
Abstract
Spinocerebellar ataxia type 7 (SCA7), a neurodegenerative disease characterized by cerebellar ataxia and retinal degeneration, is caused by a CAG repeat expansion in the ATXN7 gene coding region. Disease onset and progression are highly variable between patients, thus identification of specific/sensitive biomarkers that can improve the monitoring of disease progression is an immediate need. Because altered expression of circulating microRNAs (miRNAs) has been shown in various neurological diseases, they could be useful biomarkers for SCA7. In this study, we showed, to our knowledge for the first time, the expression profile of circulating miRNAs in SCA7. Using the TaqMan profiling low density array (TLDA), we found 71 differentially expressed miRNAs in the plasma of SCA7 patients, compared with healthy controls. The reliability of TLDA data was validated independently by quantitative real-time polymerase chain reaction in an independent cohort of patients and controls. We identified four validated miRNAs that possesses the diagnostic value to discriminate between healthy controls and patients (hsa-let-7a-5p, hsa-let7e-5p, hsa-miR-18a-5p, and hsa-miR-30b-5p). The target genes of these four miRNAs were significantly enriched in cellular processes that are relevant to central nervous system function, including Fas-mediated cell-death, heparansulfate biosynthesis, and soluble-N-ethylmaleimide-sensitive factor activating protein receptor pathways. Finally, we identify a signature of four miRNAs associated with disease severity that discriminate between early onset and adult onset, highlighting their potential utility to surveillance disease progression. In summary, circulating miRNAs might provide accessible biomarkers for disease stage and progression and help to identify novel cellular processes involved in SCA7.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxia type 7 (SCA7) is an inherited neurodegenerative disorder caused by the abnormal expansion of a cytosine-adenine-guanine (CAG) trinucleotide repeat located in the ATXN7 gene [1], which results in progressive loss of neurons within the cerebellum, brainstem, and retina [2,3,4], and consequently, in a plethora of clinical manifestations, including gait-ataxia, dysarthria, dysmetria, dysdiadochokinesia, hyperreflexia, postural tremor, sensory loss, and visual problems [2,3,4]. The polymorphic tract of CAG repeats ranges from 4 to 18 in normal chromosomes and from 36 to up to 460 repeats in expanded chromosomes [4,5,6]. SCA7 alleles present also a range of “intermediate alleles” that includes between 28 and 33 repeats; this range is not clinically significant, but is genetically unstable, whereas the presence of 34–36 CAG repeats may be provisionally defined as alleles with reduced penetrance [7]. Interestingly, the length of the CAG tract inversely correlates with age at onset, with repeats below 46 CAGs related with adult onset [4] and repeats above 70 CAGs related to infantile onset [7].
The ATXN7 gene product, named ataxin-7, harbors a polyglutamine (polyQ)-tract expansion when synthesized from the mutant allele, which confers on the protein an aberrant dominant activity that in turn alters multiple cellular processes, including transcriptional regulation, protein conformation and clearance, autophagy, and oxidative stress [6, 8,9,10,11]. One of the most studied mechanisms underlying SCA7 is transcriptional dysregulation of the SPT3-TAF9-acetyltransferase-GCN5 (STAGA) transcription coactivator complex; ataxin-7 is a core component of STAGA, while mutant ataxin-7 exerts a dominant-negative function by interfering with the nucleosomal histone acetylation activity of GCN5 [12, 13].
Carriers of mutant alleles display diverse clinical presentations that range from the asymptomatic stage to a very severe phenotype, due in part to the significant variability in age of onset and to the unpredictable progression of both cerebellar and non-cerebellar symptoms. This diversity of SCA7 stages is not fully explained by the length of the pathological CAG tract, and it is thought that gene modifiers and environmental factors might shape SCAs [14]. In this scenario, the identification of biomarkers that allow the monitoring of the natural history of the disease and that could serve for diagnosis and/or prognosis would be highly valuable.
Recently, the implication of miRNAs in the pathogenesis of different neurodegenerative disorders has been shown (Alzheimer disease [AD], Parkinson disease [PD], Huntington disease [HD], and some spinocerebellar ataxias), because their expression profile dynamically changes in these disorders and correlates with specific pathological stages [15,16,17,18,19,20,21,22,23,24]. MicroRNAs (miRNAs) are small noncoding RNAs (20–26 nucleotides in length) that regulate gene expression at the post-transcriptional level [25]. Interestingly, recent studies have demonstrated the presence of miRNAs in human body fluids, such as serum, plasma, and cerebrospinal fluid (CSF) [26,27,28,29,30,31], which opens the possibility of analyzing miRNAs in biofluids of SCA7 patients, as a proxy for inaccessible tissues. Identification of miRNAs with clinical potential might allow the timely diagnosis and surveillance of the disease progress.
In this study, we show, to our knowledge for the first time, the circulating miRNAs expression profiles in SCA7. We found altered circulating miRNAs in the plasma of SCA7 patients and identified some miRNAs that appear to be associated with specific disease phenotypes. In addition, we determined a set of four upregulated validated miRNAs with diagnostic value to differentiate healthy control from patients. Furthermore, we identified by computational analysis a second group of four miRNAs with potential prognostic value. Finally, functional enrichment analyses of the diagnostic miRNAs target genes identified several signaling pathways potentially involved in SCA7.
Materials and Methods
Subjects
The studied samples included 35 SCA7 patients and 17 healthy individuals (blood relatives and unrelated subjects), who were confirmed as non-carriers of the SCA7 mutation by molecular testing, recruited by the National Rehabilitation Institute (INR) and the Rehabilitation and Social Inclusion Center of Veracruz-DIF (CRIS-DIF) from the central region of Veracruz State, Mexico. Patients and controls have a similar range of age, nourishment, and lifestyle conditions. All subjects were volunteers and provided signed informed consent. Patients with secondary ataxias due to alcoholism, neoplasias, vascular pathology, malformations, autoimmune or inflammatory diseases, neuropharmacological treatment, and other non-genetic causes were excluded. Subjects with any of the following conditions were also excluded: infections, hepatic dysfunction, pregnancy, cancer, drug abuse, current therapy with immunosuppressive or anticonvulsant medicines or antioxidants, as well as nutritional deficiency or serum creatinine elevation.
All subjects were interviewed to obtain their clinical history and were clinically assessed with standard neurological exams following Mayo Clinic procedures: Motor control of limbs was evaluated to identify dysmetria and dysdiadochokinesia, while cerebellar signs were assessed to detect gait ataxia, tremor or dysarthria. Cranial nerve examination determined altered ocular movements, while extrapyramidal signs exploration identified involuntary movements. Muscle strength and pathological reflexes were also assessed [32]. Evaluation of ataxia-associated symptoms was determined using the Scale for the Assessment and Rating of Ataxia (SARA) [33], while exploration for extracerebellar features was carried out using the Inventory of Non-Ataxia Symptoms (INAS) [34]. SCA7 patients were categorized into two main phenotypes: adult onset (AO) and early onset (EO), based on the age at onset of the first referred symptoms (> or < of 20 years, respectively) [4]. In general, EO patients referred visual symptoms (decreased visual acuity or macular dysfunction) within the two first decades of their life, while AO referred cerebellar symptoms (moto alterations) as the first symptom until after the third decade of life. The study was approved by the INR Ethics/Research Committee.
Molecular Diagnostic of SCA7
Analysis of CAG repeats was performed by fluorescent PCR and capillary electrophoresis. Genomic DNA was extracted from peripheral blood leukocytes using the Gentra Puregene blood kit (Qiagen, Hilden, NRW, Germany). Fluorescent multiplex PCR was carried out on an Applied BioSystems Thermal Cycler using 60-ng DNA template and the chimeric primers described by Dorschner et al. [35]. PCR-amplified products were analyzed by capillary electrophoresis on the ABI PRISM 3730XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), as previously reported [36, 37].
Isolation of miRNAs from Plasma Samples
Five milliliters of peripheral blood from patients and healthy control subjects was collected into tubes containing ethylenediaminetetraacetic acid (EDTA) and processed for plasma isolation immediately after collection. Total RNA was isolated from 600 μl of plasma, using the miRNeasy Mini Kit (Cat. 217004) (Qiagen, Valencia, CA, USA), with modifications suggested by Kroh et al. [38] and following the manufacturer’s instructions. RNA was eluted in 30 μl of RNase-free water, and its concentration was determined on the NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, Inc., USA). RNA samples were stored at − 80 °C to further processing.
TaqMan Profiling Low Density Array
The miRNA expression profiles were obtained from the plasma samples of 18 SCA7 patients (nine AO and nine EO patients) and five healthy subjects (Table 1), using pre-printed Taqman profiling low density assay (TLDA) microfluidic cards (Human miR ver. 2.0; Applied Biosystems, Melbourne, Australia). This system is a 384-well PCR-based microfluidic card with embedded TaqMan primers and probes in each well for the 380-different mature human miRNA and four nucleolar RNA controls. Briefly, 3 μl out of 30 μl of total RNA, isolated from 600 μl of plasma, was reverse-transcribed into complementary DNA (cDNA) on the GeneAmp-PCR System 9700 (Applied Biosystems, Foster City, CA, USA), using the TaqMan microRNA reverse-transcription kit (Applied Biosystems, Carlsbad, CA, USA) and Megaplex RT primers (Human Pool-A; Applied Biosystems, Carlsbad, CA, USA), according to the manufacturer’s protocol. PCR conditions were as follows: 40 cycles at 16 °C for 30 min; at 42 °C for 30 min, followed by a final step at 80 °C for 5 min to inactivate the reverse transcriptase enzyme. Then, cDNA libraries were pre-amplified employing Megaplex PreAmp primer Human Pool-A and PreAmp Master Mix kits (Applied Biosystems, Carlsbad, CA, USA), according to the manufacturer’s instructions, and the amplified products were diluted in 5-μl Tris-EDTA for further real time (RT)-PCR analysis. RT-PCR reactions were carried out on the 7900 HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s protocol.
TLDA Bioinformatics Analysis
The Ct values (fractional-cycle-number at which the midpoint of logarithmic amplification was reached) of all plates were determined using an automatic baseline and a threshold of 0.2 on Data Assist software (Applied Biosystems, Carlsbad, CA, USA). miRNAs included in the analysis were filtered with a maximum Ct value fixed at 38. Furthermore, miRNAs that were not expressed in at least 80% of the samples or with more than 20% of undetermined probes were removed from the analysis. To define best internal reference miRNAs, a control stability (meanM) analysis was carried out, utilizing the geNorm algorithm (http://genomebiology.com/2002/3/7/ research/0034); miRNAs showing least variable expression across all of the samples tested were identified with the SLqPCR Library on the bioconductor platform (Supplemental Fig. 1). snRNAU6 (embedded control in TaqMan human microRNA arrays), and hsa-miR-483-5p miRNAs were employed as endogenous controls for normalization (Supplemental Fig. 1). Normalized relative expression levels of miRNAs were calculated by the comparative average threshold cycle (Ct) method (2−ΔΔCt) [39, 40], which subtracts the mean of the chosen controls from all other values in the miRNA set. Then, to determine the differential expression of normalized miRNAs, a moderated t test was implemented and Benjamini-Hochberg (BH) multiple adjusted test was performed to calculate adjusted p values with the HTqPCR-limma library implemented on R environment. Expression profiles were adjusted by the clinical characteristics age and sex in HTqPCR-limma analysis by removing continuous covariate effects. A miRNA with a p value < 0.05, adjusted p value < 0.05, and a fold change of > 1 were considered as differentially expressed. Clustering analysis using Pearson correlation and average linkage was used to define different subgroups, based on differentially expressed miRNAs. All the previous analyses were conducted using R-software (HTqPCR, limma, gplots) on the bioconductor platform.
TaqMan-Based miRNA Assay for Independent Validation of Individual miRNA
Validation of the expression of 20 selected miRNAs was performed on plasma samples from 17 SCA7 patients (seven AO and 10 EO patients) and 12 healthy control samples using TaqMan RT-qPCR assays (Applied Biosystems, Foster City, CA, USA). This set of miRNAs was chosen on the basis of both its significant fold change found in patients and its previous implication in any neurodegenerative disease, accordingly to miRNA functional annotations. Briefly, 1 μl out of 30 μl of total RNA, isolated from 400 μl of plasma, was retrotranscribed in 7.5 μl on the StepOne-Real Time PCR System (Applied Biosystems, Foster City, CA, USA) using specific miRNA primer and TaqMan small RNA assay probes (Applied Biosystems, Carlsbad, CA, USA). Pre-amplification of cDNA was conducted for 40 cycles at 16 °C for 2 min, at 42 °C for 1 min, and at 50 °C for 1 s, followed by a final step at 80 °C for 5 min for inactivation of the enzyme. Then, quantitative RT-PCR (qPCR) was carried out utilizing forward and reverse specific primers for each human miRNA (hsa-let-7a-5p, hsa-let-7e-5p, hsa-let-7g-5p, hsa-miR-10b-5p, hsa-miR-17-5p, hsa-miR-18a-5p, hsa-miR-19a-3p, hsa-miR-20a-5p, hsa-miR-26a-5p, hsa-miR-27a-3p, hsa-miR-29a-3p, hsa-miR-30b-5p, hsa-miR-132-3p, hsa-miR-148b-3p, hsa-miR-221-3p, hsa-miR-223-3p, hsa-miR-323-3p, hsa-miR-324-5p, hsa-miR-340-5p, and hsa-miR-375-3p) and two selected controls (snRNAU6 and has-miR-483-5p). These microRNAs were selected based on the differential expression between SCA7 patients and healthy controls and because of their statistical significance.
Statistical Analysis
Demographic and clinical characteristics of all subjects were presented as mean ± standard deviation (SD). Descriptive statistics were applied to determine differential individual characteristics between patients with SCA7 and control-group subjects by means of the Mann–Whitney U test for continuous variables and the chi-square (Χ2) or Fisher exact test (two-tailed) for categorical variables. The Stata ver. 10.0 statistical software package (StataCorp, College Station, TX, USA) was utilized for the calculations.
Pathway enrichment analysis was performed using: (a) Tool for Annotations of MicroRNAs (TAM; http://www.cuilab.cn/tam; version 2.0) for the set up regulated miRNAs in SCA7. Briefly, we looked for overrepresentation in family, cluster, disease, and pathway modules with the significant upregulated miRNAs found in each comparison. Through this analysis, we evaluated the statistical significance of each miRNA category and searched for enrichments with specific pathways and diseases already reported in the literature (not by gene target prediction). We evaluated the significance by a Fisher exact test p value of < 0.05, Bonferroni correction and FDR; (b) for overrepresentation analysis of the SCA7 diagnostic miRNAs signature, we used miEAA (https://ccb-compute2.cs.uni-saarland.de/ mieaa_tool/?id=www/www-ccb/html/mieaa_tool/) program. A Fisher exact test p value of < 0.05 was deemed statistically significant.
To evaluate a possible significant association of miRNA expression profiles and phenotype presence (disease), a descriptive method (univariate analysis) between these variables was computed on SAS University Edition (remote version via virtual-machine). The normalized expression values (2−ΔΔCt) were standardized by z score to define the pattern of relationships between miRNA expression distributions and the disease phenotype. The behavior was evaluated as discovery set (TLDAs analysis) and validation set (independent assay). A receiver operating characteristic (ROC) curve was performed to calculate the miRNA relationship between sensitivity and specificity, and these were plotted in R-environment. Computational analysis to identify miRNAs with potential prognostic value was carried out with varSelRF package, using the random forest gene selection method [41, 42]. The Pearson correlation coefficient was used to test for association between prognosis-related miRNAs expression and SARA and INAS. The significance level was set at p < 0.05. STATA 10.0 software (Stata Corporation, College Station, TX, USA) was used for calculations.
Results
Characteristics of Studied Subjects
Clinical characteristics of the discovery cohort as well as the demographic features of healthy control subjects are described in Table 1. A total of 35 SCA7 patients were categorized into two clinical phenotypes: EO and AO (see “Material and Methods”). EO patients exhibited a more severe form of the disease and a larger CAG repeat tract than AO subjects.
Differential Expression of Plasma miRNAs in SCA7 Patients
To determine differentially expressed circulating miRNAs in SCA7, plasma samples from 18 patients (nine EO and nine AO) and five healthy control subjects were analyzed using the TLDA, designed to detect 384 mature human miRNAs. We were able to measure 252 miRNAs, with an average threshold cycle (Ct) ≤ 38 under all experimental conditions. To optimally normalize miRNA expression, we employed both an endogenous control embedded in the TLDA system (snRNAU6) and a stable endogenous miRNA normalizer (hsa-miR-483-5p) (Supplemental Fig. 1). Furthermore, miRNA expression data were adjusted for age and gender using HTqPCR-limma analysis, to avoid potential biases caused by these covariates. Utilizing these stringent normalization criteria, 71 miRNAs were found significantly upregulated in SCA7 patients, as depicted in the heat map (Fig. 1a, b and Supplemental Table 1 [adjusted p values ≤ 0.05]). When EO or AO patients were compared with healthy control subjects separately, 47 and 60 upregulated miRNAs were found in the AO and EO groups, respectively (Fig. 1c–f and Supplemental Tables 2 and 3 [nominal p values ≤ 0.05]), implying that greater disease severity resulted in a higher number of deregulated miRNAs. Interestingly, 25 altered miRNAs were particularly found in EO patients, while only 12 miRNAs were specific for AO patients; the remaining 35 misregulated miRNAs were common to both phenotypes (Fig. 1g). Furthermore, the differentially expressed miRNAs were subjected to principal component analysis (PCA), showing separation of the different phenotype groups: SCA7 patients vs controls; AO vs controls; and EO vs controls, with a principal component (PC1, PC2, and PC3) accounting for 69.62, 68.85, and 65.49% of the variability, respectively (Supplemental Fig. 2).

Altered profile of plasma miRNAs in SCA7 patients. The TaqMan profiling low density array (TLDA) was utilized to analyze miRNA expression in plasma samples from SCA7 patients and healthy controls. Heat maps (left) and volcano plots (right) of differentially expressed miRNAs in SCA7 patients are shown: a and b SCA7 patients vs healthy controls. c and d Adult onset (AO) patients vs healthy controls. e and f Early Onset (EO) patients vs healthy controls. In the heat maps, columns correspond to the different subjects analyzed, whereas rows represent the differentially expressed miRNAs. Red color indicates high relative expression, and green denotes low relative expression. Volcano plot showing differentially expressed miRNAs in b SCA7 patients; d AO patients and f EO patients. miRNAs with expression fold change > 1 (p ≤ 0.05) were considered as upregulated. g Venn diagram showing overlapping and non-overlapping miRNAs between EO and AO patients
To confirm the accuracy of TLDA data, 19 differentially expressed miRNAs were selected for further validation by single qPCR assay in an independent cohort of 17 SCA7 patients (ten AO and seven EO patients) and 12 healthy control subjects. The altered expression of all the miRNAs tested was confirmed by this independent analysis (nominal p values ≤ 0.05, Supplemental Table 4). Supplemental Table 5 shows mirBase accesses and the sequence of all validated miRNAs.
Prediction of miRNA Target Genes and Enrichment Analysis
Single miRNA can post-transcriptionally regulate many target genes and ultimately act as a modulator for multiple cellular pathways. To obtain insight into the biological function of the mature miRNAs that were found deregulated in SCA7 patients, a gene ontology approach was applied using TAM (the Tool for Annotations of human miRNAs) and miEAA (the miRNA Enrichment Analysis and Annotation) software. We identified enrichment of the predicted miRNA targets involved in various biological categories (Bonferroni-corrected p < 0.01), including hormone regulation, adipocyte regulation, onco-miRNAs, human embryonic stem-cell regulation, angiogenesis, hematopoiesis, and cell division (Bonferroni-corrected p < 0.01, Table 2). Supplemental Table 6 depicts the miRNAs implicated in each biological category. Furthermore, overrepresentation of certain miRNA families (let-7 and mir-17) and expression of the hsa-mir-17 cluster were revealed (Table 2), implying that the modified expression of this set of miRNAs may have a common biological alteration during the establishment and progression of the disease. Also, this phenomenon pointed out the coordinate activity of these miRNAs on the regulation of common target genes or the modulation of specific pathways by the post-transcriptional regulation of different genes belonging to the same biological process. The enriched miRNA profile significantly associates with several pathological conditions, including neurodegenerative disorders, and such a miRNA signature was common to both AO and EO phenotypes (Supplemental Table 7).
Diagnostic Performance of Plasma miRNAs
The diagnostic power of validated miRNAs was evaluated by univariate logistic regression. We found five miRNAs (hsa-let-7a-5p, hsa-let7e-5p, hsa-miR-18a-5p, hsa-miR-30b-5p, and hsa-miR-132-3p) significantly associated with the disease. Except for hsa-miR-132-3p, the diagnostic value of this set of miRNAs was confirmed using random forest gene selection as a cross-validation approach (Supplemental Table 8). The prediction and the definition of final model describes in Fig. 2a–d. ROC curves showed that the average expression of this set of circulating miRNAs has indeed the power to discriminate SCA7 patients from healthy control subjects (area under the curve = 0.89 and 0.84 for discovery and validation sets, respectively) (Fig. 2e–f). Based on the random forest analysis and the univariate regression approach, we obtained a robust diagnostic miRNA-signature, which were confirmed in both discovery and validation cohorts. Interestingly, most of these predictive miRNAs appear to be involved in the same biological processes, including FAS signaling (p = 0.014), SNARE-vesicular transport (p = 0.010), and heparansulfate biosynthesis (p = 0.002) pathways, as exhibited by miEAA analysis (p values ≤ 0.05, Table 3).

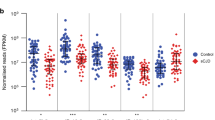
Signature of four miRNAs associated with SCA7 phenotype. a The best number of genes that lead to the smallest error rate in the class prediction model had been identified N = 4, as shown in the plot out-of-bag error rate vs. the number of genes. Original data (red line) and the 200 bootstrap samples (black lines). b Further, the probability of class membership of each sample analysis showed that most of the samples were optimal classified; indeed, the prediction error of the predicted model was 0.08821, while error rate at random was 0.22. c Mean expression of hsa-let-7a, hsa-let-7e, hsa-miR-18a, and hsa-miR-30b that showed clinically relevant to discriminate patients and controls samples in discovery set (TLDAs analysis) and d validation set (independent assay). e ROC analysis shows that the average expression of the diagnostic miRNA model accurately discriminates SCA patients from healthy controls in discovery set and f in validation set. Area under the curve (AUC) value is presented discovery set (TLDAs analysis) and validation set (independent assay)
Computational Analysis to Identify Putative miRNAs with Prognostic Value
Comparison between differentially expressed miRNAs of AO (n = 9) and EO (n = 9) patients revealed 17 upregulated miRNAs in the latter group (Supplemental Table 9). Owing to the greater severity of the EO phenotype, we next sought to identify miRNAs that may serve as prognostic markers, using a computational analysis based on random forest gene selection and univariate regression analyses. This computational model identified four prognosis-related miRNAs (hsa-miR-342-5p, hsa-miR-215-5p, hsa-miR-375-3p, and hsa-miR-365a-3p; Fig. 3a), which were found to be associated with disease severity by a univariate logistic regression (Supplemental Table 10). Interestingly, SCA7 patients were accurately assigned to the AO or EO groups with robust sensitivity using this predictive model (bootstrap estimator of prediction error: 0.22) (Fig. 3b). Furthermore, the mean expression of the selected miRNAs was greater in EO than AO patients (Fig. 3c), while ROC curves demonstrated the robust prognostic power of the prognosis-related signature of miRNAs, with an AUC of 0.84 (Fig. 3d). Finally, the expression of prognosis-related miRNAs was correlated with severity ataxia rating scales. Interestingly, most of the miRNAs showed significant correlation with SARA and INAS, including the gene module (composed by the mean expression levels of altered miRNAs) (Supplemental Table 11). It is worth to note that hsa-miR-375-3p expression significantly correlated with ataxia rating scales in the two cohorts of patients: SARA (discovery set, r = 0.39 and p = 0.052; validation set, r = 0.56 and p = 0.048) and INAS (discovery set, r = 0.32 and p = 0.049; validation set, r = 0.52 and p = 0.02) (Supplemental Table 11). However, no significant correlation was observed with the expression of this miRNA and disease evolution. Collectively, our data indicate that the prognostic miRNAs signature might be an effective predictor of SCA7 progression.

miRNAs associated with SCA7 severity. a Out-of-bag error rate vs the number of genes in the class prediction model. Original data (red line) and the 200 bootstrap samples (black lines). b Probability of class membership of each sample from SCA7 patients and healthy control group. c Mean expression of the four miRNAs associated with SCA7 severity (AO and EO) in the discovery cohort. d ROC analysis shows that the average expression of the prognostic miRNA-model accurately discriminates SCA patients from healthy controls
Discussion
Recent studies have provided solid evidence concerning the function of miRNAs in neurodegeneration [43], and lately, the analysis of circulating miRNAs in neurodegenerative diseases has gained attention [44]. However, except for HD and SCA3, studies aimed at clinically identifying relevant miRNAs in Poly-Q pathologies are lacking. In this study, we showed, to our knowledge for the first time, the expression profile of circulating miRNAs in SCA7 patients. By using TLDA analysis, we identified a set (N = 71) of differentially expressed miRNAs between SCA7 patients and healthy controls; further comparative analysis revealed that EO patients displayed a higher number of deregulated miRNAs than AO patients, implying that the most severe phenotype of the disease could be the result of a more extensive deregulation of miRNAs expression. Nineteen differentially expressed miRNAs were further validated by qPCR, supporting the reliability of TLDA data. To reduce the impact of population conformation, miRNA expression profiles were covariate adjusted by age and gender. Correcting miRNA expression allows us to remove potential confounding factors; thus, none of the reported deregulated miRNAs in patients with SCA7 are associated with dysregulation expression in accordance with age and/or gender [45, 46].
The analysis of the different stages of SCA7 is currently based on the evaluation of cerebellar and non-cerebellar features, using subjective clinical scales (SARA, BARS, and INAS) [4], which is time-consuming and requires extensive examiner training to avoid bias. Therefore, identification of corporal fluids disease-specific biomarkers, ideally at the asymptomatic stage, would improve the monitoring of both the natural history of the disease and patient progression in clinical trials. In this regard, we demonstrated that a signature of four miRNAs (hsa-let-7a-5p, hsa-let-7e-5p, hsa-miR-18a-5p, and hsa-miR-30b-5p) has the power to discriminate, with high sensitivity and specificity, between SCA7 patients and healthy control subjects. In addition, we identified many plasma miRNAs that are exclusively deregulated in AO (12) or EO (25) patients, suggesting that expression of these miRNAs correlates with the different presentations of disease and might be useful to discriminate even between EO and AO phenotypes. Although the miRNAs signature could play an important role in the pathogenesis of the disease, their efficacy to follow disease progression is unknown. In an effort to approach this, we performed a computational analysis on the differentially expressed miRNAs between AO and EO patients to identify prognosis-related miRNAs. We obtained a set of four miRNAs (hsa-miR-342-5p, hsa-miR-215-5p, hsa-miR-375-3p, and hsa-miR-365a-3p) with potential prognostic value. Remarkable, the expression of hsa-miR-375-3p positively correlated with ataxia clinical scales. It is worth to note that the homolog of this miRNA (dme-mir-375-3p) was found deregulated in a D. melanogaster model of SCA7 (ATXN7.102Q) [47], which highlights the importance of hsa-miR-375-3p in the mechanisms underlying SCA7. Unfortunately, the correlation with the evolution time of disease was not observed, suggesting that the sample size and the high variability in the evolution of the pathology among the patients were not enough to reach significant values. Therefore, the miRNA signature comprises a specific novel auxiliary marker for SCA7, because they are sampled in a relatively noninvasive manner and are readily detected by RT-qPCR assay, a technique widely utilized in clinical laboratories. It is worth to mention that determination of a miRNAs expression profile in SCA7 has no the pretension to replace classical PCR, which is currently the simplest inexpensive diagnostic method. Instead, identification of a miRNA signature with the power to discriminate between patients and healthy subjects, as described herein, is the first necessary step toward the wide range of applications of miRNAs, including identification of disease-stage miRNAs, the use of longitudinal studies to identification miRNAS with prognostic value, and eventually miRNAs that help to monitor patient’s response to therapeutic intervention for future clinical trials.
The functional link between deregulated miRNAs in the blood of patients and the neurodegenerative disease itself remains a controversial issue. It is thought that brain-derived miRNAs can pass through the blood–brain barrier (BBB) by exosome transport [48,49,50]; encapsulation of miRNAs in membrane-bound vesicles, such as exosomes, might protect them from the nuclease-rich environment present in biological fluids [51]. Alternatively, it has been proposed that the complexing of miRNAs with Ago1 and Ago2 proteins confers stability on plasma miRNAs [52,53,54,55]. In line with the idea that altered expression of circulating miRNAs in SCA7 might reflect the physiological state of distant organs, we carried enrichment analysis of the SCA7 miRNAs profile to identify mRNA targets and envisage unrecognized biological functions involved in SCA7. Such an analysis revealed 10 canonical biological processes, all of which have been previously implicated in neurodegenerative diseases [56,57,58], neurogenesis, neurodegeneration, and angiogenesis [59,60,61,62]. Particularly, we found four-miRNA signature (hsa-let-7a-5p, hsa-let7e-5p, hsa-miR-18a-5p, and hsa-miR-30b-5p) that discriminates between SCA7 patients and healthy controls. Such analysis suggests the participation of these miRNAs in various signaling pathways with relevance to central nervous system (CNS) function, including Fas-mediated cell-death, heparan sulfate (HS) biosynthesis, and soluble-N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) pathways. Fas-mediated cell death regulates the gradual loss of specific neurons [63, 64] and, in conjunction with other members of the tumor necrosis factor receptor (TNFR) family, indirectly modulates the inflammatory response and cell growth and proliferation [65], while HS, a main extracellular matrix (ECM) component, regulates cell homeostasis, acting as a growth-factor co-receptor to facilitate ligand-receptor bond [66,67,68]. With respect to the SNARE pathway, which mediates vesicle exocytosis during neuronal growth and neurotransmitter release for synaptic transmission [69], it was found that miR-153 adversely regulates the expression of SNAP-25 (the core component of the SNARE complex) in order to control motor neuron development and neurotransmission in zebrafish [70]. Remarkably, SNAP-25 dysregulation has been found in various CNS pathologies, including attention deficit hyperactivity disorder (ADHD), schizophrenia, bipolar I disorder, HD, and AD [71]. Overall, these evidence in the literature supports the hypothesis that altered expression of the circulating miRNAs observed herein in patients might be related to the neurodegenerative features of SCA7, including inflammation and the dysfunction and loss of neuronal cells. Clearly, further molecular experiments in cellular and animal models of SCA7 are necessary to sustain this idea. It has recently described that hsa-miR-124 mediates the post-transcriptional cross-talk between the long noncoding RNA lnc-SCA7-mediated negative feedback loop and ATXN7 mRNA in the cerebellum and retina of SCA7 mouse models [72, 73]. Suppression of STAGA activity by mutant ataxin-7 results in decreased expression of miR-124. We found no significant changes in miR-124 expression in the plasma samples of SCA7 patients, which suggests that miR-124 function is unrestricted to SCA7-specific organs; therefore, basal expression of miR-124 in other tissues [73,74,75] could be reflect some subtle changes in plasma.
A logical inquiry from our data is whether the SCA7 diagnostic and prognosis miRNAs signature is shared with other neurodegenerative diseases. Searching in published data sets allowed us to noted that hsa-miR-30b-5p expression was found to increase in peripheral blood of PD patients [76] and brain cortex of HD and SCA1 patients [15, 76], while hsa-let-7a-5p levels augmented in the cerebellum and brain cortex of SCA1 patients [77] and hsa-let-7e-5p in the CSF of AD patients [78]. Moreover, the expression of hsa-miR-215-5p and hsa-miR-375-3p was decreased in serum from SCA3 patients [20]. With respect to animal models of PolyQ diseases, the expression of mmu-miR-365-3p was found to increase in the cerebral cortex of N171-82Q HD mice, while that of mmu-miR-30b-5p augmented in the cerebellum of SCA1BO5 mice [18, 79]. Finally, the expression of mmu-miR-342-5p was found to augment in an immortalized striatal cell line derived from STHdhQ111/HdhQ111 mice [80]. Thus, with the exception of hsa-miR-18a-5p, which has not been involved in other polyQ diseases and appears to be specific for SCA7, the remaining SCA7-related miRNAs describe here could be involved in converging mechanisms of neurodegenerative diseases. Future studies in SCA7 mouse models are needed to support the utility of these miRNAs in preclinical studies and therapeutic intervention.
Although, our study deeply describes the circulating miRNA landscape in SCA7 condition, there were some limitations. First, cases and controls do not completely match at age and gender; nonetheless, we mitigated this imbalance by including age and gender as covariates, to filter miRNAs expression data. Another limitation is the sample size. The cohort of 35 patients was highly valuable, because of the rare incidence of SCA7 (< 1/100,000); however, further classification into EO and AO patients rendered short sample sizes. Therefore, although we identified distinctive miRNAs of each disease stage with significant nominal p values, EO and AO samples were not large enough to maintain significant adjusted p values. Future studies on larger sample sizes and longitudinal analysis are required to confirm the existence of disease stage-specific miRNAs. Finally, while enrichment analysis, based on statistical associations, pointed at some molecular mechanisms underlying SCA7, further functional studies are needed to support their possible association with the biology of the disease.
In summary, we described, to our knowledge for the first time, the expression profile of plasma miRNAs in SCA7 and identified a set of four miRNAs with potential diagnostic power. Furthermore, we identified a second set of four miRNAs with potential prognostic value, using a computational analysis. Our results will provide impetus for further evaluation of the clinical value of plasma miRNAs in SCA7 progression, therapeutic efficacy, and prognosis. Furthermore, identification of miRNA targets with potential biological significance can aid in the identification of novel cellular processes involved in SCA7.
References
David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G et al (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17(1):65–70. https://doi.org/10.1038/ng0997-65
David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, Belal S, Lebre AS et al (1998) Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum Mol Genet 7(2):165–170
Horton LC, Frosch MP, Vangel MG, Weigel-DiFranco C, Berson EL, Schmahmann JD (2013) Spinocerebellar ataxia type 7: clinical course, phenotype-genotype correlations, and neuropathology. Cerebellum 12(2):176–193. https://doi.org/10.1007/s12311-012-0412-4
Velazquez-Perez L, Cerecedo-Zapata CM, Hernandez-Hernandez O, Martinez-Cruz E, Tapia-Guerrero YS, Gonzalez-Pina R, Salas-Vargas J, Rodriguez-Labrada R et al (2015) A comprehensive clinical and genetic study of a large Mexican population with spinocerebellar ataxia type 7. Neurogenetics 16(1):11–21. https://doi.org/10.1007/s10048-014-0424-y
van de Warrenburg BP, Frenken CW, Ausems MG, Kleefstra T, Sinke RJ, Knoers NV, Kremer HP (2001) Striking anticipation in spinocerebellar ataxia type 7: the infantile phenotype. J Neurol 248(10):911–914
Garden GA, La Spada AR (2008) Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum 7(2):138–149. https://doi.org/10.1007/s12311-008-0027-y
Michalik A, Martin JJ, Van Broeckhoven C (2004) Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet 12(1):2–15. https://doi.org/10.1038/sj.ejhg.5201108
Chou AH, Chen CY, Chen SY, Chen WJ, Chen YL, Weng YS, Wang HL (2010) Polyglutamine-expanded ataxin-7 causes cerebellar dysfunction by inducing transcriptional dysregulation. Neurochem Int 56(2):329–339. https://doi.org/10.1016/j.neuint.2009.11.003
Alves S, Cormier-Dequaire F, Marinello M, Marais T, Muriel MP, Beaumatin F, Charbonnier-Beaupel F, Tahiri K et al (2014) The autophagy/lysosome pathway is impaired in SCA7 patients and SCA7 knock-in mice. Acta Neuropathol 128(5):705–722. https://doi.org/10.1007/s00401-014-1289-8
Ajayi A, Yu X, Lindberg S, Langel U, Strom AL (2012) Expanded ataxin-7 cause toxicity by inducing ROS production from NADPH oxidase complexes in a stable inducible spinocerebellar ataxia type 7 (SCA7) model. BMC Neurosci 13:86. https://doi.org/10.1186/1471-2202-13-86
Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T et al (2006) Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci 9(10):1302–1311. https://doi.org/10.1038/nn1750
Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, Miguet L, Potier N et al (2004) Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet 13(12):1257–1265. https://doi.org/10.1093/hmg/ddh139
Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y, Chait BT, La Spada AR et al (2005) Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U S A 102(24):8472–8477. https://doi.org/10.1073/pnas.0503505102
Tezenas du Montcel S, Durr A, Bauer P, Figueroa KP, Ichikawa Y, Brussino A, Forlani S, Rakowicz M et al (2014) Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain 137(Pt 9):2444–2455. https://doi.org/10.1093/brain/awu174
Marti E, Pantano L, Banez-Coronel M, Llorens F, Minones-Moyano E, Porta S, Sumoy L, Ferrer I et al (2010) A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res 38(20):7219–7235. https://doi.org/10.1093/nar/gkq575
Salta E, De Strooper B (2012) Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol 11(2):189–200. https://doi.org/10.1016/S1474-4422(11)70286-1
Roshan R, Ghosh T, Gadgil M, Pillai B (2012) Regulation of BACE1 by miR-29a/b in a cellular model of spinocerebellar ataxia 17. RNA Biol 9(6):891–899. https://doi.org/10.4161/rna.19876
Rodriguez-Lebron E, Liu G, Keiser M, Behlke MA, Davidson BL (2013) Altered Purkinje cell miRNA expression and SCA1 pathogenesis. Neurobiol Dis 54:456–463. https://doi.org/10.1016/j.nbd.2013.01.019
Muller S (2014) In silico analysis of regulatory networks underlines the role of miR-10b-5p and its target BDNF in Huntington’s disease. Transl Neurodegener 3:17. https://doi.org/10.1186/2047-9158-3-17
Shi Y, Huang F, Tang B, Li J, Wang J, Shen L, Xia K, Jiang H (2014) MicroRNA profiling in the serums of SCA3/MJD patients. Int J Neurosci 124(2):97–101. https://doi.org/10.3109/00207454.2013.827679
Huang F, Zhang L, Long Z, Chen Z, Hou X, Wang C, Peng H, Wang J et al (2014) miR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett 588(24):4791–4798. https://doi.org/10.1016/j.febslet.2014.11.013
Koscianska E, Krzyzosiak WJ (2014) Current understanding of the role of microRNAs in spinocerebellar ataxias. Cerebellum Ataxias 1:7. https://doi.org/10.1186/2053-8871-1-7
Hoss AG, Lagomarsino VN, Frank S, Hadzi TC, Myers RH, Latourelle JC (2015) Study of plasma-derived miRNAs mimic differences in Huntington’s disease brain. Mov Disord 30(14):1961–1964. https://doi.org/10.1002/mds.26457
Pan Y, Liu R, Terpstra E, Wang Y, Qiao F, Wang J, Tong Y, Pan B (2016) Dysregulation and diagnostic potential of microRNA in Alzheimer’s disease. Am J Alzheimers Dis 49(1):1–12. https://doi.org/10.3233/JAD-150451
Flynt AS, Lai EC (2008) Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet 9(11):831–842. https://doi.org/10.1038/nrg2455
Danborg PB, Simonsen AH, Waldemar G, Heegaard NH (2014) The potential of microRNAs as biofluid markers of neurodegenerative diseases—a systematic review. Biomarkers 19(4):259–268. https://doi.org/10.3109/1354750X.2014.904001
Muller M, Kuiperij HB, Versleijen AA, Chiasserini D, Farotti L, Baschieri F, Parnetti L, Struyfs H et al (2016) Validation of microRNAs in cerebrospinal fluid as biomarkers for different forms of dementia in a multicenter study. Am J Alzheimers Dis 52(4):1321–1333. https://doi.org/10.3233/JAD-160038
Sheinerman KS, Toledo JB, Tsivinsky VG, Irwin D, Grossman M, Weintraub D, Hurtig HI, Chen-Plotkin A et al (2017) Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimers Res Ther 9(1):89. https://doi.org/10.1186/s13195-017-0316-0
Lusardi TA, Phillips JI, Wiedrick JT, Harrington CA, Lind B, Lapidus JA, Quinn JF, Saugstad JA (2017) MicroRNAs in human cerebrospinal fluid as biomarkers for Alzheimer’s disease. Am J Alzheimers Dis 55(3):1223–1233. https://doi.org/10.3233/JAD-160835
Chen L, Yang J, Lu J, Cao S, Zhao Q, Yu Z (2018) Identification of aberrant circulating miRNAs in Parkinson’s disease plasma samples. Brain Behav 8(4):e00941. https://doi.org/10.1002/brb3.941
Zi Y, Yin Z, Xiao W, Liu X, Gao Z, Jiao L, Deng L (2015) Circulating MicroRNA as potential source for neurodegenerative diseases biomarkers. Mol Neurobiol 52(3):1494–1503. https://doi.org/10.1007/s12035-014-8944-x
Denny-Brown D, Dawson DM, Tyler HR (1982) Handbook of neurological examination and case recording, 3rd edn. Harvard University Press, Cambridge
Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, Giunti P, Globas C et al (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66(11):1717–1720. https://doi.org/10.1212/01.wnl.0000219042.60538.92
Schmitz-Hubsch T, Coudert M, Bauer P, Giunti P, Globas C, Baliko L, Filla A, Mariotti C et al (2008) Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology 71(13):982–989. https://doi.org/10.1212/01.wnl.0000325057.33666.72
Dorschner MO, Barden D, Stephens K (2002) Diagnosis of five spinocerebellar ataxia disorders by multiplex amplification and capillary electrophoresis. J Mol Diagn 4(2):108–113. https://doi.org/10.1016/S1525-1578(10)60689-7
Magana JJ, Tapia-Guerrero YS, Velazquez-Perez L, Cerecedo-Zapata CM, Maldonado-Rodriguez M, Jano-Ito JS, Leyva-Garcia N, Gonzalez-Pina R et al (2014) Analysis of CAG repeats in five SCA loci in Mexican population: epidemiological evidence of a SCA7 founder effect. Clin Genet 85(2):159–165. https://doi.org/10.1111/cge.12114
Magana JJ, Gomez R, Maldonado-Rodriguez M, Velazquez-Perez L, Tapia-Guerrero YS, Cortes H, Leyva-Garcia N, Hernandez-Hernandez O et al (2013) Origin of the spinocerebellar ataxia type 7 gene mutation in Mexican population. Cerebellum 12(6):902–905. https://doi.org/10.1007/s12311-013-0505-8
Kroh EM, Parkin RK, Mitchell PS, Tewari M (2010) Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR). Methods 50(4):298–301. https://doi.org/10.1016/j.ymeth.2010.01.032
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3(6):1101–1108
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Diaz-Uriarte R, Alvarez de Andres S (2006) Gene selection and classification of microarray data using random forest. BMC Bioinf 7:3. https://doi.org/10.1186/1471-2105-7-3
Diaz-Uriarte R (2007) GeneSrF and varSelRF: a web-based tool and R package for gene selection and classification using random forest. BMC Bioinf 8:328. https://doi.org/10.1186/1471-2105-8-328
Maciotta S, Meregalli M, Torrente Y (2013) The involvement of microRNAs in neurodegenerative diseases. Front Cell Neurosci 7:265. https://doi.org/10.3389/fncel.2013.00265
Pereira P, Queiroz JA, Figueiras A, Sousa F (2017) Current progress on microRNAs-based therapeutics in neurodegenerative diseases. Wiley Interdiscip Rev RNA 8(3). doi:https://doi.org/10.1002/wrna.1409
Ameling S, Kacprowski T, Chilukoti RK, Malsch C, Liebscher V, Suhre K, Pietzner M, Friedrich N et al (2015) Associations of circulating plasma microRNAs with age, body mass index and sex in a population-based study. BMC Med Genet 8:61. https://doi.org/10.1186/s12920-015-0136-7
Kumar S, Vijayan M, Bhatti JS, Reddy PH (2017) MicroRNAs as peripheral biomarkers in aging and age-related diseases. Prog Mol Biol Transl Sci 146:47–94. https://doi.org/10.1016/bs.pmbts.2016.12.013
Reinhardt A, Feuillette S, Cassar M, Callens C, Thomassin H, Birman S, Lecourtois M, Antoniewski C et al (2012) Lack of miRNA misregulation at early pathological stages in Drosophila neurodegenerative disease models. Front Genet 30(3):226. https://doi.org/10.3389/fgene.2012.00226. eCollection 2012.
Andras IE, Toborek M (2016) Extracellular vesicles of the blood-brain barrier. Tissue Barriers 4(1):e1131804. https://doi.org/10.1080/21688370.2015.1131804
Haqqani AS, Delaney CE, Tremblay TL, Sodja C, Sandhu JK, Stanimirovic DB (2013) Method for isolation and molecular characterization of extracellular microvesicles released from brain endothelial cells. Fluids Barriers CNS 10(1):4. https://doi.org/10.1186/2045-8118-10-4
Cichon C, Sabharwal H, Ruter C, Schmidt MA (2014) MicroRNAs regulate tight junction proteins and modulate epithelial/endothelial barrier functions. Tissue Barriers 2(4):e944446. https://doi.org/10.4161/21688362.2014.944446
Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Madrid F, Mittelbrunn M (2013) Analysis of microRNA and protein transfer by exosomes during an immune synapse. Methods Mol Biol 1024:41–51. https://doi.org/10.1007/978-1-62703-453-1_4
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF et al (2011) Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A 108(12):5003–5008. https://doi.org/10.1073/pnas.1019055108
Turchinovich A, Burwinkel B (2012) Distinct AGO1 and AGO2 associated miRNA profiles in human cells and blood plasma. RNA Biol 9(8):1066–1075. https://doi.org/10.4161/rna.21083
Wagner J, Riwanto M, Besler C, Knau A, Fichtlscherer S, Roxe T, Zeiher AM, Landmesser U et al (2013) Characterization of levels and cellular transfer of circulating lipoprotein-bound microRNAs. Arterioscler Thromb Vasc Biol 33(6):1392–1400. https://doi.org/10.1161/ATVBAHA.112.300741
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT (2011) MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 13(4):423–433. https://doi.org/10.1038/ncb2210
Eacker SM, Dawson TM, Dawson VL (2009) Understanding microRNAs in neurodegeneration. Nat Rev Neurosci 10(12):837–841. https://doi.org/10.1038/nrn2726
Su W, Aloi MS, Garden GA (2016) MicroRNAs mediating CNS inflammation: small regulators with powerful potential. Brain Behav Immun 52:1–8. https://doi.org/10.1016/j.bbi.2015.07.003
Procaccini C, Santopaolo M, Faicchia D, Colamatteo A, Formisano L, de Candia P, Galgani M, De Rosa V et al (2016) Role of metabolism in neurodegenerative disorders. Metab Clin Exp 65(9):1376–1390. https://doi.org/10.1016/j.metabol.2016.05.018
Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, Armstrong DL, Wu SM et al (2003) SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 37(3):383–401
Wang S, Tang Y, Cui H, Zhao X, Luo X, Pan W, Huang X, Shen N (2011) Let-7/miR-98 regulate Fas and Fas-mediated apoptosis. Genes Immun 12(2):149–154. https://doi.org/10.1038/gene.2010.53
Jin J, Kim SN, Liu X, Zhang H, Zhang C, Seo JS, Kim Y, Sun T (2016) miR-17-92 cluster regulates adult hippocampal neurogenesis, anxiety, and depression. Cell Rep 16(6):1653–1663. https://doi.org/10.1016/j.celrep.2016.06.101
Roush S, Slack FJ (2008) The let-7 family of microRNAs. Trends Cell Biol 18(10):505–516. https://doi.org/10.1016/j.tcb.2008.07.007
Su JH, Anderson AJ, Cribbs DH, Tu C, Tong L, Kesslack P, Cotman CW (2003) Fas and Fas ligand are associated with neuritic degeneration in the AD brain and participate in beta-amyloid-induced neuronal death. Neurobiol Dis 12(3):182–193
Choi C, Benveniste EN (2004) Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Res Rev 44(1):65–81
Kragh CL, Fillon G, Gysbers A, Hansen HD, Neumann M, Richter-Landsberg C, Haass C, Zalc B et al (2013) FAS-dependent cell death in alpha-synuclein transgenic oligodendrocyte models of multiple system atrophy. PLoS One 8(1):e55243. https://doi.org/10.1371/journal.pone.0055243
Mikami T, Kitagawa H (2016) Sulfated glycosaminoglycans: their distinct roles in stem cell biology. Glycoconj J 34:725–735. https://doi.org/10.1007/s10719-016-9732-9
Properzi F, Lin R, Kwok J, Naidu M, van Kuppevelt TH, Ten Dam GB, Camargo LM, Raha-Chowdhury R et al (2008) Heparan sulphate proteoglycans in glia and in the normal and injured CNS: expression of sulphotransferases and changes in sulphation. Eur J Neurosci 27(3):593–604. https://doi.org/10.1111/j.1460-9568.2008.06042.x
Maeda N (2015) Proteoglycans and neuronal migration in the cerebral cortex during development and disease. Front Neurosci 9:98. https://doi.org/10.3389/fnins.2015.00098
Hong W (2005) SNAREs and traffic. Biochim Biophys Acta 1744(3):493–517
Wei C, Thatcher EJ, Olena AF, Cha DJ, Perdigoto AL, Marshall AF, Carter BD, Broadie K et al (2013) miR-153 regulates SNAP-25, synaptic transmission, and neuronal development. PLoS One 8(2):e57080. https://doi.org/10.1371/journal.pone.0057080
Noor A, Zahid S (2017) A review of the role of synaptosomal-associated protein 25 (SNAP-25) in neurological disorders. Int J Neurosci 127(9):805–811. https://doi.org/10.1080/00207454.2016.1248240
Saraiva C, Esteves M, Bernardino L (2017) MicroRNA: basic concepts and implications for regeneration and repair of neurodegenerative diseases. Biochem Pharmacol 141:118–131. https://doi.org/10.1016/j.bcp.2017.07.008
Tan JY, Vance KW, Varela MA, Sirey T, Watson LM, Curtis HJ, Marinello M, Alves S et al (2015) Corrigendum: cross-talking noncoding RNAs contribute to cell-specific neurodegeneration in SCA7. Nat Struct Mol Biol 22(3):272. https://doi.org/10.1038/nsmb0315-272b
Lee Y, Kim HJ, Park CK, Kim YG, Lee HJ, Kim JY, Kim HH (2013) MicroRNA-124 regulates osteoclast differentiation. Bone 56(2):383–389. https://doi.org/10.1016/j.bone.2013.07.007
Kang K, Peng X, Zhang X, Wang Y, Zhang L, Gao L, Weng T, Zhang H et al (2013) MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J Biol Chem 288(35):25414–25427. https://doi.org/10.1074/jbc.M113.460287
Serafin A, Foco L, Zanigni S, Blankenburg H, Picard A, Zanon A, Giannini G, Pichler I et al (2015) Overexpression of blood microRNAs 103a, 30b, and 29a in L-dopa-treated patients with PD. Neurology 84(7):645–653. https://doi.org/10.1212/WNL.0000000000001258
Persengiev S, Kondova I, Otting N, Koeppen AH, Bontrop RE (2011) Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol Aging 32(12):2316 e2317–2316 e2327. https://doi.org/10.1016/j.neurobiolaging.2010.03.014
Derkow K, Rossling R, Schipke C, Kruger C, Bauer J, Fahling M, Stroux A, Schott E et al (2018) Distinct expression of the neurotoxic microRNA family let-7 in the cerebrospinal fluid of patients with Alzheimer’s disease. PLoS One 13(7):e0200602. https://doi.org/10.1371/journal.pone.0200602
Jin J, Cheng Y, Zhang Y, Wood W, Peng Q, Hutchison E, Mattson MP, Becker KG et al (2012) Interrogation of brain miRNA and mRNA expression profiles reveals a molecular regulatory network that is perturbed by mutant huntingtin. J Neurochem 123(4):477–490. https://doi.org/10.1111/j.1471-4159.2012.07925.x
Sinha M, Ghose J, Das E, Bhattarcharyya NP (2010) Altered microRNAs in STHdh(Q111)/Hdh(Q111) cells: miR-146a targets TBP. Biochem Biophys Res Commun 396(3):742–747. https://doi.org/10.1016/j.bbrc.2010.05.007
Acknowledgments
The authors thank Emilio Martínez and Nelson Pérez for their contribution to this study. Our paper is dedicated to the patients and the members of SCA-affected families.
Funding
This work was supported by the CONACyT grant (grant number 258043) to JJM. CV-V was a recipient of Doctoral fellowship under CONACyT grant (263396).
Author information
Authors and Affiliations
Contributions
The reported work was performed with collaboration between all co-authors:
Conceptual design: JJM and BC.
Experimental execution: VBC and CVV.
Data acquisition: VBC and CVV.
Data analysis: SRC, AHM, VBC, CVV, JJM, and BC.
Molecular diagnosis of patients: YTG.
Technical Support: YTG, SRC, CCZ, and OHH.
Management and clinical evaluation of patients: CCZ and OHH.
Manuscript preparation and approval: JJM, BC, VBC, and SRC.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Rights and permissions
About this article

Cite this article
Borgonio-Cuadra, V.M., Valdez-Vargas, C., Romero-Córdoba, S. et al. Wide Profiling of Circulating MicroRNAs in Spinocerebellar Ataxia Type 7. Mol Neurobiol 56, 6106–6120 (2019). https://doi.org/10.1007/s12035-019-1480-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-1480-y