event is stretching and disrupting of neuronal and axonal cell membranes [46], resulting in a deregulated flux of ions, including an efflux of potassium and influx of calcium, which accelerate glutamate release as an excitatory neurotransmitter. Glutamate binds to N-methyl-d-aspartate (NMDA) receptors and leads to calcium influx and neurons suppression with glucose hypometabolism [47, 48]. Increased glucose uptake followed by increased activity of membrane pumps causes calcium entry into mitochondria and oxidative metabolism impairment with lactate production, which leads to acidosis, and edema. Calcium influx triggers microtubule disassembly and some kinase activation [47, 48].
Calcium is uptake by the endoplasmic reticulum (ER) and mitochondria, which may result in increased ROS levels, significant structural damage, and dysfunction of organelles. Dysfunctional mitochondria release cytochrome C, which activates caspases, and throws out of calcium, which activates calpain [49,50,51]. Cytoskeletal proteins split by calpain and cause cytoskeletal degradation. Neuroinflammation, dysfunction of ER, and mitochondria, activation of ROS, caspase, and calpain, and cytoskeletal disruption result in tau pathology (Fig. 1). In return, pathologic tau contributes to neuroinflammation, ER/mitochondrial dysfunction, ROS generation, and cytoskeletal disruption [49,50,51,52].

TBI pathophysiology. TBI induces axonal injury, neuroinflammation, increases in intracellular Ca2+ and hyperphosphorylation of tau. Ca2+ uptake by the ER and mitochondria leads to organelle dysfunction and increases in ROS. Releasing of cytochrome c upon mitochondria dysfunction activates caspases and throws out of Ca2+ into the cytosol, which activates calpain. Cytoskeletal proteins (spectrin) splits by calpain. Neuroinflammation, ER/mitochondrial dysfunction, ROS, activation of caspase/calpain, and cytoskeletal disruption contribute to tau pathology. TBI: traumatic brain injury; ER: endoplasmic reticulum; ROS: reactive oxygen species
Tau protein is a family of microtubule-associated proteins (MAPs) predominantly located in CNS axons, where it maintains the stability of microtubules and participates in anterograde transport within axons [53, 54]. Hyperphosphorylation of tau (P-tau) has been demonstrated in several neurodegenerative disorders, together known as tauopathies, such as Alzheimer’s disease (AD), CTE [53,54,55,56,57], TBI [58,59,60,61,62,63,64], and SCI [65,66,67] in animals and humans, and it is clear that neurodegeneration upon CNS injury spreads to other regions of the brain and/or spinal cord [68]. TBI and SCI studies showed an increase in cerebrospinal fluid (CSF) and serum P-tau as a biomarker [69,70,71,72,73], but there are a few studies on changes in P-tau levels following different severities of SCI.
SCI Pathological Hallmarks
SCI causes a breach in the BBB; resulting in various protein secretion, such as neurofilaments (NF) [71], S100 calcium-binding protein β (S100β) [74], neuron-specific enolase (NSE) [74], and glial fibrillary acidic protein (GFAP) [71] and tau [75]. Tau is moderately phosphorylated in physiological conditions but its abnormal hyperphosphorylation reflects pathogenicity. The human brain is enriched with lower molecular weight isoforms (48–68 kDa), while the peripheral nervous system (PNS) and spinal cord are full of high molecular weight species (110–120 kDa) [76]. Tau phosphorylated at pathogenic residues such as Tau-1, AT8 [Ser202/Thr205], AT180 [Thr231/Ser235], AT100 [Thr212/Ser214], 12E8, and paired helical filament 1 (PHF1) [77]. It has been suggested that neuronal cell death primes to tangle formation, meaning that some earlier intermediates are responsible for tau toxicity [78]. Soluble and oligomeric forms of hyperphosphorylated tau (P-tau) are the most pathologic tau species during neurodegenerative diseases [79]. Such oligomers are secreted and transmitted into the cells, reflecting tau pathology propagation [78, 80, 81]. In addition, they can lead to synaptic and mitochondrial impairment linked to memory deficits [82, 83].
Tau Hyperphosphorylation Cascades
Several protein kinases involved in hyperphosphorylation of tau that are divided into two groups of proline and non-proline-directed kinases [84,85,86]. Among the tau kinases, cyclin-dependent kinase 5 (CDK5), and glycogen synthase kinase 3β (GSK3β) are important. They phosphorylate tau at serine/threonine residues, so they cause tau pathology [87, 88]. Tau hyperphosphorylation at Thr231 and Ser262 is associated with its loss of functions [89], highly reduces tau microtubule-binding ability [90, 91]. Protein phosphatase 1 (PP1), PP2A, PP2B, and PP5 dephosphorylate tau protein [92]. PP2A has the most potent effect on tau dephosphorylating [93]. Tau phosphorylation can modulate with cellular stress such as thermal stress [94, 95] or oxidative stress [94, 96, 97]. Oxidative stress is earlier stages in tauopathies [98] that increases the activity of tau kinases and decreases the activity of tau phosphatase, which leads to aggregation of tau protein [94, 96, 97, 99].
Tau Abnormalities Triggered by SCI
Tau pathology triggered by traumatic SCI could be divided into two processes: the initial mechanical injury, which results in the axonal degenerations, and subsequent secondary events including ischemia, oxidative stress, edema, inflammation, and loss of ionic homeostasis, resulting in extensive neurodegeneration over the course of minutes to weeks [15, 100, 101]. The secondary events, particularly ischemia, result in excessive calcium entry into cells through NMDA receptors [102,103,104,105]. Activation of calcium-dependent proteases (calpains) and kinases is a critical step [106]. Activation of calpain results in loss of membrane potential, increased membrane permeability, and cell death [107]. Also, increased intracellular calcium levels activate various kinases, such as CDK5, Ca2+/calmodulin-dependent protein kinase (CaMK), microtubule affinity-regulating kinase (MARK), cyclic AMP-dependent protein kinase (PKA), GSK3, and casein kinase 1 [77, 108, 109]. CDK5 can phosphorylate the tau protein, and p35, and p25 as tau endogenous activators regulate its activity [110]. Conversion of p35 to p25 by calpain causes prolonged activation and mislocalization of CDK5. Improper calpain activation mediates neuronal death. Also, the CDK5-p25 complex causes tau hyperphosphorylation and tau pathology [111].
Tau in Animal Models of SCI
There is profound tau hyperphosphorylation upon SCI in a rat model of thoracic spinal cord hemisection, which phosphorylated at pathogenic residues, Tau-1 [112], and AT8 [Ser202/Thr205] [66, 67]. Based on the Hung et al. report, P-tau amounts in rats with traumatic spinal cord tissue showed a decrease following treatment with a calpain inhibitor; likely through attenuating p25 formation [66]. It has been reported that insulin-like growth factor I (IGF-I) (Box 1) reduces tau hyperphosphorylation, suppresses microglia and astrocyte activation, and prevents neurodegeneration upon SCI [67], so it can be addressed to p25 downregulation.
Box 1 IGF-I is a strong neurotrophic factor promoting projection neurons development, dendritic arborization, and synaptogenesis [113, 114]. IGF-I can act in the brain in an endocrine, paracrine, or autocrine manner for prooting glucose use, by phosphatidylinositol 3–kinase/Akt and the downstream GSK-3β routes. IGF-I has a neuroprotective effect that is coordinated through activating Akt, inhibiting GSK-3β, and subsequently inhibiting tau phosphorylation [115, 116]. |
Total-tau was assessed in a study of 51 dogs following intervertebral disc herniation (IVDH), which shows that CSF tau levels increased based on the trauma severity [72]. Therefore, CSF tau is possibly an appropriate biomarker for SCI. Rat models of spinal cord injuries were developed by Qi et al. [117]; they were injected with mouse hippocampal neural stem cells (NSC) via the tail vein. The results indicated that increased tau levels in the outer chamber did not affect NSC migration, but it showed a reduction after alterations in intracellular tau phosphorylation state. Also, migration at the trauma site was targeted by NSC. Tau is not a chemokine for targeted migration of NSC, but intracellular tau phosphorylation/dephosphorylation inhibits cell migration [117]. P-tau as a biomarker has been studied by Caprelli et al. [65]. They reported that severe impact compression SCI at T8 indicated P-tau existence in injured axons with the same time-course and distribution model to β–amyloid precursor protein (β-APP), a biomarker for axonal damage. P-tau and β-APP positive axons extended no more than 5000 μm rostral and caudal toward the injury epicenter and were at their maximum 1 day after SCI. CSF concentrations of P-tau and total tau showed a significant increase 1 day after the injury, but only serum levels of P-tau were significantly increased in SCI rats than naive rats. Accordingly, CSF and serum P-tau are probably appropriate biomarkers for severe traumatic SCI. Furthermore, at 1-day post-injury, CSF analysis of P-tau and total tau in SCI rats showed a 7.5-fold and 550-fold increase in P-tau and total tau levels, respectively, while the serum analysis showed a 7-fold and 1.2-fold increase in P-tau and total tau levels, respectively [65]. This indicates that total tau protein is a more sensitive biomarker for axonal injury within CSF; however, P-tau appeared to be a more sensitive biomarker within the serum.
In another experimental study, rats were subcutaneously inoculated with saline or allogeneic spinal cord tissue on 1 or 2 occasions. Serum samples collected following the Morris water maze (MWM) test and animals were sacrificed after 8 weeks. Their findings indicate inflammation in the spinal cord and brain upon exposure to spinal cord antigens resulting in CNS cell degeneration, memory impairment, and tau production especially when this exposure is repeated [118]. There is one study of changes in P-tau levels upon different severities of SCI. Briefly, 160 female rats following a spinal cord trauma with dropping a 10-g rod upon the exposed spinal cord from different heights: 12.5 mm (mild SCI), 25 mm (moderate SCI), and 50 mm (severe SCI). Tau protein concentration was evaluated in serum and CSF specimens following surgery. There was a significant positive linear correlation between tau protein concentration and SCI severity in groups, as well as between the tau protein concentration and Basso, Beattie, and Bresnahan locomotor rating scale scores [119] (Table 1).
Tau in SCI Human
Pouw et al. measured tau concentrations in CSF obtained from 3 to 24 h after injury in motor-complete and motor-incomplete cases. Based on the results, tau levels were lower in the American Spinal Injury Association (ASIA) Impairment Scale (AIS) (Box 2) A patients that improved in their AIS scores than cases who remained AIS A. Interestingly, cases who remained AIS A during follow-up showed a higher tau level in AIS A than AIS A cases who neurologically converted to an AIS B [71]. Kwon et al. have reported that tau levels were significantly increased depending on the severity of injury in CSF sampled from complete or incomplete cases 24 h after injury [69, 70]. They plotted the tau level in the CSF between 8 and 120 h after SCI [69]. Surprisingly, the tau level remained higher in subjects from to 48 h following injury [69] (Table 3, Fig. 2).
Box 2 ASIA was established in 1973 for facilitating the exchange of investigations, information, and opinions among practitioners associated with the treatment of SCI cases. The Frankel scale had been introduced previously for categorizing spinal cord injuries, but it had several limitations [120]. In 1982, the ASIA published the International Standards for Neurological Classification of Spinal cord Injury (ISNCSCI), a scoring and categorizing model that would evolve into the present AIS (Table 2) [121]. The ISNCSCI classification is helpful in identifying crucial muscle groups and sensory points improving practitioners’ precision to identify the neurologic degree of injury. Moreover, it was reproducibly characterized by a comprehensive explanation about every sensory and motor grade, which leads to precise characterization of incomplete and complete spinal cord injuries [122]. Unfortunately, in the very acute stages after injury, it is often impossible to conduct the ISNCSCI examination due to associated comorbidities including TBI, pharmacologic sedation, intoxication, or multi-system trauma [123, 124]. In performing clinical research in acute SCI patients, because of this variability, large numbers of patients should be enrolled to obtain enough statistical power. |

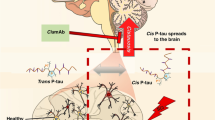
Tau in healthy neurons and in tauopathies after SCI. Tau facilitates microtubule stabilization within cells and is particularly abundant in neurons. Tau function is compromised in SCI and other tauopathies. This probably results from both tau hyperphosphorylation, which reduces the binding of tau to microtubules, and the sequestration of hyperphosphorylated tau into NFTs, which reduces the amount of tau that is available to bind microtubules. SCI: spinal cord injury; CSF: cerebrospinal fluid; NFTs: neurofibrillary tangles
Cistauosis: Novel Neurodegeneration Mechanism
It has been demonstrated some post phosphorylation modifications may lead to tau pathogenicity. In particular, phosphorylated tau at Thr-Pro motifs may exist in two distinct cis and trans conformations in which that cis P-tau is almost neurotoxic and prone to aggregation [125, 126]. Cis to trans isomerization is being conducted by peptidyl-prolyl cis/trans isomerase (Pin1). It is clear that Pin1 suppression reflects cis P-tau accumulation and tau pathology [127,128,129]. Moreover, it has been recently demonstrated that phosphorylated tau at Th231 in the cis conformation is extremely neurotoxic and early driver of neurodegeneration. Cis pT231-tau has a prion nature and spreads into brain areas and CSF. Cis P-tau levels increase as Alzheimer’s disease develops. Also, cis pT231-tau is being increased and spread upon TBI in a timely manner. Moreover, the more severe trauma, the more cis pT231-tau. Cis P-tau accumulates and makes aggregates inside the neurons and consequently, induces cell death upon either TBI, oxidative stress, or nutrition depletion; a process termed cistauosis [68]. In TBI, cis P-tau appears earlier than other P-tau epitopes such as PHF [68, 130, 131]. Furthermore, cis P-tau can cause and spread axonal degeneration in the acute and chronic stages following TBI. TBI induces cis P-tau in a dose-dependent fashion [68]. Although mild TBI is able to cause transient and modest cis P-tau induction, repetitive mild (rmTBI) or single severe TBI (ssTBI) causes ongoing and potent cis P-tau induction prior to tau oligomerization and tangle formation [68]. Cis P-tau is diffusely localized to axons, which leads to microtubule impairment and mitochondrial transport deficiency, apoptosis, and it expands within the brain that all subsequently cause and spread cistauosis phenotypes in the brain [68]. In addition, like in vitro studies, cistauosis induction is blocked after treatment with cis P-tau monoclonal antibody (mAb) following ssTBI in murine brains that completely suppresses the growth and expansion of TBI-oriented tau pathologic events, deposition, impaired long-term potentiation, and anxiety or risk-taking behaviors, and brain atrophy, whereas using cis mAb can improve cognitive impairment [68, 125, 126].
Albayram et al. studied cis P-tau in autopsy samples and indicated cis P-tau importance in developing and treatment of short-term and long-term outcomes of TBI, such as several CTE-like neurodegenerative features, like axonal, tau, and cerebellar pathology, neuroinflammation, neuronal loss, white matter degeneration, and also common clinical functional impairments, such as imbalanced sensorimotor coordination, urinary incontinence, and cognitive deficit. However, humans exposed to repetitive head trauma detected by CTE at autopsy showed robust cis P-tau in the cerebral cortex and deeper brain areas, such as the thalamus. Six months following injury, cis mAb has found to eliminate and block the spread of cis P-tau, pathology in axons, and astrogliosis into the hippocampus, as well as prevent other mechanisms of neuropathology, including tau oligomerization, tangle generation, gliosis, and APP assembly, and preventing sensorimotor coordination impairments and urinary incontinence [130] (Fig. 3). Therefore, cis P-tau is probably a precursor of tau pathology and a major driver of neurodegeneration.

A model for the roles of cis P-tau and its mAb in the development and treatment of TBI. Upon phosphorylation of tau, Pin1 converts cis to trans of P-tau. Cis P-tau mainly localizes to axons and causes and spreads axonal pathology, contributing to the development and progression of a range of neuropathological and functional outcomes. Treatment of TBI mice with cis mAb not only eliminates cis P-tau and blocks its spreading, but also prevents the development and progression of neuropathological and functional outcomes, but cistauosis after SCI is unknown. TBI: traumatic brain injury; Pin1: peptidyl-prolyl cis/trans isomerase; SCI: spinal cord injury
Conclusion
In recent years, aggregation intermediates, like soluble cis pT231-tau has been considered a possible option for the toxic species in frequent tauopathies [68, 125, 126, 130, 132]. Several studies have been conducted on tau aggregation intermediates making its prion-like nature apparent. Cases with TBI have high levels of CSF cis pT231-tau and toxic tau can seed and spread within connected areas of the brain and displays systematic and predictable models of diffusion [68, 130]. Using mouse monoclonal antibodies, which distinguishes trans from cis P-tau, can be an appropriate strategy to characterize and treat tau pathology. Cis pT231-tau removal suppressed neurodegeneration in TBI models [68].
On the other hand, clinical and animal research has found that serum and CSF P-tau is a feasible candidate biomarker for axonal injury upon SCI. These data indicate that serum and CSF levels of tau have a positive correlation with the severity of the injury and negative correlation with the locomotor function [65,66,67, 69,70,71,72]. Therefore, tau protein levels have been suggested as a novel biomarker for assessing SCI severity.
This review has shown that the identification of the prion-like behaviors of the tau protein is a critical step to discover novel and beneficial therapeutic methods for a large number of prevalent neurodegenerative disorders. Secondary injury cascades, as active pathological processes, seem to be a promising option for treating SCI, and future studies on cistauosis upon SCI are therefore recommended.
References
Krueger H, Noonan VK, Trenaman LM, Joshi P, Rivers CS (2013) The economic burden of traumatic spinal cord injury in Canada. Chronic Dis Inj Can 33(3):113–122
Noonan VK, Fingas M, Farry A, Baxter D, Singh A, Fehlings MG, Dvorak MF (2012) Incidence and prevalence of spinal cord injury in Canada: a national perspective. Neuroepidemiology 38(4):219–226. https://doi.org/10.1159/000336014
Priebe MM, Chiodo AE, Scelza WM, Kirshblum SC, Wuermser LA, Ho CH (2007) Spinal cord injury medicine. 6. Economic and societal issues in spinal cord injury. Arch Phys Med Rehabil 88(3 Suppl 1):S84–S88. https://doi.org/10.1016/j.apmr.2006.12.005
Singh A, Tetreault L, Kalsi-Ryan S, Nouri A, Fehlings MG (2014) Global prevalence and incidence of traumatic spinal cord injury. Clin Epidemiol 6:309–331. https://doi.org/10.2147/CLEP.S68889
Carlson SL, Parrish ME, Springer JE, Doty K, Dossett L (1998) Acute inflammatory response in spinal cord following impact injury. Exp Neurol 151(1):77–88. https://doi.org/10.1006/exnr.1998.6785
Trivedi A, Olivas AD, Noble-Haeusslein LJ (2006) Inflammation and spinal cord injury: infiltrating leukocytes as determinants of injury and repair processes. Clin Neurosci Res 6(5):283–292. https://doi.org/10.1016/j.cnr.2006.09.007
Biglari B, Swing T, Child C, Buchler A, Westhauser F, Bruckner T, Ferbert T, Jurgen Gerner H et al (2015) A pilot study on temporal changes in IL-1beta and TNF-alpha serum levels after spinal cord injury: the serum level of TNF-alpha in acute SCI patients as a possible marker for neurological remission. Spinal Cord 53(7):510–514. https://doi.org/10.1038/sc.2015.28
Janssen O, Qian J, Linkermann A, Kabelitz D (2003) CD95 ligand--death factor and costimulatory molecule? Cell Death Differ 10(11):1215–1225. https://doi.org/10.1038/sj.cdd.4401305
Beattie MS (2004) Inflammation and apoptosis: linked therapeutic targets in spinal cord injury. Trends Mol Med 10(12):580–583. https://doi.org/10.1016/j.molmed.2004.10.006
Davies SJ, Field PM, Raisman G (1996) Regeneration of cut adult axons fails even in the presence of continuous aligned glial pathways. Exp Neurol 142(2):203–216. https://doi.org/10.1006/exnr.1996.0192
Schmitt AB, Buss A, Breuer S, Brook GA, Pech K, Martin D, Schoenen J, Noth J et al (2000) Major histocompatibility complex class II expression by activated microglia caudal to lesions of descending tracts in the human spinal cord is not associated with a T cell response. Acta Neuropathol 100(5):528–536. https://doi.org/10.1007/s004010000221
Stichel CC, Muller HW (1998) The CNS lesion scar: new vistas on an old regeneration barrier. Cell Tissue Res 294(1):1–9. https://doi.org/10.1007/s004410051151
Bethea JR, Dietrich WD (2002) Targeting the host inflammatory response in traumatic spinal cord injury. Curr Opin Neurol 15(3):355–360
Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nat Rev Neurosci 5(2):146–156. https://doi.org/10.1038/nrn1326
Tator CH, Fehlings MG (1991) Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg 75(1):15–26. https://doi.org/10.3171/jns.1991.75.1.0015
Nardone R, Holler Y, Brigo F, Seidl M, Christova M, Bergmann J, Golaszewski S, Trinka E (2013) Functional brain reorganization after spinal cord injury: systematic review of animal and human studies. Brain Res 1504:58–73. https://doi.org/10.1016/j.brainres.2012.12.034
Cavdar S, Onat FY, Cakmak YO, Yananli HR, Gulcebi M, Aker R (2008) The pathways connecting the hippocampal formation, the thalamic reuniens nucleus and the thalamic reticular nucleus in the rat. J Anat 212(3):249–256. https://doi.org/10.1111/j.1469-7580.2008.00858.x
Hulsebosch CE, Hains BC, Crown ED, Carlton SM (2009) Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev 60(1):202–213. https://doi.org/10.1016/j.brainresrev.2008.12.010
Zhao P, Waxman SG, Hains BC (2007) Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci 27(33):8893–8902. https://doi.org/10.1523/JNEUROSCI.2209-07.2007
Hains BC, Saab CY, Waxman SG (2005) Changes in electrophysiological properties and sodium channel Nav1.3 expression in thalamic neurons after spinal cord injury. Brain 128(Pt 10):2359–2371. https://doi.org/10.1093/brain/awh623
Hubscher CH, Johnson RD (2006) Chronic spinal cord injury induced changes in the responses of thalamic neurons. Exp Neurol 197(1):177–188. https://doi.org/10.1016/j.expneurol.2005.09.007
Knerlich-Lukoschus F, Noack M, von der Ropp-Brenner B, Lucius R, Mehdorn HM, Held-Feindt J (2011) Spinal cord injuries induce changes in CB1 cannabinoid receptor and C-C chemokine expression in brain areas underlying circuitry of chronic pain conditions. J Neurotrauma 28(4):619–634. https://doi.org/10.1089/neu.2010.1652
Yoon EJ, Kim YK, Shin HI, Lee Y, Kim SE (2013) Cortical and white matter alterations in patients with neuropathic pain after spinal cord injury. Brain Res 1540:64–73. https://doi.org/10.1016/j.brainres.2013.10.007
Wu J, Raver C, Piao C, Keller A, Faden AI (2013) Cell cycle activation contributes to increased neuronal activity in the posterior thalamic nucleus and associated chronic hyperesthesia after rat spinal cord contusion. Neurotherapeutics 10(3):520–538. https://doi.org/10.1007/s13311-013-0198-1
Crawley AP, Jurkiewicz MT, Yim A, Heyn S, Verrier MC, Fehlings MG, Mikulis DJ (2004) Absence of localized grey matter volume changes in the motor cortex following spinal cord injury. Brain Res 1028(1):19–25. https://doi.org/10.1016/j.brainres.2004.08.060
Mason MR, Lieberman AR, Anderson PN (2003) Corticospinal neurons up-regulate a range of growth-associated genes following intracortical, but not spinal, axotomy. Eur J Neurosci 18(4):789–802. https://doi.org/10.1046/j.1460-9568.2003.02809.x
Nielson JL, Sears-Kraxberger I, Strong MK, Wong JK, Willenberg R, Steward O (2010) Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J Neurosci 30(34):11516–11528. https://doi.org/10.1523/JNEUROSCI.1433-10.2010
Bonatz H, Rohrig S, Mestres P, Meyer M, Giehl KM (2000) An axotomy model for the induction of death of rat and mouse corticospinal neurons in vivo. J Neurosci Methods 100(1–2):105–115. https://doi.org/10.1016/s0165-0270(00)00238-7
Feringa ER, Vahlsing HL (1985) Labeled corticospinal neurons one year after spinal cord transection. Neurosci Lett 58(3):283–286. https://doi.org/10.1016/0304-3940(85)90067-9
Giehl KM, Tetzlaff W (1996) BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur J Neurosci 8(6):1167–1175. https://doi.org/10.1111/j.1460-9568.1996.tb01284.x
Hains BC, Black JA, Waxman SG (2003) Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J Comp Neurol 462(3):328–341. https://doi.org/10.1002/cne.10733
Hammond EN, Tetzlaff W, Mestres P, Giehl KM (1999) BDNF, but not NT-3, promotes long-term survival of axotomized adult rat corticospinal neurons in vivo. Neuroreport 10(12):2671–2675. https://doi.org/10.1097/00001756-199908200-00043
Klapka N, Hermanns S, Straten G, Masanneck C, Duis S, Hamers FP, Muller D, Zuschratter W et al (2005) Suppression of fibrous scarring in spinal cord injury of rat promotes long-distance regeneration of corticospinal tract axons, rescue of primary motoneurons in somatosensory cortex and significant functional recovery. Eur J Neurosci 22(12):3047–3058. https://doi.org/10.1111/j.1460-9568.2005.04495.x
Cohen ML, Tulsky DS, Holdnack JA, Carlozzi NE, Wong A, Magasi S, Heaton RK, Heinemann AW (2017) Cognition among community-dwelling individuals with spinal cord injury. Rehabil Psychol 62(4):425–434. https://doi.org/10.1037/rep0000140
Collins-Praino LE, Corrigan F (2017) Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun 60:369–382. https://doi.org/10.1016/j.bbi.2016.09.027
Coulthart MB, Jansen GH, Cashman NR (2016) Evidence for transmissibility of Alzheimer disease pathology: Cause for concern? CMAJ 188(10):E210–E212. https://doi.org/10.1503/cmaj.151257
Craig A, Guest R, Tran Y, Middleton J (2017) Cognitive impairment and mood states after spinal cord injury. J Neurotrauma 34(6):1156–1163. https://doi.org/10.1089/neu.2016.4632
Dowler RN, Harrington DL, Haaland KY, Swanda RM, Fee F, Fiedler K (1997) Profiles of cognitive functioning in chronic spinal cord injury and the role of moderating variables. J Int Neuropsychol Soc 3(5):464–472
Jure I, Labombarda F (2017) Spinal cord injury drives chronic brain changes. Neural Regen Res 12(7):1044–1047. https://doi.org/10.4103/1673-5374.211177
Wu J, Stoica BA, Luo T, Sabirzhanov B, Zhao Z, Guanciale K, Nayar SK, Foss CA et al (2014) Isolated spinal cord contusion in rats induces chronic brain neuroinflammation, neurodegeneration, and cognitive impairment. Involvement of cell cycle activation. Cell Cycle 13(15):2446–2458. https://doi.org/10.4161/cc.29420
Wu J, Zhao Z, Sabirzhanov B, Stoica BA, Kumar A, Luo T, Skovira J, Faden AI (2014) Spinal cord injury causes brain inflammation associated with cognitive and affective changes: role of cell cycle pathways. J Neurosci 34(33):10989–11006. https://doi.org/10.1523/JNEUROSCI.5110-13.2014
Hayes KC, Hull TC, Delaney GA, Potter PJ, Sequeira KA, Campbell K, Popovich PG (2002) Elevated serum titers of proinflammatory cytokines and CNS autoantibodies in patients with chronic spinal cord injury. J Neurotrauma 19(6):753–761. https://doi.org/10.1089/08977150260139129
Zhang Z, Zoltewicz JS, Mondello S, Newsom KJ, Yang Z, Yang B, Kobeissy F, Guingab J et al (2014) Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PLoS One 9(3):e92698. https://doi.org/10.1371/journal.pone.0092698
Noble LJ, Wrathall JR (1989) Distribution and time course of protein extravasation in the rat spinal cord after contusive injury. Brain Res 482(1):57–66. https://doi.org/10.1016/0006-8993(89)90542-8
Blennow K, Hardy J, Zetterberg H (2012) The neuropathology and neurobiology of traumatic brain injury. Neuron 76(5):886–899. https://doi.org/10.1016/j.neuron.2012.11.021
Spain A, Daumas S, Lifshitz J, Rhodes J, Andrews PJ, Horsburgh K, Fowler JH (2010) Mild fluid percussion injury in mice produces evolving selective axonal pathology and cognitive deficits relevant to human brain injury. J Neurotrauma 27(8):1429–1438. https://doi.org/10.1089/neu.2010.1288
Barkhoudarian G, Hovda DA, Giza CC (2011) The molecular pathophysiology of concussive brain injury. Clin Sports Med 30(1):33–48, vii-iii. https://doi.org/10.1016/j.csm.2010.09.001
Giza CC, Hovda DA (2001) The neurometabolic cascade of concussion. J Athl Train 36(3):228–235
Galluzzi L, Blomgren K, Kroemer G (2009) Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 10(7):481–494. https://doi.org/10.1038/nrn2665
Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE (2005) Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res 79(1–2):231–239. https://doi.org/10.1002/jnr.20292
Wang KK (2000) Calpain and caspase: can you tell the difference? Trends Neurosci 23(1):20–26. https://doi.org/10.1016/s0166-2236(99)01479-4
Kampfl A, Posmantur RM, Zhao X, Schmutzhard E, Clifton GL, Hayes RL (1997) Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy: a review and update. J Neurotrauma 14(3):121–134. https://doi.org/10.1089/neu.1997.14.121
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A 91(12):5562–5566. https://doi.org/10.1073/pnas.91.12.5562
Gendron TF, Petrucelli L (2009) The role of tau in neurodegeneration. Mol Neurodegener 4:13. https://doi.org/10.1186/1750-1326-4-13
Geschwind DH (2003) Tau phosphorylation, tangles, and neurodegeneration: the chicken or the egg? Neuron 40(3):457–460. https://doi.org/10.1016/s0896-6273(03)00681-0
Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I (2009) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol 118(1):53–69. https://doi.org/10.1007/s00401-009-0486-3
McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS et al (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68(7):709–735. https://doi.org/10.1097/NEN.0b013e3181a9d503
Chen LJ, Wang YJ, Tseng GF (2010) Compression alters kinase and phosphatase activity and tau and MAP2 phosphorylation transiently while inducing the fast adaptive dendritic remodeling of underlying cortical neurons. J Neurotrauma 27(9):1657–1669. https://doi.org/10.1089/neu.2010.1308
Hoshino S, Tamaoka A, Takahashi M, Kobayashi S, Furukawa T, Oaki Y, Mori O, Matsuno S et al (1998) Emergence of immunoreactivities for phosphorylated tau and amyloid-beta protein in chronic stage of fluid percussion injury in rat brain. Neuroreport 9(8):1879–1883. https://doi.org/10.1097/00001756-199806010-00039
Lv Q, Lan W, Sun W, Ye R, Fan X, Ma M, Yin Q, Jiang Y et al (2014) Intranasal nerve growth factor attenuates tau phosphorylation in brain after traumatic brain injury in rats. J Neurol Sci 345(1–2):48–55. https://doi.org/10.1016/j.jns.2014.06.037
Shultz SR, Wright DK, Zheng P, Stuchbery R, Liu SJ, Sashindranath M, Medcalf RL, Johnston LA et al (2015) Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 138(Pt 5):1297–1313. https://doi.org/10.1093/brain/awv053
Tran HT, LaFerla FM, Holtzman DM, Brody DL (2011) Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci 31(26):9513–9525. https://doi.org/10.1523/JNEUROSCI.0858-11.2011
Tran HT, Sanchez L, Brody DL (2012) Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J Neuropathol Exp Neurol 71(2):116–129. https://doi.org/10.1097/NEN.0b013e3182456aed
Yang WJ, Chen W, Chen L, Guo YJ, Zeng JS, Li GY, Tong WS (2017) Involvement of tau phosphorylation in traumatic brain injury patients. Acta Neurol Scand 135(6):622–627. https://doi.org/10.1111/ane.12644
Caprelli MT, Mothe AJ, Tator CH (2018) Hyperphosphorylated tau as a novel biomarker for traumatic axonal injury in the spinal cord. J Neurotrauma 35(16):1929–1941. https://doi.org/10.1089/neu.2017.5495
Hung KS, Hwang SL, Liang CL, Chen YJ, Lee TH, Liu JK, Howng SL, Wang CH (2005) Calpain inhibitor inhibits p35-p25-Cdk5 activation, decreases tau hyperphosphorylation, and improves neurological function after spinal cord hemisection in rats. J Neuropathol Exp Neurol 64(1):15–26. https://doi.org/10.1093/jnen/64.1.15
Hung KS, Tsai SH, Lee TC, Lin JW, Chang CK, Chiu WT (2007) Gene transfer of insulin-like growth factor-I providing neuroprotection after spinal cord injury in rats. J Neurosurg Spine 6(1):35–46. https://doi.org/10.3171/spi.2007.6.1.35
Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM et al (2015) Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523(7561):431–436. https://doi.org/10.1038/nature14658
Kwon BK, Stammers AM, Belanger LM, Bernardo A, Chan D, Bishop CM, Slobogean GP, Zhang H et al (2010) Cerebrospinal fluid inflammatory cytokines and biomarkers of injury severity in acute human spinal cord injury. J Neurotrauma 27(4):669–682. https://doi.org/10.1089/neu.2009.1080
Kwon BK, Streijger F, Fallah N, Noonan VK, Belanger LM, Ritchie L, Paquette SJ, Ailon T et al (2017) Cerebrospinal fluid biomarkers to stratify injury severity and predict outcome in human traumatic spinal cord injury. J Neurotrauma 34(3):567–580. https://doi.org/10.1089/neu.2016.4435
Pouw MH, Kwon BK, Verbeek MM, Vos PE, van Kampen A, Fisher CG, Street J, Paquette SJ et al (2014) Structural biomarkers in the cerebrospinal fluid within 24 h after a traumatic spinal cord injury: a descriptive analysis of 16 subjects. Spinal Cord 52(6):428–433. https://doi.org/10.1038/sc.2014.26
Roerig A, Carlson R, Tipold A, Stein VM (2013) Cerebrospinal fluid tau protein as a biomarker for severity of spinal cord injury in dogs with intervertebral disc herniation. Vet J 197(2):253–258. https://doi.org/10.1016/j.tvjl.2013.02.005
Rubenstein R, Chang B, Yue JK, Chiu A, Winkler EA, Puccio AM, Diaz-Arrastia R, Yuh EL et al (2017) Comparing plasma phospho tau, total tau, and phospho tau-total tau ratio as acute and chronic traumatic brain injury biomarkers. JAMA Neurol 74(9):1063–1072. https://doi.org/10.1001/jamaneurol.2017.0655
Wolf H, Krall C, Pajenda G, Leitgeb J, Bukaty AJ, Hajdu S, Sarahrudi K (2014) Alterations of the biomarker S-100B and NSE in patients with acute vertebral spine fractures. Spine J 14(12):2918–2922. https://doi.org/10.1016/j.spinee.2014.04.027
Mori H, Hosoda K, Matsubara E, Nakamoto T, Furiya Y, Endoh R, Usami M, Shoji M et al (1995) Tau in cerebrospinal fluids: establishment of the sandwich ELISA with antibody specific to the repeat sequence in tau. Neurosci Lett 186(2–3):181–183. https://doi.org/10.1016/0304-3940(95)11291-4
Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M et al (1999) Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol 155(6):2153–2165. https://doi.org/10.1016/S0002-9440(10)65533-2
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17(1):5–21. https://doi.org/10.1038/nrn.2015.1
Gerson JE, Kayed R (2013) Formation and propagation of tau oligomeric seeds. Front Neurol 4:93. https://doi.org/10.3389/fneur.2013.00093
Gerson JE, Mudher A, Kayed R (2016) Potential mechanisms and implications for the formation of tau oligomeric strains. Crit Rev Biochem Mol Biol 51(6):482–496. https://doi.org/10.1080/10409238.2016.1226251
Castillo-Carranza DL, Nilson AN, Van Skike CE, Jahrling JB, Patel K, Garach P, Gerson JE, Sengupta U et al (2017) Cerebral microvascular accumulation of tau oligomers in Alzheimer’s disease and related tauopathies. Aging Dis 8(3):257–266. https://doi.org/10.14336/AD.2017.0112
Goedert M, Eisenberg DS, Crowther RA (2017) Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci 40:189–210. https://doi.org/10.1146/annurev-neuro-072116-031153
Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP et al (2011) Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol 179(4):2071–2082. https://doi.org/10.1016/j.ajpath.2011.07.004
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6:39. https://doi.org/10.1186/1750-1326-6-39
Fontaine SN, Sabbagh JJ, Baker J, Martinez-Licha CR, Darling A, Dickey CA (2015) Cellular factors modulating the mechanism of tau protein aggregation. Cell Mol Life Sci 72(10):1863–1879. https://doi.org/10.1007/s00018-015-1839-9
Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Terro F (2013) Tau protein phosphatases in Alzheimer’s disease: the leading role of PP2A. Ageing Res Rev 12(1):39–49. https://doi.org/10.1016/j.arr.2012.06.008
Morris M, Maeda S, Vossel K, Mucke L (2011) The many faces of tau. Neuron 70(3):410–426. https://doi.org/10.1016/j.neuron.2011.04.009
Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF (1999) Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol 58(9):1010–1019. https://doi.org/10.1097/00005072-199909000-00011
Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF (1998) Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res 797(2):267–277. https://doi.org/10.1016/s0006-8993(98)00296-0
Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 103(1):26–35. https://doi.org/10.1007/s004010100423
Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E (1993) Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11(1):153–163. https://doi.org/10.1016/0896-6273(93)90279-z
Hyman BT (2014) Tau propagation, different tau phenotypes, and prion-like properties of tau. Neuron 82(6):1189–1190. https://doi.org/10.1016/j.neuron.2014.06.004
Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22(8):1942–1950. https://doi.org/10.1111/j.1460-9568.2005.04391.x
Qian W, Shi J, Yin X, Iqbal K, Grundke-Iqbal I, Gong CX, Liu F (2010) PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J Alzheimers Dis 19(4):1221–1229. https://doi.org/10.3233/JAD-2010-1317
Galas MC, Dourlen P, Begard S, Ando K, Blum D, Hamdane M, Buee L (2006) The peptidylprolyl cis/trans-isomerase Pin1 modulates stress-induced dephosphorylation of Tau in neurons. Implication in a pathological mechanism related to Alzheimer disease. J Biol Chem 281(28):19296–19304. https://doi.org/10.1074/jbc.M601849200
Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K et al (2004) Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J Neurosci 24(10):2401–2411. https://doi.org/10.1523/JNEUROSCI.5561-03.2004
Davis DR, Anderton BH, Brion JP, Reynolds CH, Hanger DP (1997) Oxidative stress induces dephosphorylation of tau in rat brain primary neuronal cultures. J Neurochem 68(4):1590–1597. https://doi.org/10.1046/j.1471-4159.1997.68041590.x
Zambrano CA, Egana JT, Nunez MT, Maccioni RB, Gonzalez-Billault C (2004) Oxidative stress promotes tau dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Radic Biol Med 36(11):1393–1402. https://doi.org/10.1016/j.freeradbiomed.2004.03.007
Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova NN, Yang L, Starkov AA et al (2011) Behavioral deficit, oxidative stress, and mitochondrial dysfunction precede tau pathology in P301S transgenic mice. FASEB J 25(11):4063–4072. https://doi.org/10.1096/fj.11-186650
Su B, Wang X, Lee HG, Tabaton M, Perry G, Smith MA, Zhu X (2010) Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci Lett 468(3):267–271. https://doi.org/10.1016/j.neulet.2009.11.010
Taoka Y, Okajima K (1998) Spinal cord injury in the rat. Prog Neurobiol 56(3):341–358. https://doi.org/10.1016/s0301-0082(98)00049-5
Young W (1993) Secondary injury mechanisms in acute spinal cord injury. J Emerg Med 11(Suppl 1):13–22
Happel RD, Smith KP, Banik NL, Powers JM, Hogan EL, Balentine JD (1981) Ca2+-accumulation in experimental spinal cord trauma. Brain Res 211(2):476–479. https://doi.org/10.1016/0006-8993(81)90976-8
Liu D, Thangnipon W, McAdoo DJ (1991) Excitatory amino acids rise to toxic levels upon impact injury to the rat spinal cord. Brain Res 547(2):344–348. https://doi.org/10.1016/0006-8993(91)90984-4
Moriya T, Hassan AZ, Young W, Chesler M (1994) Dynamics of extracellular calcium activity following contusion of the rat spinal cord. J Neurotrauma 11(3):255–263. https://doi.org/10.1089/neu.1994.11.255
Young W (1992) Role of calcium in central nervous system injuries. J Neurotrauma 9(Suppl 1):S9–S25
Banik NL, Matzelle DC, Gantt-Wilford G, Osborne A, Hogan EL (1997) Increased calpain content and progressive degradation of neurofilament protein in spinal cord injury. Brain Res 752(1–2):301–306. https://doi.org/10.1016/s0006-8993(96)01488-6
Wingrave JM, Schaecher KE, Sribnick EA, Wilford GG, Ray SK, Hazen-Martin DJ, Hogan EL, Banik NL (2003) Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J Neurosci Res 73(1):95–104. https://doi.org/10.1002/jnr.10607
Brunden KR, Trojanowski JQ, Lee VM (2009) Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov 8(10):783–793. https://doi.org/10.1038/nrd2959
Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH (1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18(1):29–42. https://doi.org/10.1016/s0896-6273(01)80044-1
Maccioni RB, Otth C, Concha II, Munoz JP (2001) The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer’s pathology. Eur J Biochem 268(6):1518–1527. https://doi.org/10.1046/j.1432-1033.2001.02024.x
Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405(6784):360–364. https://doi.org/10.1038/35012636
Zhang SX, Underwood M, Landfield A, Huang FF, Gison S, Geddes JW (2000) Cytoskeletal disruption following contusion injury to the rat spinal cord. J Neuropathol Exp Neurol 59(4):287–296. https://doi.org/10.1093/jnen/59.4.287
Cheng CM, Mervis RF, Niu SL, Salem N Jr, Witters LA, Tseng V, Reinhardt R, Bondy CA (2003) Insulin-like growth factor 1 is essential for normal dendritic growth. J Neurosci Res 73(1):1–9. https://doi.org/10.1002/jnr.10634
Niblock MM, Brunso-Bechtold JK, Riddle DR (2000) Insulin-like growth factor I stimulates dendritic growth in primary somatosensory cortex. J Neurosci 20(11):4165–4176
Cheng CM, Reinhardt RR, Lee WH, Joncas G, Patel SC, Bondy CA (2000) Insulin-like growth factor 1 regulates developing brain glucose metabolism. Proc Natl Acad Sci U S A 97(18):10236–10241. https://doi.org/10.1073/pnas.170008497
Hong M, Lee VM (1997) Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem 272(31):19547–19553. https://doi.org/10.1074/jbc.272.31.19547
Qi ZP, Wang GX, Xia P, Hou TT, Zhou HL, Wang TJ, Yang XY (2016) Effects of microtubule-associated protein tau expression on neural stem cell migration after spinal cord injury. Neural Regen Res 11(2):332–337. https://doi.org/10.4103/1673-5374.177744
Schneider L, Reichert E, Faulkner J, Reichert B, Sonnen J, Hawryluk GWJ (2019) CNS inflammation and neurodegeneration: sequelae of peripheral inoculation with spinal cord tissue in rat. J Neurosurg 132:1–12. https://doi.org/10.3171/2018.10.JNS181517
Tang Y, Liu HL, Min LX, Yuan HS, Guo L, Han PB, Lu YX, Zhong JF et al (2019) Serum and cerebrospinal fluid tau protein level as biomarkers for evaluating acute spinal cord injury severity and motor function outcome. Neural Regen Res 14(5):896–902. https://doi.org/10.4103/1673-5374.249238
Frankel HL, Hancock DO, Hyslop G, Melzak J, Michaelis LS, Ungar GH, Vernon JD, Walsh JJ (1969) The value of postural reduction in the initial management of closed injuries of the spine with paraplegia and tetraplegia. I. Paraplegia 7(3):179–192. https://doi.org/10.1038/sc.1969.30
Kirshblum S, Waring W 3rd (2014) Updates for the international standards for neurological classification of spinal cord injury. Phys Med Rehabil Clin N Am 25(3):505–517, vii. https://doi.org/10.1016/j.pmr.2014.04.001
Marino RJ, Barros T, Biering-Sorensen F, Burns SP, Donovan WH, Graves DE, Haak M, Hudson LM et al (2003) International standards for neurological classification of spinal cord injury. J Spinal Cord Med 26(Suppl 1):S50–S56. https://doi.org/10.1080/10790268.2003.11754575
Burns AS, Lee BS, Ditunno JF Jr, Tessler A (2003) Patient selection for clinical trials: the reliability of the early spinal cord injury examination. J Neurotrauma 20(5):477–482. https://doi.org/10.1089/089771503765355540
Lee RS, Noonan VK, Batke J, Ghag A, Paquette SJ, Boyd MC, Fisher CG, Street J et al (2012) Feasibility of patient recruitment into clinical trials of experimental treatments for acute spinal cord injury. J Clin Neurosci 19(10):1338–1343. https://doi.org/10.1016/j.jocn.2012.02.015
Nakamura K, Zhen Zhou X, Ping Lu K (2013) Cis phosphorylated tau as the earliest detectable pathogenic conformation in Alzheimer disease, offering novel diagnostic and therapeutic strategies. Prion 7(2):117–120. https://doi.org/10.4161/pri.22849
Nakamura K, Zhou XZ, Lu KP (2013) Distinct functions of cis and trans phosphorylated tau in Alzheimer’s disease and their therapeutic implications. Curr Mol Med 13(7):1098–1109. https://doi.org/10.2174/1566524011313070001
Lu KP, Hanes SD, Hunter T (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380(6574):544–547. https://doi.org/10.1038/380544a0
Ranganathan R, Lu KP, Hunter T, Noel JP (1997) Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89(6):875–886. https://doi.org/10.1016/s0092-8674(00)80273-1
Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J et al (1997) Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278(5345):1957–1960. https://doi.org/10.1126/science.278.5345.1957
Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, Herbert MK, Qiu J et al (2017) Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 8(1):1000. https://doi.org/10.1038/s41467-017-01068-4
Farhadi A, Vosough M, Zhang JS, Tahamtani Y, Shahpasand K (2019) A possible neurodegeneration mechanism triggered by diabetes. Trends Endocrinol Metab 30(10):692–700. https://doi.org/10.1016/j.tem.2019.07.012
Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP (2012) Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 149(1):232–244. https://doi.org/10.1016/j.cell.2012.02.016
Funding
The grant of this study was provided by the Neurosciences Research Center of Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran (code of ethics: IR.TBZMED.VCR.REC.1398.067) and Council for Stem Cell Sciences and Technologies, Tehran, Iran (grant number: 11/35721).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Elnaz Nakhjiri expressed the idea of the article and performed the literature search. The first draft of the manuscript was written by Elnaz Nakhjiri, Koorosh Shahpasand, and Parviz Shahabi. All authors edited the manuscript and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nakhjiri, E., Vafaee, M.S., Hojjati, S.M.M. et al. Tau Pathology Triggered by Spinal Cord Injury Can Play a Critical Role in the Neurotrauma Development. Mol Neurobiol 57, 4845–4855 (2020). https://doi.org/10.1007/s12035-020-02061-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02061-7