
Abstract
Studies have suggested the involvement of inflammatory processes in the physiopathology of bipolar disorder. Preclinical evidences have shown that histone deacetylase inhibitors may act as mood-stabilizing agents and protect the brain in models of mania and depression. The aim of the present study was to evaluate the effects of sodium butyrate (SB) and valproate (VPA) on behavioral changes, histone deacetylase activity, and the levels of cytokines in an animal model of mania induced by dextroamphetamine (d-AMPH). Wistar rats were first given d-AMPH or saline (Sal) for a period of 14 days, and then, between the 8th and 14th days, the rats were treated with SB, VPA, or Sal. The activity of histone deacetylase and the levels of cytokines (interleukin (IL) IL-4, IL-6, and IL-10 and tumor necrosis factor-alpha (TNF-α)) were evaluated in the frontal cortex and striatum of the rats. The administration of d-AMPH increased the activity of histone deacetylase in the frontal cortex. Administration of SB or VPA decreased the levels of histone deacetylase activity in the frontal cortex and striatum of rats. SB per se increased the levels of cytokines in both of the brain structures evaluated. AMPH increased the levels of cytokines in both of the brain structures evaluated, and VPA reversed this alteration. The effects of SB on d-AMPH-induced cytokine alterations were dependent on the brain structure and the cytokine evaluated. Despite VPA and SB having a similar mechanism of action, both being histone deacetylase inhibitors, they showed different effects on the levels of cytokines. The present study reinforces the need for more research into histone deacetylase inhibitors being used as a possible target for new medications in the treatment of bipolar disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bipolar disorder (BD) is a chronic mental disorder which affects about 1–3% of the global population. BD is associated with multiple medical and psychiatric comorbidities, such as cardiovascular disease, diabetes mellitus, obesity, and thyroid dysfunction. Furthermore, this medical condition is accompanied by significant functional impairment, deterioration of both the patients and the patients’ families’ quality of life, and higher rates of suicide [1–3].
Although BD contributes to dominate as the global burden of mental disorders, little is known about the precise neurobiological underpinnings of BD [4]. Some theories about the pathophysiology of BD have been discussed in the literature, such as alterations to the monoamine systems [5, 6], oxidative stress [7, 8], and mitochondrial dysfunctions [9–11]. More recently, evidences have suggested that inflammatory mechanisms have an important role in the pathophysiology of BD [12, 13].
In the central nervous system (SNC), the cells responsible for the activation of immune responses are called microglial cells. These cells respond to the binding of pathogen proteins or even to specific endogenous biomolecules from damage and cellular death binding to its membrane receptors, which triggers an intracellular cascade, leading to the activation of a local inflammatory response. Cytokines are inflammatory mediators, released by microglial cells, and are responsible for the modulation of the inflammatory response [14, 15]. Cytokines play an important role in inflammatory signaling, inducing molecular adhesion to neutrophils, increasing vascular permeability, and the attraction and activation of neutrophils [16].
In fact, several studies have demonstrated that there are alterations in the levels of inflammatory mediators present in BD [17]. Clinical studies have shown that peripheral levels of IL-1β, IL-6, IL-18, IL-23, and IL-33; TGF; sIL-2R and sIL-6R; CRP; sTNF-R1; and MCP-1 are increased in patients with BD when compared with control subjects [17–24]. Genetic studies with bipolar patients have shown that they have alterations in their TNF-α gene, which has been related to higher levels of TNF-α production [25, 26]. In addition, it was demonstrated that there is a genetic variant of the IL-1 and RIL-1 genes in BD [27, 28]. It was also discovered that there is mRNA overexpression of TNF-훼, IL-1훽, IL-6, and CCL2 in the monocytes of bipolar patients and also in the offspring of bipolar parents [29].
Mood-stabilizing agents may produce their therapeutic effects by regulating gene expression within neuronal circuits. Valproate (VPA) is an anticonvulsant that is used as a mood stabilizer in the treatment of BD. One of the mechanisms of action of VPA is that it regulates gene expression by acting as a histone deacetylase (HDAC) inhibitor [30]. Based on the mechanism of action for VPA, other HADC inhibitors, such as sodium butyrate (SB), have also been studied as possible mood-stabilizing drugs. In fact, treatment with SB was able to reverse both the manic- and depressive-like symptoms of BD in preclinical studies [31–35].
One of the main features of BD is its manic symptoms, which can be related to dopaminergic hyperactivity. Thus, even though the animal model of mania induced by dextroamphetamine (d-AMPH) only addresses one pole of the disorder, it is still the most useful model currently used in studying BD [36, 39, 40]. A previous study has shown that BD patients exhibit modifications in an allele of the dopamine transporter encoder, which may enhance dopamine levels in the brains of BD patients [37]. Recently, a study from our research laboratory evaluated the levels of cytokines in the frontal cortex, striatum, hippocampus, serum, and cerebrospinal fluid in an animal model of mania induced by d-AMPH. In this previous study, it was shown that d-AMPH increased the levels of IL-4, IL-6, IL-10, and TNF-α in the frontal cortex, striatum, and serum of rats, suggesting that dopamine can be related to the alterations in the inflammatory system observed in BD [38].
Several clinical neuroimaging studies have demonstrated morphological and physiological alterations in the frontal cortex and striatum of patients with BD [41–43]. In addition, previous studies have demonstrated that there are alterations in the levels of cytokines in the postmortem frontal cortex of patients with BD [44]. However, most studies evaluate the levels of cytokines in patients’ blood [18–24], and few studies have evaluated these molecules in the central nervous system (CNS) of BD patients [44].
Considering that inflammatory signaling plays a pivotal role in the pathophysiology of BD, and that HDAC inhibitors show a potential effect as mood stabilizer; the present study aims to investigate the effects of BS and VPA treatments on the levels of cytokines in the frontal cortex and striatum of rats submitted to the animal model of mania induced by d-AMPH, in order to clarify its use as a new therapeutic approach.
Experimental Methods
Animals
All of the animals used in the present study were adult male Wistar rats (weighing 250–350 g) which were obtained from our breeding colony. They were housed five animals to a cage, with food and water available ad libitum, and were maintained on a 12-h light/dark cycle (lights on at 7:00 a.m.) at a temperature of 22 ± 1 °C. All experimental procedures were performed in accordance with, and with the approval of, the local ethics committee for the use of animals at the Universidade do Extremo Sul Catarinense under protocol number 66/2010. All experiments were performed at the same time during the day to avoid circadian variations.
Drugs and Pharmacological Procedures
The animals received one daily intraperitoneal (i.p.) injection of d-AMPH at a dose of 2 mg/kg or saline (Sal) for a period of 14 days (48 animals per group). On the eighth day of treatment, the animals in both the saline and d-AMPH groups were divided into three subgroups (eight animals per group); the first group was treated with VPA (200 mg/kg i.p.), the second group was treated with SB (600 mg/kg i.p.), and the third group received Sal (NaCl 0.9%, i.p). The animals treated with VPA, SB, or Sal received the drugs twice a day for a period of 7 days. On the 15th day of treatment, the animals received a single injection of d-AMPH or Sal, and their locomotor activity was assessed 2 h after the last injection using an open field test (described below).
The doses of VPA and SB were based on previous studies from our laboratory, showing that VPA (200 mg/kg) and SB (600 mg/kg) prevented and reversed the hyperactivity induced by amphetamine in rats [38, 39].
Note
d-AMPH (Sigma®), SB (Sigma®), and VPA (Sanofi Aventis®) were diluted in saline solution (NaCl 0.9%). The drugs were administered intraperitoneally (i.p.) at a volume of 1 ml/kg body weight. The control groups received an equivalent volume of saline.
Locomotor Activity
Locomotor activity was assessed using the open field task. This task was performed in a 40 × 60-cm open field surrounded by 50-cm-high walls made of brown plywood, with the floor divided into 12 equal rectangles by black lines. The animals were gently placed on the left rear rectangle, and they were left to freely explore the arena for a period of 5 min. Crossings (locomotor activity/horizontal activity measured by the number of times the animals crossed the black lines) and rearings (exploratory activity/vertical activity) were counted.
Preparation of the Samples
Immediately after the open field test, the rats were sacrificed by decapitation, and the brains were transferred within 1 min to ice-cold isolation buffer (0.23 M mannitol, 0.07 M sucrose, 10 mM Tris–HCl, and 1 mM EDTA, pH 7.4). The frontal cortex and striatum were dissected in ice-cold buffer in a Petri dish.
Note
The striatum and frontal cortex were dissected according to the stereotaxic atlas from Paxinos and Watson [45]. Coordinates of the striatum—anteroposterior 1.6 mm, lateral 1.8 mm, and ventral 7.5 mm. Coordinates of the frontal cortex—3.2 mm anteroposterior, lateral 0.6 mm, and ventral 1.5 mm.
Nuclear Extraction
The obtained samples were flash-frozen and stored at −80 °C until nuclear proteins could be extracted. The frontal cortex and striatum from both hemispheres were subjected to a nuclear extraction protocol using a commercial Nuclear Extraction kit (Chemicon, USA). The nuclear extraction was used to evaluate the levels of HDAC activity.
HDAC Activity
Nuclear extracts from the frontal cortex and striatum were subjected to an HDAC activity assay using a commercial HDAC Assay kit (Fluorometric Detection) according to the manufacturer’s instructions (Upstate, USA). Briefly, 5 μL of nuclear extract was mixed with 5 μL of HDAC Assay Buffer and 5 μL of HDAC Assay Substrate in a 96-well plate, and then incubated at 30 °C for 45 min. Concomitantly, a standard curve was performed with serial dilutions of deacetylated substrate, with both positive and negative controls also being added to the plate. Subsequently, 10 μL of activator solution was added to the wells, and the plate was incubated at room temperature for a period of 15 min. A fluorescence reading was obtained using a fluorescence plate reader, with 360 nm for excitation and 460 nm for emission. HDAC activity was calculated on the basis of the standard curve, and values are presented as μM/μg protein. Total protein was measured using a modified version of Lowry’s method [46] using bovine serum albumin as a standard. HDAC activity was calculated as the micromolar concentration of deacetylated standard substrate per microgram of protein.
Assessment of IL-4, IL-6, IL-10, and TNF-α Levels
In order to evaluate the levels of cytokines, the frontal cortex and striatum from both hemispheres were homogenized in extraction solution containing aprotinin (100 mg of tissue per 1 mL). The concentrations of cytokines were determined using a commercially available enzyme-linked immunosorbent assay (ELISAs), following the instructions supplied by the manufacturer (DuoSet kits, R&D Systems; Minneapolis). The results are shown in pg/100 mg of tissue. Total protein was measured using a modified version of Lowry’s method [46] using bovine serum albumin as a standard.
Statistical Analysis
Data were analyzed using the two-way analysis of variance (ANOVA) test, followed by the Tukey’s post hoc test. The data are expressed as the mean ± standard deviation. All analyses were performed using the Statistical Package for the Social Sciences software (SPSS). In all experiments, the level of significance was p < 0.05.
Results
Behavioral Test
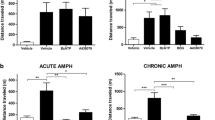
Results for locomotor activity are shown in Fig. 1. Two-way ANOVA revealed that there was a significant effect for d-AMPH (F(1.24) = 29.516; p < 0.001), treatments with SB and VPA (F(1.24) = 30.978; p < 0.001), and d-AMPH versus treatment interaction (F(2.24) = 20.395; p < 0.001) in relation to the number of crossings. Further analysis with the Tukey post hoc test showed that the administration of d-AMPH increased the number of crossings in the rats treated with saline, which is considered a manic-like behavior. Treatments with SB or VPA reversed this behavioral alteration. Lone treatments with SB or VPA did not alter the behavioral measures, indicating that the effects of SB or VPA on rats treated with d-AMPH were not associated with sedation.

Effects of VPA or SB administration on locomotor activity (number of crossings) in the animal model of mania induced by AMPH. Bars represent means; error bars represent standard deviations; *p < 0.05 vs. Sal + Sal group, according to a two-way ANOVA followed by Tukey’s test; #p < 0.05 vs. AMPH+Sal group, according to a two-way ANOVA followed by Tukey’s test
HDAC Activity
Results for HDAC activity in the frontal cortex and striatum are shown in Fig. 2. The levels of HDAC activity were found to be significantly increased within the frontal cortex, but not in the striatum, of rats in the AMPH + Sal group, when compared to the control group. Treatments with VPA reversed this alteration. The animals in the AMPH + SB group showed reductions in HDAC activity within the frontal cortex and striatum, when compared to the control group. The administration of VPA or SB per se decreased the levels of HDAC activity in the frontal cortex and striatum.

Effects of VPA or SB administration on HDAC activity in the animal model of mania induced by AMPH. Bars represent means; error bars represent standard deviations; *p < 0.05 vs. Sal + Sal group, according to a two-way ANOVA followed by Tukey’s test; #p < 0.05 vs. AMPH + Sal group, according to a two-way ANOVA followed by Tukey’s test
Data from the two-way ANOVA for AMPH administration (frontal cortex F(1.24) = 12.9, p = 0.0015; striatum F(1.24) = 53.17, p < 0.001), treatment with VPA or SB (frontal cortex F(2.24) = 51.8, p < 0.001; striatum F(2.24) = 186.02, p < 0.001), and AMPH × treatment interaction (frontal cortex F(2.24) = 6.67, p = 0.005; striatum F(2.24) = 8.23, p = 0.0019).
Cytokines Levels
Results for interleukin levels within the frontal cortex and striatum are shown in Fig. 3. The administration of AMPH increased the levels of IL-4 in the frontal cortex and striatum of rats; however, treatment with VPA reversed this alteration. The administration of SB per se increased the levels of IL-4 in the frontal cortex and striatum of rats (Fig. 3a).

Effects of VPA or SB administration on IL-4 (a), IL-6 (b), IL-10 (c), and TNF-α (d) levels in the animal model of mania induced by AMPH. Bars represent means; error bars represent standard deviations; *p < 0.05 vs. Sal + Sal group, according to a two-way ANOVA followed by Tukey’s test; #p < 0.05 vs. AMPH + Sal group, according to a two-way ANOVA followed by Tukey’s test
Data from the two-way ANOVA for AMPH administration (frontal cortex F(1.24) = 22.33, p < 0.001; striatum F(1.24) = 6.14, p = 0.02), treatment with VPA or SB (frontal cortex F(2.24) = 37.36, p < 0.001; striatum F(2.24) = 9.09, p = 0.0011), and AMPH × treatment interaction (frontal cortex F(2.24) = 18.46, p < 0.001; striatum F(2.24) = 19.61, p < 0.001).
The administration of AMPH increased the levels of IL-6 in the frontal cortex and striatum of rats. Treatment with VPA reversed this alteration in the frontal cortex and partially reversed this alteration in the striatum. In addition, SB partially reversed the increase of AMPH-induced IL-6 in the striatum of rats. The administration of SB per se increased the levels of IL-6 in the frontal cortex and striatum of rats (Fig. 3b).
Data from the two-way ANOVA for AMPH administration (frontal cortex F(1.24) = 20, p < 0.001; striatum F(1.24) = 95, p < 0.001), treatment with VPA or SB (frontal cortex F(2.24) = 55.81, p < 0.001; striatum F(2.24) = 57, p < 0.001), and AMPH × treatment interaction (frontal cortex F(2.24) = 17.76, p < 0.001; striatum F(2.24) = 74, p < 0.001).
The administration of AMPH increased the levels of IL-10 in the frontal cortex and striatum of rats, and treatment with VPA reversed this interleukin alteration. SB partially reversed the AMPH-induced increase in the levels of IL-10 in the frontal cortex of rats. The administration of SB per se increased the levels of IL-10 in the frontal cortex and striatum of rats (Fig. 3c).
Data from the two-way ANOVA for AMPH administration (frontal cortex F(1.24) = 32, p < 0.001; striatum F(1.24) = 40, p < 0.001), treatment with VPA or SB (frontal cortex F(2.24) = 85, p < 0.001; striatum F(2.24) = 53, p < 0.001), and AMPH × treatment interaction (frontal cortex F(2.24) = 70, p < 0.001; striatum F(2.24) = 23, p < 0.001).
The administration of AMPH increased the levels of TNF-α in the frontal cortex and striatum of rats, and treatment with VPA reversed this cytokine alteration. In addition, SB partially reversed the increase in AMPH-induced TNF-α in the frontal cortex. SB alone increased the levels of TNF-α in the frontal cortex and striatum of rats (Fig. 3d).
Data from the two-way ANOVA for AMPH administration (frontal cortex F(1.24) = 59, p < 0.001; striatum F(1.24) = 53, p < 0.001), treatment with VPA or SB (frontal cortex F(2.24) = 98, p < 0.001; striatum F(2.24) = 62, p < 0.001), and AMPH × treatment interaction (frontal cortex F(2.24) = 77, p < 0.001; striatum F(2.24) = 14, p < 0.001).
Discussion
The present study was able to reproduce previous results from our research laboratory, in which VPA and SB reversed the hyperlocomotion induced by d-AMPH, a potent central nervous system dopaminergic stimulant [36, 47]. Indeed, dopaminergic system dysfunctions have been implicated in the pathophysiology of BD [37, 48, 49]. It is important to emphasize that amphetamine can induce manic-like symptoms in healthy subjects and exacerbate symptoms or induce manic episodes in bipolar patients [50, 51]. Thus, amphetamine-induced hyperactivity is considered a good animal model of mania because psychomotor agitation is generally observed in manic episodes [52, 53]. Besides this, a significant number of studies have shown that the administration of amphetamine in rats causes a wide range of neurochemical changes that are also seen in BD patients, like oxidative stress [39], mitochondrial dysfunction [38], decreases in BDNF [39], and increases in the levels of cytokines in the brain [38], reinforcing the significance of this model in studying the pathophysiology of BD and in screening new mood stabilizers.
In the present study, SB as well as VPA reversed the d-AMPH-induced hyperactivity in rats. Indeed, in recent studies from our laboratory, it was reported that treatments with SB prevented and reversed the manic-like behaviors induced by ouabain and d-amphetamine [29, 31, 33–35, 39, 54]. Wu and colleagues [55] demonstrated that HDAC inhibitors upregulate GDNF and BDNF expression in astrocytes, protecting DA neurons. In fact, some studies from our laboratory have shown that VPA and SB increased the levels of BDNF, NGF, and GDNF in the brain of rats [33, 34, 56]. Together with our findings, these studies suggest that HDAC inhibitors can protect DA neurons, which should decrease the levels of AMPH-induced hyperactivity.
Moreover, AMPH increased the levels of HDAC activity in the frontal cortex, but not in the striatum. Treatment with VPA and SB reversed this enzyme alteration in the frontal cortex. In accordance with the present results, previous studies from our laboratory have demonstrated that AMPH-induced manic-like behaviors are associated with alterations in HDAC activity within the frontal cortex, which was reversed by VPA and SB [33]. In a preclinical study evaluating the neuroanatomical profile of the antimanic effects of HDAC inhibitors, it was shown that the antimanic effects of SB and VPA are related to the amygdala, striatum, and frontal cortex [54]. Contrary to the present study, Arent and colleagues [54] demonstrated that acute injections of AMPH did not alter the levels of HDAC activity in the frontal cortex, hippocampus, striatum, and amygdala. This discrepancy may be explained by the differences in the administration of AMPH—acute (in the previous study) [54] and subchronic (in the present study). It is well described in the literature that AMPHs act by increasing extraneuronal dopamine in this projection, as measured by in vivo microdialysis and voltammetry [57]. Although there is no evidence of a direct association between AMPH and HDAC, the ability of AMPH to increase dopamine levels in the synaptic cleft may have consequences in the nucleus.
It is important to emphasize that in the present study, the levels of HDAC activity did not explain the antimanic effects of VPA and SB. Indeed, there are no concrete studies proving that either BD or the administration of AMPH can change the levels of HDAC; nevertheless, studies show that the administration of HDAC inhibitors improves both the manic symptoms in humans and the manic-like symptoms induced by psychostimulants in rats [47, 54, 58]. Therefore, it can be suggested that there is a yet unknown underlying pathway in which HDAC inhibitors may be providing an antimanic effect.
The present study was able to reproduce previous results from our research laboratory, in which AMPH increased the levels of IL-4, IL-6, IL-10, and TNFα in the frontal cortex and striatum of rats [37]. It is important to note that the increase of cytokines induced by AMPH does not seem to be directly related to changes in HDAC activity, since the AMPH-induced increase in cytokine levels was observed in all of the brain structures evaluated; however, this drug only altered HDAC activity in the frontal cortex of rats. There is a body of data indicating that AMPH induces serious damage to dopaminergic neurons [38, 59]. It is well described that AMPH induces the over production of reactive oxygen species (ROS), increasing protein, lipid, and DNA oxidative damage in the brain of rats [31, 60, 61], which can lead to subsequent inflammation [62]. It is important to emphasize that abnormalities in the inflammatory system appear to play an important role in the pathophysiology of BD. Recently, Kim and colleagues [44] demonstrated that the levels of IL-1β, IL-6, TNF-α, and IL-10 are increased in the postmortem frontal cortex from patients with BD. Therefore, studies attempting to explain this system and the search for new drugs that target the inflammatory system in the CNS are very important for the treatment of BD.
In the present study, it was shown that VPA is able to reverse the increase in AMPH-induced interleukin levels in the frontal cortex and striatum of rats. In vitro and in vivo experiments show that VPA significantly inhibited the production of TNF-α and IL-6 in human monocytic leukemia cells [63]. A preclinical study also showed that VPA was able to reverse the increase in the levels of IL-10 induced by maternal separation in rats [64]. Additionally, it was demonstrated that VPA downregulates lipopolysaccharide (LPS)-induced proinflammatory cytokine production in mouse RAW 264.7 macrophage-like cells [65]. Jambalganiin and colleagues [65] found that VPA inhibits the phosphorylation of PI3K/Akt/MDM2 signaling, enhancing p53 expression and decreasing NF-κB transcriptional activation, which has a central role in the production of proinflammatory cytokines. Therefore, it can be suggested that in the present study, VPA reversed the increase in AMPH-induced cytokines levels via the inhibition of the PI3K/Akt/MDM2 signaling pathway.
On the other hand, in the present study, SB was unable to reverse the increases in the levels of cytokines induced by amphetamine seen in most of the analysis (frontal cortex: IL-4 and IL-6 and striatum: IL-4, IL-10, and TNFα). In addition, the administration of SB per se increased the levels of cytokines in all of the brain structures evaluated, when compared to the control group. However, the literature shows controversial data, as discussed below. In accordance with our results, SB increased the levels of IL-4 and IL-10 in human monocytes [66]. Berndt and colleagues [67] showed that SB increased IL-23 by stimulating dendritic cells. In the same study, the same authors [67] also demonstrated that SB did not alter the increase in the levels of TNF-α and IL-6 that was induced by LPS. In an in vitro study, it was found that SB enhanced both the secretion of LPS-induced IL-6 and the expression of protein in transformed N9 microglial cells. However, in the same study, it was found that SB decreased the levels of IL-6 and TNF-α in rat primary microglia [68]. Additionally, a previous study has shown that trichostatin A, a specific inhibitor of class I and II HDAC, also induced a strong potentiation of LPS-induced secretion of IL-6 in transformed N9 microglial cells [69].
In the present study, it was also demonstrated that SB partially reversed the AMPH-induced increases of IL-10 and TNF-α in the frontal cortex and IL-6 in the striatum. Indeed, several sources of data from the literature have also demonstrated the anti-inflammatory effects of SB. In a previous study, pretreatment with SB or tricostatin A (TSA) caused a robust decrease in LPS-induced proinflammatory responses and protected DA neurons from damage in mesencephalic neuron-glia cultures [70]. Furthermore, SB-treated animals in a severe acute pancreatitis model showed decreases in their levels of IL-6 and TNF-α [71]. In a study using a rat-based permanent ischemic model of stroke, it was demonstrated that postinsult treatment with VPA or SB suppressed microglial activation, reduced the number of microglia, and inhibited inflammatory markers in the ischemic brain [72]. By treating isolated synovial cells from patients with RA (which is a systemic inflammatory disease) with TSA, it was possible to decrease the levels of proinflammatory cytokines, and also to increase apoptosis [73]. Inhibitors of HDAC induced microglial apoptosis, which could be related to their neuroprotective effects in response to proinflammatory stimuli [70, 73]. Additionally, it is known that HDAC inhibitors prevent the activation of NF-kappaB [74], which has an important role in the proinflammatory signaling pathway via the expression of proinflammatory genes including cytokines, chemokines, and adhesion molecules [75].
Our data showed that VPA reversed d-AMPH-induced cytokine dysfunction in all of the brain regions analyzed. However, SB presented less effects against d-AMPH-induced cytokine dysfunction. This discrepancy can be explained, at least in part, by the fact that although VPA and SB are HDAC inhibitors, they have differing chemical structures and abilities to inhibit the various HDAC isoforms [76]. In addition, a review article from Akimova and colleagues [77] highlights an important discussion relating to some controversial data from the literature regarding the effects of HDAC inhibitors on the inflammatory system. In the review, the authors suggested that the effects of HDAC inhibitors can be cell-, tissue-, or context-dependent and can involve the modulation of specific inflammatory signaling pathways as well as epigenetic mechanisms. In addition, besides the inhibition of HDAC, VPA has other molecular targets. It is known that VPA enhances GABA activity within the brain by inhibiting its degradation, stimulating its synthesis and release, and directly enhancing its postsynaptic effects [78]. VPA also acts as an inhibitor on the protein kinase C, decreasing the release of neurotransmitters [70, 79, 80].
In conclusion, we demonstrated that VPA reversed d-AMPH-induced cytokine dysfunction in all of the brain regions analyzed. However, SB presented less effects against d-AMPH-induced cytokine dysfunction. Furthermore, the effects of SB can vary depending on the brain region and cytokines analyzed. Therefore, we can suggest that VPA could provide a better level of protection against inflammatory dysfunction, as observed in bipolar patients.
Limitations of the Study
(1) It is important to note that no animal model developed to date can fully mimic the “corresponding” human psychiatric disorder; therefore, the administration of d-AMPH in rats did not mimic all of the manic symptoms seen in humans. (2) The present study did not evaluate the levels of cytokines in the blood, despite most studies with BD evaluating the levels of cytokines in the patients’ blood. (3) The results of the present study suggest that the antimanic effects of SB are not related to the modulation of cytokines; however, this does not explain the specific mechanisms of action involved in this effect.
References
Bauer MS, Altshuler L, Evans DR, Beresford T, Williford WO, Hauger R (2005) Prevalence and distinct correlates of anxiety, substance, and combined comorbidity in a multi-site public sector sample with bipolar disorder. J Affect Disord 85:301–315. doi:10.1016/j.jad.2004.11.009
Leboyer M, Soreca I, Scott J, Frye M, Henry C, Tamouza R, Kupfer DJ (2012) Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord 141(1):1–10. doi:10.1016/j.jad.2011.12.049
Brenner CJ, Shyn SI (2014) Diagnosis and management of bipolar disorder in primary care: a DSM-5 update. Med Clin North Am 98(5):1025–1048. doi:10.1016/j.mcna.2014.06.004
Maletic V, Raison C (2014) Integrated neurobiology of bipolar disorder. Front Psychiatry 5:98. doi:10.3389/fpsyt.2014.00098
Haase J, Brown E (2014) Integrating the monoamine, neurotrophin and cytokine hypotheses of depression—a central role for the serotonin transporter? Pharmacol Ther 147:1–11. doi:10.1016/j.pharmthera.2014.10.002
Jun C, Choi Y, Lim SM, Bae S, Hong YS, Kim JE, Lyoo IK (2014) Disturbance of the glutamatergic system in mood disorders. Exp Neurobiol 23(1):28–35. doi:10.5607/en.2014.23.1.28
Pfaffenseller B, Fries GR, Wollenhaupt-Aguiar B, Colpo GD, Stertz L, Panizzutti B, Magalhães PV, Kapczinski F (2013) Neurotrophins, inflammation and oxidative stress as illness activity biomarkers in bipolar disorder. Expert Rev Neurother 13(7):827–842. doi:10.1586/14737175.2013.811981
Siwek M, Sowa-Kućma M, Dudek D, Styczeń K, Szewczyk B, Kotarska K, Misztakk P, Pilc A et al (2013) Oxidative stress markers in affective disorders. Pharmacol Rep 65(6):1558–1571
Mansur RB, Cha DS, Asevedo E, McIntyre RS, Brietzke E (2013) Selfish brain and neuroprogression in bipolar disorder. Prog Neuro-Psychopharmacol Biol Psychiatry 43:66–71. doi:10.1016/j.pnpbp.2012.12.004
de Sousa RT, Machado-Vieira R, Zarate CA Jr, Manji HK (2014) Targeting mitochondrially mediated plasticity to develop improved therapeutics for bipolar disorder. Expert Opin Ther Targets 18(10):1131–1147. doi:10.1517/14728222.2014.940893
Streck EL, Gonçalves CL, Furlanetto CB, Scaini G, Dal-Pizzol F, Quevedo J (2014) Mitochondria and the central nervous system: searching for a pathophysiological basis of psychiatric disorders. Rev Bras Psiquiatr 36(2):156–167. doi:10.1590/1516-4446-2013-1224
Bauer IE, Pascoe MC, Wollenhaupt-Aguiar B, Kapczinski F, Soares JC (2014) Inflammatory mediators of cognitive impairment in bipolar disorder. J Psychiatr Res 56:18–27. doi:10.1016/j.jpsychires.2014.04.017
Baumeister D, Russell A, Pariante CM, Mondelli V (2014) Inflammatory biomarker profiles of mental disorders and their relation to clinical, social and lifestyle factors. Soc Psiquiatria Psychiatr Epidemiol 49(6):841–849. doi:10.1007/s00127-014-0887-z
Mallucci G, Peruzzotti-Jametti L, Bernstock JD, Pluchino S (2015) The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog Neurobiol 127-128C:1–22. doi:10.1016/j.pneurobio.2015.02.003
Kang JW, Kim SJ, Cho HI, Lee SM (2015) DAMPs activating innate immune responses in sepsis. Ageing Res Rev 24(Pt A):54–65. doi:10.1016/j.arr.2015.03.003
Sava P, Cook IO, Mahal RS, Gonzalez AL (2015) Human microvascular pericyte basement membrane remodeling regulates neutrophil recruitment. Microcirculation 22(1):54–67. doi:10.1111/micc.12173
Barbosa IG, Bauer ME, Machado-Vieira R, Teixeira AL (2014a) Cytokines in bipolar disorder: paving the way for neuroprogression. Neural Plast 2014:360481. doi:10.1155/2014/360481
Li H, Hong W, Zhang C, Wu Z, Wang Z, Yuan C, Li Z, Huang J et al (2015) IL-23 and TGF-β1 levels as potential predictive biomarkers in treatment of bipolar I disorder with acute manic episode. J Affect Disord 174:361–366. doi:10.1016/j.jad.2014.12.033
Munkholm K, Weikop P, Kessing LV, Vinberg M (2015) Elevated levels of IL-6 and IL-18 in manic and hypomanic states in rapid cycling bipolar disorder patients. Brain Behav Immun 43:205–213. doi:10.1016/j.bbi.2014.09.021
Bai YM, Su TP, Tsai SJ, Wen-Fei C, Li CT, Pei-Chi T, Mu-Hong C (2014) Comparison of inflammatory cytokine levels among type I/type II and manic/ hypomanic/euthymic/depressive states of bipolar disorder. J Affect Disord 166:187–192. doi:10.1016/j.jad.2014.05.009
Bai YM, Su TP, Li CT, Tsai SJ, Chen MH, Tu PC, Chiou WF (2015) Comparison of pro-inflammatory cytokines among patients with bipolar disorder and unipolar depression and normal controls. Bipolar Disord 17(3):269–277. doi:10.1111/bdi.12259
Wieck A, Grassi-Oliveira R, do Prado CH, Rizzo LB, de Oliveira AS, Kommers-Molina J, Viola TW, Marciano Vieira EL et al (2014) Pro-inflammatory cytokines and soluble receptors in response to acute psychosocial stress: differential reactivity in bipolar disorder. Neurosci Lett 580:17–21. doi:10.1016/j.neulet.2014.07.040
Monfrim X, Gazal M, De Leon PB, Quevedo L, Souza LD, Jansen K, Oses JP, Pinheiro RT et al (2014) Immune dysfunction in bipolar disorder and suicide risk: is there an association between peripheral corticotropin-releasing hormone and interleukin-1β? Bipolar Disord 16(7):741–747. doi:10.1111/bdi.12214
Barbosa IG, Morato IB, de Miranda AS, Bauer ME, Soares JC, Teixeira AL (2014b) A preliminary report of increased plasma levels of IL-33 in bipolar disorder: further evidence of pro-inflammatory status. J Affect Disord 157:41–44. doi:10.1016/j.jad.2013.12.042
Pae CU, Lee KU, Han H, Serretti A, Jun TY (2004) Tumor necrosis factor alpha gene-G308A polymorphism associated with bipolar I disorder in the Korean population. Psychiatry Res 125(1):65–68. doi:10.1016/j.psychres.2003.06.002
Clerici M, Arosio B, Mundo E et al (2009) Cytokine polymorphisms in the pathophysiology of mood disorders. CNS Spectrums 14(8):419–425
Papiol S, Molina V, Desco M, Rosa A, Reig S, Sanz J, Palomo T, Fañanás L (2008) Gray matter deficits in bipolar disorder are associated with genetic variability at interleukin-1 beta gene (2q13). Genes Brain Behav 7(7):796–801. doi:10.1111/j.1601-183X.2008.00421.x
Rafiei A, Hosseini SH, Taheri M, Hosseni-Khah Z, Hajilooi M, Mazaheri Z (2013) Influence of IL-1RN intron 2 variable number of tandem repeats (VNTR) polymorphism on bipolar disorder. Neuropsychobiology 67(2):116–121. doi:10.1159/000346112
Padmos RC, Hillegers MH, Knijff EM, Vonk R, Bouvy A, Staal FJ, de Ridder D, Kupka RW et al (2008) A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry 65(4):395–407. doi:10.1001/archpsyc.65.4.395
Grunze H, Azorin JM (2014) Clinical decision making in the treatment of mixed states. World J Biol Psychiatry 15(5):355–368. doi:10.3109/15622975.2014.908238
Moretti M, Valvassori SS, Varela RB, Ferreira CL, Rochi N, Benedet J, Scaini G, Kapczinski F et al (2011) Behavioral and neurochemical effects of sodium butyrate in an animal model of mania. Behav Pharmacol 22(8):766–772. doi:10.1097/FBP.0b013e32834d0f1b
Steckert A, Valvassori SS, Varela RB, Mina F, Resende WR, Bavaresco DV, Ornell F, Dal-Pizzol F et al (2013) Effects of sodium butyrate on oxidative stress and behavioral changes induced by administration of D-AMPH. Neurochem Int 62(4):425–432. doi:10.1016/j.neuint.2013.02.001
Lopes-Borges J, Valvassori SS, Varela RB, Tonin PT, Vieira JS, Gonçalves CL, Streck EL, Quevedo J (2015) Histone deacetylase inhibitors reverse manic-like behaviors and protect the rat brain from energetic metabolic alterations induced by ouabain. Pharmacol Biochem Behav 128:89–95. doi:10.1016/j.pbb.2014.11.014
Stertz L, Fries GR, Aguiar BW, Pfaffenseller B, Valvassori SS, Gubert C, Ferreira CL, Moretti M et al (2014) Histone deacetylase activity and brain-derived neurotrophic factor (BDNF) levels in a pharmacological model of mania. Rev Bras Psiquiatr 36(1):39–46. doi:10.1590/1516-4446-2013-1094
Varela RB, Valvassori SS, Lopes-Borges J, Mariot E, Dal-Pont GC, Amboni RT, Bianchini G, Quevedo J (2015) Sodium butyrate and mood stabilizers block ouabain-induced hyperlocomotion and increase BDNF, NGF and GDNF levels in brain of Wistar rats. J Psychiatr Res 61:114–121. doi:10.1016/j.jpsychires.2014.11.003
Valvassori SS, Calixto KV, Budni J, Resende WR, Varela RB, de Freitas KV, Gonçalves CL, Streck EL et al (2013) Sodium butyrate reverses the inhibition of Krebs cycle enzymes induced by amphetamine in the rat brain. J Neural Transm 120(12):1737–1742. doi:10.1007/s00702-013-1056-3
Pinsonneault JK, Han DD, Burdick KE, Kataki M, Bertolino A, Malhotra AK, Gu HH, Sadee W (2011) Dopamine transporter gene variant affecting expression in human brain is associated with bipolar disorder. Neuropsychopharmacology 36(8):1644–1655. doi:10.1038/npp.2011.45
Valvassori SS, Tonin PT, Varela RB, Carvalho AF, Mariot E, Amboni RT, Bianchini G, Andersen ML et al (2015) Lithium modulates the production of peripheral and cerebral cytokines in an animal model of mania induced by dextroamphetamine. Bipolar Disord 17(5):507–517. doi:10.1111/bdi.12299
Frey BN, Valvassori SS, Réus GZ, Martins MR, Petronilho FC, Bardini K, Dal-Pizzol F, Kapczinski F et al (2006) Effects of lithium and valproate on amphetamine-induced oxidative stress generation in an animal model of mania. J Psychiatry Neurosci 31(5):326–332
Cappeliez P, Moore E (1990) Effects of lithium on an amphetamine animal model of bipolar disorder. Prog Neuro-Psychopharmacol Biol Psychiatry 14(3):347–358
Bearden CE, Glahn DC, Monkul ES, Barrett J, Najt P, Villarreal V, Soares JC (2006a) Patterns of memory impairment in bipolar disorder and unipolar major depression. Psychiatry Res 142(2–3):139–150
Bearden CE, Glahn DC, Monkul ES, Barrett J, Najt P, Kaur S, Sanches M, Villarreal V et al (2006b) Sources of declarative memory impairment in bipolar disorder: mnemonic processes and clinical features. J Psychiatr Res 40(1):47–58
Nugent AC, Milham MP, Bain EE, Mah L, Cannon DM, Marrett S, Zarate CA, Pine DS et al (2006) Cortical abnormalities in bipolar disorder investigated with MRI and voxel-based morphometry. NeuroImage 30(2):485–497
Kim HK, Andreazza AC, Elmi N, Chen W, Young LT (2016) Nod-like receptor pyrin containing 3 (NLRP3) in the post-mortem frontal cortex from patients with bipolar disorder: a potential mediator between mitochondria and immune-activation. J Psychiatr Res 72:43–50
Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Academic Press, San Diego
Lowry OH, Rosebrough NJ, Farr A, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Resende WR, Valvassori SS, Réus GZ, Varela RB, Arent CO, Ribeiro KF, Bavaresco DV, Andersen ML et al (2013) Effects of sodium butyrate in animal models of mania and depression: implications as a new mood stabilizer. Behav Pharmacol 24(7):569–579. doi:10.1097/FBP.0b013e32836546fc
Joyce PR, Fergusson DM, Woollard G, Abbott RM, Horwood LJ, Upton J (1995) Urinary catecholamines and plasma hormones predict mood state in rapid cycling bipolar affective disorder. J Affect Disord 33(4):233–243. doi:10.1016/0165-0327(94)00094-P
Lee SY, Chen SL, Chen SH, Chu CH, Chang YH, Lin SH, Huang SY, Tzeng NS et al (2012) Interaction of the DRD3 and BDNF gene variants in subtyped bipolar disorder. Prog Neuro-Psychopharmacol Biol Psychiatry 39(2):382–387. doi:10.1016/j.pnpbp.2012.07.015
Meyendorff E, Lerer B, Moore NC, Bow J, Gershon S (1985) Methylphenidate infusion in euthymic bipolars: effect of carbamazepine pretreatment. Psychiatry Res 16(4):303–308. doi:10.1016/0165-1781(85)90121-0
Peet M, Peters S (1995) Drug-induced mania. Drug Saf 12(2):146–153. doi:10.2165/00002018-199512020-00007
Gould TJ, Keith RA, Bhat RV (2001) Differential sensitivity to lithium’s reversal of amphetamine-induced open-field activity in two inbred strains of mice. Behav Brain Res 118(1):95–105. doi:10.1016/S0166-4328(00)00318-1
Young JW, Henry BL, Geyer MA (2011) Predictive animal models of mania: hits, misses and future directions. Br J Pharmacol 164(4):1263–1284. doi:10.1111/j.1476-5381.2011.01318.x Mol Neurobiol. 2011 Jun;43(3):207-14. doi: 10.1007/s12035-011-8178-0
Arent CO, Valvassori SS, Fries GR, Stertz L, Ferreira CL, Lopes-Borges J, Mariot E, Varela RB et al (2011) Neuroanatomical profile of antimaniac effects of histone deacetylases inhibitors. Mol Neurobiol 43(3):207–214. doi:10.1007/s12035-011-8178-0
Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N et al (2008) Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol 11(8):1123–1134. doi:10.1017/S1461145708009024
Valvassori SS, Varela RB, Arent CO, Dal-Pont GC, Bobsin TS, Budni J, Reus GZ, Quevedo J (2014) Sodium butyrate functions as an antidepressant and improves cognition with enhanced neurotrophic expression in models of maternal deprivation and chronic mild stress. Curr Neurovasc Res 11(4):359–366. doi:10.2174/156720261166614082 9162158
Sulzer D, Rayport S (1990) Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron Dezembro 5(6):797–808. doi:10.1016/0896-6273(90)90339-H
Sharma RP, Grayson DR, Gavin DP (2008) Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophr Res 98(1–3):111–117
Yamamoto BK, Moszczynska A, Gudelsky GA (2010) Amphetamine toxicities: classical and emerging mechanisms. Ann N Y Acad Sci 1187:101–121. doi:10.1111/j.1749-6632.2009.05141.x
Andreazza AC, Kauer-Sant’Anna M, Frey BN, Stertz L, Zanotto C, Ribeiro L, Giasson K, Valvassori SS et al (2008) Effects of mood stabilizers on DNA damage in an animal model of mania. J Psychiatry Neurosci 33(6):516–524
da Rosa DD, Valvassori SS, Steckert AV, Arent CO, Ferreira CL, Lopes-Borges J, Varela RB, Mariot E et al (2012) Differences between dextroamphetamine and methamphetamine: behavioral changes and oxidative damage in brain of Wistar rats. J Neural Transm (Vienna) 119(1):31–38. doi:10.1007/s00702-011-0691-9
Northrop NA, Yamamoto BK (2012) Persistent neuroinflammatory effects of serial exposure to stress and methamphetamine on the blood-brain barrier. J NeuroImmune Pharmacol 7(4):951–968. doi:10.1007/s11481-012-9391-y
Andrzejczak D (2011) Epilepsy and pro-inflammatory cytokines. Immunomodulating properties of antiepileptic drugs. Neurol Neurochir Pol 45(3):275–285 . doi:10.1016/S0028-3843(14)60080-3
Pinheiro RM, de Lima MN, Portal BC, Busato SB, Falavigna L, Ferreira RD, Paz AC, de Aguiar BW et al (2015) Long-lasting recognition memory impairment and alterations in brain levels of cytokines and BDNF induced by maternal deprivation: effects of valproic acid and topiramate. J Neural Transm (Vienna) 122(5):709–719. doi:10.1007/s00702-014-1303-2
Jambalganiin U, Tsolmongyn B, Koide N, Odkhuu E, Naiki Y, Komatsu T, Yoshida T, Yokochi T (2014) A novel mechanism for inhibition of lipopolysaccharide-induced proinflammatory cytokine production by valproic acid. Int Immunopharmacol 20(1):181–187. doi:10.1016/j.intimp.2014.02.032
Säemann MD, Böhmig GA, Osterreicher CH, Burtscher H, Parolini O, Diakos C, Stöckl J, Hörl WH et al (2000) Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J 14(15):2380–2382. doi:10.1096/fj.00-0359fje
Berndt BE, Zhang M, Owyang SY, Cole TS, Wang TW, Luther J, Veniaminova NA, Merchant JL et al (2012) Butyrate increases IL-23 production by stimulated dendritic cells. Am J Physiol Gastrointest Liver Physiol 303(12):G1384–G1392. doi:10.1152/ajpgi.00540.2011
Huuskonen J, Suuronen T, Nuutinen T, Kyrylenko S, Salminen A (2004) Regulation of microglial inflammatory response by sodium butyrate and short-chain fatty acids. Br J Pharmacol 141(5):874–880. doi:10.1038/sj.bjp.0705682
Suuronen T, Huuskonen J, Pihlaja R, Kyrylenko S, Salminen A (2003) Regulation of microglial inflammatory response by histone deacetylase inhibitors. J Neurochem 87(2):407–416 2003Chen et al. 2007
Chen G, Manji HK, Hawver DB, Wright CB, Potter WZ (1994) Chronic sodium valproate selectively decreases protein kinase C alpha and epsilon in vitro. J Neurochem 63:2361–2364. doi:10.1046/j.1471-4159.1994.63062361.x
Zhang T, Xia M, Zhan Q, Zhou Q, Lu G, An F (2015) Sodium butyrate reduces organ injuries in mice with severe acute pancreatitis through inhibiting HMGB1 expression. Dig Dis Sci 60(7):1991–1999. doi:10.1007/s10620-015-3586-z
Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM (2007) Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther 321(3):892–901. doi:10.1124/jpet.107.120188
Nakamura C, Matsushita I, Kosaka E, Kondo T, Kimura T (2008) Anti-arthritic effects of combined treatment with histone deacetylase inhibitor and low-intensity ultrasound in the presence of microbubbles in human rheumatoid synovial cells. Rheumatology (Oxford) 47(4):418–424. doi:10.1093/rheumatology/ken003
Lührs H, Gerke T, Müller JG, Melcher R, Schauber J, Boxberge F, Scheppach W, Menzel T (2002) Butyrate inhibits NF-kappaB activation in lamina propria macrophages of patients with ulcerative colitis. Scand J Gastroenterol 37(4):458–466
Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1(6):a001651. doi:10.1101/cshperspect.a001651
Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y et al (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol 29(3):255–265. doi:10.1038/nbt.1759
Akimova T, Beier UH, Liu Y, Wang L, Hancock WW (2012) Histone/protein deacetylases and T-cell immune responses. Blood 119(11):2443–2451. doi:10.1182/sangue-2011-10-292003
Owens MJ, Nemerroff CB (2003) Pharmacology of valproate. Psychopharmacol Bull 37:17–24
Kurita M, Nishino S, Ohtomo K, Rai M, Shirakawa H, Mashiko H, Niwa S, Nakahata N (2007) Sodium valproate at therapeutic concentrations changes Ca2+ response accompanied with its weak inhibition of protein kinase C in human astrocytoma cells. Prog Neuro-Psychopharmacol Biol Psychiatry 31(3):600–604. doi:10.1016/j.pnpbp.2006.11.019
DiazGranados N, Zarate CA Jr (2008) A review of the preclinical and clinical evidence for protein kinase C as a target for drug development for bipolar disorder. Curr Psychiatry Rep 10(6):510–519
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Valvassori, S.S., Resende, W.R., Varela, R.B. et al. The Effects of Histone Deacetylase Inhibition on the Levels of Cerebral Cytokines in an Animal Model of Mania Induced by Dextroamphetamine. Mol Neurobiol 55, 1430–1439 (2018). https://doi.org/10.1007/s12035-017-0384-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0384-y