
Abstract
Inflammatory cytokines are related to impaired learning and memory processes in the central nervous system, contributing to the cognitive dysfunction present in sepsis survivors. In sepsis, brain of survivors presented increased deposition of amyloid-beta (Aβ) peptide and this was associated with cognitive impairment. However, it is not known if the upregulation of secretase pathway is involved the deposition of Aβ peptide and consequent development of cognitive impairment in survivors. The aim of the study is to evaluate the effects of secretase inhibitors on behavioral, Aβ accumulation, and neuroinflammatory parameters in rats submitted to sepsis. Sepsis was induced by cecal ligation and perforation in Wistar rats, and the activity of alpha-, beta-, and gamma-secretases was determined in the hippocampus and prefrontal at different times. Additionally, in a different cohort of animal’s epigallocatechin gallate, a beta-secretase inhibitor or a gamma-secretase inhibitor was administrated once a day for three consecutive days. Fifteen or 30 days after sepsis induction, Aβ content, TNF-α, IL-1β, and IL-6 and cognitive performance were determined. There was no increase in alpha-secretase activity. Both beta- and gamma-secretase activities increased, mainly late after sepsis. The inhibition of beta- or gamma-secretases improved cognitive performance 10 days after sepsis induction, and beta-secretase inhibition improved cognitive performance up to 30 days after sepsis induction. Furthermore, beta-secretase inhibition decreased IL-1β and Aβ brain levels. It was demonstrated that during sepsis development there was an increase in the amyloidogenic route, and the inhibition of this pathway promoted attenuation of neuroinflammation, Aβ peptide content, and improvement of cognitive impairment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sepsis can be defined as the life-threatening organ dysfunction due to a dysregulated host response to infection [1]. Acute systemic inflammation can activate the innate immune system, launching a cascade of physiological changes ultimately affecting the central nervous system (CNS) [2]. The CNS is particularly vulnerable to damage, mediated by microglia activation [3, 4], in response to systemic inflammation [5]. The reactive microglia can cause neuronal dysfunction and damage through the release of inflammatory cytokines and reactive oxygen species [3, 6]. A role for pro-inflammatory cytokines in cognitive decline has been proposed in a variety of models for acute and chronic diseases [2, 6,7,8]. Inflammatory cytokines and the pro-inflammatory environment are related to impaired learning and memory processes in the CNS, contributing to the transient, longer lasting, and in some cases permanent cognitive dysfunction present in sepsis survivors [2, 7]. Some of these CNS alterations resemble the pathophysiological mechanisms of neurodegenerative diseases [9]. Alzheimer’s disease (AD) is characterized by extracellular amyloid-beta (Aβ) peptide deposition, which activates microglia, induces neuroinflammation, and drives neurodegeneration [10]. In sepsis induced by cecal ligation and puncture (CLP), brain of survivor animals presented increased deposition of Aβ peptide and this was associated with cognitive impairment, supporting the hypothesis that Aβ accumulation in brains of sepsis survivors is related with long-term cognitive dysfunction [11]. This can be more relevant in the hippocampus and prefrontal due to the vulnerability of these regions [12]. Additionally, it is known that the density of resident microglia varies in brain regions but predominates in gray matter of the hippocampus and prefrontal cortex [13]. However, it is not known if the upregulation of secretase-beta pathway is involved the deposition of Aβ peptide and consequent development of cognitive impairment in sepsis survivors.
In this context, the aim of this study was to evaluate the effects of secretase inhibitors on behavioral, Aβ accumulation, and neuroinflammatory parameters in rats submitted to sepsis.
Materials and Methods
Drugs
Epigallocatechin gallate (EGCG)-ECGC 4524 was purchased from Tocris Bioscience. EGCG was reported to inhibit Aβ fibrillization and redirect Aβ aggregation into unstructured, off-pathway oligomers [14].
β-secretase inhibitor I–7501 (H-Lys-Thr-Glu-Glu-Ile-Ser-Glu-Val-Asn-Stat-Val-Ala-Glu-Phe-OH) was purchased from BioVision.
γ-Secretase inhibitor–SCP0004-(N-benzyloxycarbonyl-Leu-Leu-norleucinal) was purchased from Sigma-Aldrich.
All drugs were purchased based in literature and datasheet information [14, 29, 39, 41, 42].
Ethics
The experimental procedures involving animals were performed in accordance with the National Institutes of Health (Bethesda, MD, USA) Guide for Care and Use of Laboratory Animals and with the approval of our institutional ethics committee. Protocol number: 041/2016-1
Animals
Adult male Wistar rats (220 to 300 g) were obtained from our breeding colony. They were housed five to a cage with food and water available ad libitum and were maintained on a 12-h light/dark cycle (lights on at 7 a.m.). Behavioral procedures were conducted between 8 a.m. and noon.
Sepsis Induction—Cecal Ligation and Perforation Model
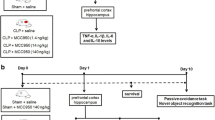
Rats were subjected to CLP as previously described by Fink and Heard (1990) [15]. Briefly, animals were anesthetized using a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg), given intraperitoneally. Under aseptic conditions, a 3-cm midline laparotomy was performed to expose the cecum and adjoining intestine. The cecum was ligated with a 3.0 silk suture at its base, below the ileocecal valve, and was perforated once with a 14-gauge needle. The cecum was then squeezed gently to extrude a small amount of feces through the perforation site. The cecum was then returned to the peritoneal cavity, and the laparotomy was closed with 4.0 silk sutures. Animals were resuscitated with regular saline (30 mL/kg) subcutaneously (s.c.) immediately after and 12 h after CLP. All animals received antibiotic (ceftriaxone at 30 mg/kg) every 6 h s.c. for a maximum of 3 days. In sham operated group, rats were subjected to all surgical procedures, but the cecum was neither ligated nor perforated. To minimize variability between different experiments, the CLP procedure was always performed by the same investigator. The mortality of this model is around 40% that is consistent with severe sepsis. We extensively characterized long-term cognitive impairment using this animal model [16,17,18].
Treatment with Secretase Inhibitors or Epigallocatechin Gallate
The animals were treated with a β-secretase inhibitor (1 μg/rat), a γ-secretase inhibitor (5 μg/rat), or EGCG (1 μg/rat) by intrathecal injection [19, 20]. These treatments were administrated once a day for three consecutive days (7, 8, and 9 days after sepsis induction). These time points were chosen based on kinetics studies demonstrating that secretase activities peaked at 10 days after sepsis. So, it was decided to inhibit secretase activities just before this time point.
Secretase Activity
After sepsis induction the activity of α, β, and γ-secretase was determined in the hippocampus and prefrontal cortex at different times (24 h, 72 h, 10 days, and 30 days).
Samples were homogenized in PBS buffer PH 7.0, centrifuged 5000 rpm for 3 min, and 100 μL of supernatant was used for each assay. The activity of α (cod FP001) and γ-secretase (cod. FP003) was determined by commercial fluorometric assay kit (R&D System), and the activity of β-secretase was determined by commercial fluorometric assay kit (ABCAM–cod. ab65357). All assay types were enzyme activity. Secretase activities were expressed by relative fluorescence units/mg protein. Five animals for each group were used for the determination of secretase activity.
Cytokine Levels
Samples were homogenized in PBS buffer PH 7.0, centrifuged 5000 rpm for 3 min, and 100 μL of supernatant was used for each assay. Concentrations of hippocampal TNF-α (cod. DY510), IL-1β (cod. DY501), and IL-6 (cod. DY506) were determined by ELISA on a microplate reader using a commercial kit (R&D System) 10 and 30 days after sepsis induction surgery. Five animals for each group were used for the determination of cytokine levels.
β-Amyloid Content
Aβ content was determined by Western blotting in samples from hippocampus and prefrontal cortex. Briefly, samples were homogenized in Laemmli-sample buffer (62.5 mM Tris–HCl, pH 6.8, 1 % (w/v) SDS, and 10 % (v/v) glycerol) and equal amounts of protein (30 μg/well) were fractionated by SDS-PAGE and electro-blotted onto nitrocellulose membranes. Protein loading and electro-blotting efficiency were verified through Ponceau S staining, and the membrane was blocked in Tween–Tris-buffered saline (TTBS; 100 mM Tris–HCl, pH 7.5, containing 0.9 % NaCl and 0.1 % Tween-20) containing 5 % albumin. Membranes were incubated overnight at 4 °C with primary antibody diluted at 1:1000 in TTBS (1:1000; anti-Aβ 68896 Abcam®-USA) and washed with TTBS. Anti-IgG from mouse or rabbit (according the species that originated the primary antibody) linked to a peroxidase was incubated with the membrane for additional 2 h at room temperature (1:5000 dilution range), the membrane was washed, and the immunoreactivity was detected by enhanced chemiluminescence using ECL Chemiluminescence kit. Densitometric analysis of the films was performed with ImageJ software. Blots were developed to be linear in the range used for densitometry.
Behavior Test
The animals were subjected to inhibitory avoidance test 10 and 30 days after sepsis induction. All behavioral tests were performed by the same person who was blind to the animal group. For each behavioral task, a total of 10 animals per group was used.
The inhibitory avoidance procedure was described in a previous report [15]. The apparatus was an acrylic box (50 × 25 × 25 cm) whose floor consisted of parallel-caliber stainless-steel bars (1-mm diameter) spaced 1 cm apart and a platform that was 7 cm wide and 2.5 cm high. Animals were placed on the platform, and their latency to step down on the grid with all four paws was measured with an automatic device. Training sessions were performed 10 or 30 days after surgery. Immediately after stepping down on the grid, animals received a foot shock of 0.3 mA for 2 s. In test sessions carried out 24 h after training, no foot shock was given and the step-down latency (maximum of 180 s) was used as a measure of memory retention.
Statistical Analysis
Data from secretase activity, Aβ-peptide, and inflammation were analyzed by one-way analysis of variance (ANOVA) followed by Tukey post hoc test and expressed as the mean ± standard deviation. Data from the inhibitory avoidance task was reported as median and interquartile ranges, and comparisons among groups were performed using Mann–Whitney U tests. The differences within individual groups (training and test) were analyzed by Wilcoxon tests. In all, comparisons p < 0.05 indicated statistical significance.
Results
Amiloidogenic Secretase Activity Is Increased Late After Sepsis
To evaluate the pathway involved in the enzymatic cleavage of amyloid precursor protein (APP) the activity of α, β, and γ-secretase was measured at 24 h, 72 h, 10 days, and 30 days after sepsis induction. There was no increase in the α-secretase activity in any of the brain structures in the evaluated times (Fig. 1a, b). In the prefrontal cortex, a significant increase was found for β-secretase 24 h, 10 days, and 30 days after sepsis (Fig. 1c), and for γ-secretase activity at 10 days and 30 days (Fig. 1e). In the hippocampus it was observed a significantly increase in both β and γ-secretase activity 10 and 30 days after sepsis (Fig. 1d, f), when compared with sham. These results support the hypothesis that systemic inflammation stimulates APP degradation by the amyloidogenic pathway.

Secretase activity in prefrontal cortex and hippocampus of rats submitted to sepsis. Sepsis was induced by cecal ligation and perforation (CLP), and the activity of alpha (a, b), beta (c, d), and gamma-secretases (e, f) was measured in the prefrontal cortex (a, c, e) and hippocampus (b, d, f) 24 h, 72 h, 10 days, and 30 days after surgery. n = 5. *Statistically different from sham, same time point
Secretase Inhibitor and Epigallocatechin Gallate Treatment Prevents Long-term Cognitive Impairment
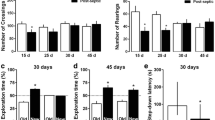
Since late after sepsis induction, when animals were completely free of infection and recovered from the acute phase (12-14), the enzymes related to the amyloidogenic pathway were upregulated it was assessed the effect of secretase inhibitors and EGCG on cognitive impairment associated with sepsis (10 days after CLP). Animals were treated for 3 days (7, 8, and 9 days after sepsis induction) just before the observed increase in secretase activity (at day 10). In the test section the animals treated with inhibitors or EGCG showed a significant increase in the latency time in relation to the training section, and this was not observed in the untreated CLP group (Fig. 2). The protective effect is more pronounced by the inhibition of β-secretase, since the latency time in the training test was significative higher when compared with both γ-secretase inhibitor and EGCG. In this context it was also determined the effect of β-secretase inhibition longer after sepsis induction. Animals received the three doses of the inhibitor (7, 8, and 9 days after induction of sepsis), and inhibitory avoidance test was performed 30 days after sepsis. In the test section there was a significant increase in latency time both in sham and treated group, but not CLP untreated animals demonstrating that inhibitor treatment attenuated cognitive impairment (Fig. 3). These results demonstrate that cognitive impairment can be minimized by secretase inhibitors.

Effect of secretase inhibitors or epigallocatechin gallate in long-term cognitive impairment in submitted to sepsis. Sepsis was induced by cecal ligation and perforation (CLP), and animals were treated on the 7th, 8th, and 9th day after induction with gamma-secretase inhibitor, beta-secretase inhibitor, or epigallocatechin gallate (EGCG). Ten days after sepsis induction animals were submitted to the inhibitory avoidance test. Data are presented as median ± interquartile range of 8–12 animals per group *Different from the training section. &Different from CLP (test section); #Different from CLP+EGCG and from CLP+ gamma-secretase inhibitor (test section)

Effect of beta-secretase inhibitor in long-term cognitive impairment in submitted to sepsis. Sepsis was induced by cecal ligation and perforation (CLP), and animals were treated on the 7th, 8th, and 9th day after induction with beta-secretase inhibitor. Thirty days after sepsis, induction animals were submitted to the inhibitory avoidance test. Data are presented as median ± interquartile range of 8–12 animals per group. *Different from the training section. &Different from CLP (test section)
Secretase Inhibitors and Epigallocatechin Gallate Treatment Reduced Brain Proinflammatory Cytokine Levels
To investigate the protective effects of secretase inhibitors and EGCG in brain inflammation, it was measured the levels of TNF-α, IL-1β, and IL-6 in the hippocampus and prefrontal cortex, 10 (Fig. 4) and 30 (Fig. 5) days after sepsis induction.

Effect of secretase inhibitors or epigallocatechin gallate in the levels of cytokines in the brain of rats submitted to sepsis. Sepsis was induced by cecal ligation and perforation (CLP), and animals were treated on the 7th, 8th, and 9th day after induction with gamma-secretase inhibitor, beta-secretase inhibitor, or epigallocatechin gallate (EGCG). Eleven days after TNF-α (a, b), IL-1β (c, d), and IL-6 (e, f) were measured in pre-frontal cortex (a, c, e) and hippocampus (b, d, f). Data were expressed as pg/mL and expressed as mean ± SD, n = 5 each group, measures were performed in duplicate. *Different from sham. #Different from CLP

Effect of beta-secretase inhibitor in the levels of cytokines in the brain of rats submitted to sepsis. Sepsis was induced by cecal ligation and perforation (CLP), and animals were treated on the 7th, 8th, and 9th day after induction with a beta-secretase inhibitor. Thirty days after IL-1β (a, b), IL-6 (c, d), and TNF-α (e, f) were measured in pre-frontal cortex (a, c, e) and hippocampus (b, d, f). Data were expressed as pg/mL and expressed as mean ± SD, n = 5 each group, measures were performed in duplicate. *Different from CLP
Ten days after sepsis TNF-α levels did not have any significant variation between groups in the evaluated brain structures (Fig. 4a, b). The levels of IL-1β were significantly increased in CLP when compared with the sham group both in the hippocampus and prefrontal cortex 10 days after sepsis (Fig. 4c, d). In the prefrontal cortex β and γ-secretase inhibitors significantly reduced IL-1β levels when compared with the CLP group (Fig. 4c). In the hippocampus, there was an increase of IL-1β after CLP, and this was not observed in animals treated with secretase inhibitors or EGCG (Fig. 4d); however, there were no significant differences between CLP untreated and treated animals. This same pattern was observed to IL-6 levels in both structures (Fig. 4e, f).
Brain cytokines were also measured 30 days after sepsis induction in animals treated with a β-secretase inhibitor (Fig. 5). Treatment with β-secretase inhibitor promoted a significant reduction of IL-1β (Fig. 5a, b), but not IL-6 (Fig. 5c, d) or TNF-α levels (Fig. 5e, f).
β-Secretase Inhibitor Reduces Aβ Levels in Brain Structures Late After Sepsis Induction
It has been previously shown that animals subjected to sepsis had increased Aβ peptide expression in brain structures [9, 11, 21] from 15 to 30 days, but not 10 days, after sepsis. Animals treated with beta-secretase inhibitor had lower levels of Aβ peptide only in the hippocampus at 15 days (Fig. 6b). There was a trend (p = 0.09) to a reduction of Aβ peptide levels in prefrontal cortex 30 days after sepsis induction (Fig. 6a).

Effect of beta-secretase inhibitor in the levels of Aβ peptide in the brain of rats submitted to sepsis. Sepsis was induced by cecal ligation and perforation (CLP), and animals were treated on the 7th, 8th, and 9th day after induction with a beta-secretase inhibitor. Fifteen and 30 days after Aβ peptide levels were measured in pre-frontal cortex (a) and hippocampus (b). Data were expressed as mean ± SD, n = 5 each group
Discussion
The findings of the present study suggest that inhibition of secretases of the amyloidogenic pathway prior to formation of the Aβ peptide during sepsis may lead to reduced brain cytokines and Aβ peptide levels and attenuation of long-term cognitive deficits in sepsis-surviving animals.
Our group demonstrated previously that during sepsis there was an increase Aβ peptide deposition in the prefrontal cortex and hippocampus 30 days after sepsis induction and this was associated with cognitive impairment in sepsis survivor animals [9]. Release of neurotoxic forms Aβ peptide results from the proteolytic processing from a large type I trans-membrane protein, the APP, by sequential cleavage of precursor by β and γ-secretase, whereas α-secretase prevents its generation by cleaving within the middle of the amyloid domain [22]. In our study, it was observed an increase in β and γ-secretase activity in prefrontal cortex and hippocampus 10 and 30 days after induction of sepsis, demonstrating activation of the amyloidogenic degradation pathway of the APP late after sepsis development, which results in generation of Aβ peptide showed in hippocampus and prefrontal cortex of survivor’s animals.
The Aβ peptide is produced naturally in small amounts throughout life, being found in the brain and CSF of healthy individuals [23, 24], but over-production or alterations of expression or processing of APP [25] under pathological conditions can lead to aggregation, forming Aβ oligomers that induce neuronal damage, including plasticity and synaptic structures and activation of caspases [25], related to cognitive and behavioral dysfunction. Critical illnesses, including sepsis, are related to significant cognitive impairment months to years after intensive care unit discharge, including alterations in memory, attention, concentration, and/or global loss of cognitive function [26,27,28,29]. We supposed that such dysfunctions may be in some way associated with the deposition of the Aβ peptide in the brain; thus, it was hypothesized that inhibiting the enzymes involved in its release could reverse the damage. β-secretase enzyme has been considered an important therapeutic target, presenting advantages over other anti-amyloid strategies, since its inhibition does not only reduce Aβ peptide levels but also prevent the generation of other APP metabolites as carboxy-terminal fragments (APP- βCTF) and soluble derivatives (sAPPβ) [22] associated with high neurotoxicity [30,31,32]. In AD and other neurodegenerative disorders, small oligomers of Aβ peptide, pre-fibrillar diffusible assemblies are now considered primary neurotoxic species [33]. Jiang & cols (2010) findings that APP-βCTF increased are linked by endocytic dysfunction and cholinergic neurodegeneration in Down syndrome and AD [34]. Amyloid oligomers are prone to form pore-like assemblies in the plasma membrane of brain cells; these annular protofibrils, formed by Aβ peptide, were recognized as a new type of “amyloid” assembly and referred to as “amyloid pores” [21]. Joshi et al (2013) demonstrated by in vitro study that reactive microglial cells release microvesicles that can promote formation of soluble Aβ species from extracellular insoluble aggregates [10], and this soluble forms are most toxic forms of Aβ, being associated with neuronal injury and loss of synapses [21, 22]. Possibly, this mechanism leading to cell dysfunction is also involved in the neurodegeneration and cognitive dysfunction observed in septic patients. In this context, we had previously demonstrated that the blockade of Aβ-RAGE pathway improved long-term cognitive deficits observed in septic survivors’ animals [21].
The results using inhibitors of amyloidogenic pathway enzymes demonstrated a benefit in cognitive dysfunction evaluated by the inhibitory avoidance test when compared with the group that did not receive treatment. Huang and colleagues (2014) using a γ-secretase inhibitor in septic rats also demonstrated improvement of cognitive impairment, as well as reduced TNF-α release and decreased apoptosis of hippocampal neurons [35]. In present study, animals treated with secretase inhibitors had a significative decrease only in IL-1β, but not IL-6 or TNF-α levels, 10 and 30 days after sepsis. The reduction in the levels of proinflammatory cytokines promoted by the treatments can contribute to a reduction in Aβ peptide generation, since studies suggest that the inflammation stimulates both β and γ-secretase [36, 37] thus promoting acceleration in Aβ peptide formation, which in turn appears to induce the increase of β-secretase itself [37], generating a vicious cycle. Memory improvement in animals treated with secretase inhibitors may be related to reduced neuroinflammation. This observation is based on the fact that proinflammatory cytokines, notably IL-1β, are indicated as mediators of long-term cognitive and behavioral changes in sepsis [38]. Additionally, IL-1β was associated with delirium occurrence in critically ill patients [39, 40], and some inflammation-related markers were associated with long-term cognitive impairment after critical illness [41, 42].
Ours results show that the use of β-secretase inhibitor prior to the formation of Aβ peptide in sepsis course promoted a partial reduction in Aβ peptide deposition. Lai & cols. (2012) observed in preclinical and clinical studies that a β-secretase inhibitor decreased the levels of Aβ in the CSF and/or plasma in healthy volunteers [43]. Another study evaluating a β-secretase inhibitor (NB-360) also demonstrated a significant reduction in Aβ peptide deposition in brain structures of both rats and dogs, suggesting that the findings related to inhibitor use are translational among species and could be extrapolated to humans [44]. Our demonstration that the β-secretase inhibitor reduces cognitive decline in septic animals, supports to previous evidence that elevation of Aβ peptide and other Aβ fragments can be associated by neurodegenerative damage. Jiang et al. (2016), in an animal model of Down syndrome, demonstrated that partial genetic reduction of β-secretase prevented AD-related pathological features such as endocytic abnormalities and cholinergic neurodegeneration by reducing APP-βCTF levels [45]. These results reinforce that observed in the present study that in addition to the reduction in Aβ levels and inflammation, there was a partial protection from long-term memory damage in the animals treated with β-secretase inhibitor.
In present study, treatment with β-secretase inhibitor promoted reduction of IL-1β 30 days after induction of sepsis, demonstrating the benefit of the use of treatment on sepsis-induced long-term neuroinflammation. In a study conducted with NB-360, Neumann and colleagues (2015) demonstrated a substantial reduction in the activation of microglial cells in the cerebral cortex, showing that the inhibitor contributed to the reduction of neuroinflammation [44]. Evidence suggests that Aβ fragments activate innate immunity by promoting release of proinflammatory cytokines that contribute to injury, dysfunction, and probably neuronal death [38, 41, 46]. The structural similarity between Aβ fragments and bacterial extracellular protein components present in enterobacteria adhesion fimbriae could explain the strong recognition of Aβ fragments as harmful by the immune system [47].
Some limitations must be pointed regarding the results presented here. First, inhibition of secretase pathway did not completely reversed brain inflammation and long-term cognitive deficits. Since one of the main pathways related to Aβ toxicity is the activation of RAGE [48], and several molecules besides Aβ could bind RAGE, this can partially explain our results. In fact, it was previously demonstrated that RAGE neutralization completely reversed cognitive impairment and brain inflammation in an animal model of sepsis [21]. This was similar to the neutralization of another RAGE ligand, HMGB1 [49]. Second, it is possible that the pharmacological inhibition of secretase pathway was not complete, or the dose regimen of drug administration was not enough to completely reverse Aβ formation, and this could also explain the partial protection. Third, only male rats were included in this study, and it is recognized that this could represent a scientific gap in animal studies. However, gonadal steroids modulate immune responses and sepsis severity [50]; thus, it is critical to understand how to incorporate females in future studies. Fourth, secretase inhibitors were administered via intracerebroventricular injection. While these limit the clinical translation of our results, from a mechanistic point of view, it decreases the probability of a systemic, non-specific effect.
In conclusion, it was demonstrated that during sepsis development there was an increase in the degradation of APP by the amyloidogenic route, and the inhibition of this pathway promoted attenuation of neuroinflammation, Aβ peptide content, and of cognitive impairment.
References
Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A et al (2016) Assessment of clinical criteria for sepsis: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315(8):762–774
Fu HQ, Yang T, Xiao W, Fan L, Wu Y, Terrando N et al (2014) Prolonged neuroinflammation after lipopolysaccharide exposure in aged rats. PLoS One 9(8)
Hannestad J, Gallezot J-D, Schafbauer T, Lim K, Kloczynski T, Morris ED, Carson RE, Ding YS et al (2012) Endotoxin-Induced Systemic inflammation activates microglia: [ 11 C]PBR28 positron emission tomography in nonhuman primates. Neuroimage 63(1):232–239
Hoogland ICM, Houbolt C, Van Westerloo DJ, Van Gool WA, Van De Beek D (2015) Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 12(1):114
Sankowski R, Mader S, Valdés-Ferrer SI, Campos V, Iván V-FS (2015) Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosc 9:28
Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonça B, Mina F et al (2015) The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun 43:54–59
Cunningham C, Hennessy E (2011) Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Res Ther 7
Singer BH, Newstead MW, Zeng X, Cooke CL, Thompson RC, Singer K, Ghantasala R, Parent JM et al (2016) Cecal ligation and puncture results in long- term central nervous system myeloid inflammation. Plos One 11(2):e0149136
Schwalm MT, Pasquali M, Miguel SP, Dos Santos JPA, Vuolo F, Comim CM, Petronilho F, Quevedo J et al (2014) Acute brain inflammation and oxidative damage are related to long-term cognitive deficits and markers of neurodegeneration in sepsis-survivor rats. Mol Neurobiol 49(1):380–385
Joshi P, Turola E, Ruiz A, Bergami A, Libera D, Benussi L et al (2013) Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ 21(10):582–593
Gasparotto J, Girardi CS, Somensi N, Ribeiro CT, Moreira JCF, Michels M, Sonai B, Rocha M et al (2018) Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. J Biol Chem. 293(1):226–244
Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT (2005) Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat. 30:144–157
Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R (2001) Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 101:249–255
Sadleir KR, Vassar R (2012) Cdk5 protein inhibition and Aβ42 increase BACE1 protein level in primary neurons by a post-transcriptional mechanism. J Biol Chem 287(10):7224–7235
Fink MP, Heard SO (1990) Laboratory models of sepsis and septic shock. J Surg Res 49(2):186–196
Barichello T, Martins MR, Reinke A, Feier G, Ritter C, Quevedo J et al (2005) Cognitive impairment in sepsis survivors from cecal ligation and perforation. Crit Care Med 33(1):221–223
Tuon L, Comim CM, Petronilho F, Barichello T, Izquierdo I, Quevedo J, Dal-Pizzol F (2008) Time-dependent behavioral recovery after sepsis in rats. Intensive Care Med 34(9):1724–1731
Petronilho F, Prico SR, Vuolo F, Mina F, Constantino L, Comim CM et al (2012) Protective effects of guanosine against sepsis-induced damage in rat brain and cognitive impairment. Brain Behav Immun 26(6):904–910
Davson H (1969) The Cerebrospinal Fluid. Handbook of Neurochemistry 2:23–48
Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, Merrill WH, Hinton RB et al (2011) Inhibitory role of Notch1 in calcific aortic valve disease. PLoS One. 6(11):e27743
Roesler R, Vianna MR, de Paris F, Quevedo J (1999) Memory-enhancing treatments do not reverse the impairment of inhibitory avoidance retention induced by NMDA receptor blockade. Neurobiol Learn Mem 72(3):252–258
Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2(5):1–25
Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA (1998) Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med 4(7):827–831
Walsh DM, Selkoe DJ (2007) A beta oligomers - a decade of discovery. J Neurochem 101(5):1172–1184
Niederst ED, Reyna SM, Goldstein LSB (2015) Axonal amyloid precursor protein and its fragments undergo somatodendritic endocytosis and processing. Mol Biol Cell 26(2):205–217
Comim CM, Constantino LC, Barichello T, Streck EL, Quevedo JQ, Dal-Pizzol F (2009) Cognitive Impairment in the Septic Brain [Internet]. Vol. 6. Current Neurovascular Research 6(3):194–203
Girard T, Pun BT, Thompson JL, Shintani AK, Gordon SM, Canonico AE et al (2013) Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med 38(7):1513–1520
Michels M, Steckert AV, Quevedo J, Barichello T, Dal-Pizzol F (2011) Mechanisms of long-term cognitive dysfunction of sepsis: from blood-borne leukocytes to glial cells. Intensive Care Med Exp 3(1):30
Barichello T, Generoso JS, Goularte JA, Collodel A, Pitcher MR, Simões LR et al (2016) Does infection-induced immune activation contribute to dementia? Aging Dis 6:342–348
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8(2):101–112
Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ (2002) Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans 30(4):552–557
Tanokashira D, Mamada N, Yamamoto F, Taniguchi K, Tamaoka A, Lakshmana MK et al (2017) The neurotoxicity of amyloid β-protein oligomers is reversible in a primary neuron model. Mol Brain 10(1):5–36
Di Scala C, Yahi N, Boutemeur S, Flores A, Rodriguez L, Chahinian H et al (2016) Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Nat Publ Gr 6:28781
Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A et al (2010) Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A 107(4):1630–1635
Huang M, Liu C, Hu Y, Wang P, Ding M (2014) alfa-secretase inhibitor DAPT prevents neuronal death and memory impairment in sepsis associated encephalopathy in septic rats. Chin Med J (Engl) 127(5):924–928
Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao F-F, Xu H, Zhang YW (2007) Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem 282(15):10873–10880
Chami L, Checler F (2012) BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and β-amyloid production in Alzheimer’s disease. Mol Neurodegener 7:52
Mina F, Comim CM, Dominguini D, Cassol OJ Jr, Dall Igna DM, Ferreira GK, Silva MC, Galant LS et al (2014) IL-1 beta involvment in cognitive impairment after sepsis. Mol Neurobiol 49(2):1069–1076
Van Den Boogaard M, Kox M, Quinn KL, Van Achterberg T, Van Der Hoeven JG, Schoonhoven L et al (2011) Biomarkers associated with delirium in critically ill patients and their relation with long-term subjective cognitive dysfunction; indications for different pathways governing delirium in inflamed and noninflamed patients. Crit Care 15:R297
Ritter C, Tomasi CD, Dal-Pizzol F, Pinto BB, Dyson A, de Miranda AS, Comim CM, Soares M et al (2014) Inflammation biomarkers and delirium in critically ill patients. Crit Care 18:R106
Hughes CG, Patel MB, Brummel NE, Thompson JL, McNeil JB, Pandharipande PP, Jackson JC, Chandrasekhar R et al (2018) Relationships between markers of neurologic and endothelial injury during critical illness and long-term cognitive impairment and disability. Intensive Care Med 44:345–355
Maciel M, Benedet SR, Lunardelli EB, Delziovo H, Domingues RL, Vuolo F, Tomasi CD, Walz R et al (2019) Predicting long-term cognitive dysfunction in survivors of critical illness with plasma inflammatory markers: a retrospective cohort study. Mol Neurobiol 56(1):763–767
Lai R, Albala B, Kaplow JM, Aluri J, Yen M, Satlin A (2012) First-in-human study of E2609, a novel BACE1 inhibitor, demonstrates prolonged reductions in plasma beta-amyloid levels after single dosing. Alzheimer’s Dement 8(4, Supplement):P96
Neumann U, Rueeger H, MacHauer R, Veenstra SJ, Lueoend RM, Tintelnot-Blomley M et al (2015) A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol Neurodegener 10(1):1–15
Jiang Y, Rigoglioso A, Peterhoff CM, Pawlik M, Sato Y, Bleiwas C, Stavrides P, Smiley JF et al (2016) Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol Aging 39:90–98
Ferretti MT, Bruno MA, Ducatenzeiler A, Klein WL, Cuello AC (2012) Intracellular Aβ-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol Aging 33(7):1329–1342
Tükel C, Nishimori JH, Wilson RP, Winter MG, Keestra AM, van Putten JPM, Bäumler AJ (2010) Toll-like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms. Cell Microbiol 12(10):1495–1505
Rapsinski GJ, Wynosky-Dolfi MA, Oppong GO, Tursi SA, Wilson RP, Brodsky IE et al (2015) Toll-like receptor 2 and NLRP3 cooperate to recognize a functional bacterial amyloid, curli. Infect Immun 83(2):693–701
Cai Z, Liu N, Wang C, Qin B, Zhou Y, Xiao M, Chang L, Yan LJ et al (2016) Role of RAGE in Alzheimer’s disease. Cell Mol Neurobiol 36:483–495
Chavan SS, Huerta PT, Robbiati S, Valdes-Ferrer SI, Ochani M, Dancho M, Frankfurt M, Volpe BT et al (2012) HMGB1 mediates cognitive impairment in sepsis survivors. Mol Med 18:930–937
Funding
This work was supported by CAPES-001, FAPESC, CNPQ and UNESC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The experimental procedures involving animals were performed in accordance with the National Institutes of Health (Bethesda, MD, USA) Guide for Care and Use of Laboratory Animals and with the approval of our institutional ethics committee. Protocol number: 041/2016-1.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Michelon, C., Michels, M., Abatti, M. et al. The Role of Secretase Pathway in Long-term Brain Inflammation and Cognitive Impairment in an Animal Model of Severe Sepsis. Mol Neurobiol 57, 1159–1169 (2020). https://doi.org/10.1007/s12035-019-01808-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-01808-1