
Abstract
There is increasing interest in the possibility that mitochondrial impairment may play an important role in bipolar disorder (BD). The Krebs cycle is the central point of oxidative metabolism, providing carbon for biosynthesis and reducing agents for generation of ATP. Recently, studies have suggested that histone deacetylase (HDAC) inhibitors may have antimanic effects. The present study aims to investigate the effects of sodium butyrate (SB), a HDAC inhibitor, on Krebs cycle enzymes activity in the brain of rats subjected to an animal model of mania induced by d-amphetamine (d-AMPH). Wistar rats were first given d-AMPH or saline (Sal) for 14 days, and then, between days 8 and 14, rats were treated with SB or Sal. The citrate synthase (CS), succinate dehydrogenase (SDH), and malate dehydrogenase (MDH) were evaluated in the prefrontal cortex, hippocampus, and striatum of rats. The d-AMPH administration inhibited Krebs cycle enzymes activity in all analyzed brain structures and SB reversed d-AMPH-induced dysfunction analyzed in all brain regions. These findings suggest that Krebs cycle enzymes’ inhibition can be an important link for the mitochondrial dysfunction seen in BD and SB exerts protective effects against the d-AMPH-induced Krebs cycle enzymes’ dysfunction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bipolar disorder (BD) is a leading cause of morbidity and mortality, yet its underlying pathophysiology remains unclear (Zarate and Manji 2006). There is increasing interest in the possibility that mitochondrial impairment may play an important role in BD (Gigante et al. 2011), resulting in the decreased production of ATP, which can lead to neuronal dysfunction (Barnes and Weitzman 1986). Brains from BD patients are under oxidative stress, which typically accompanies mitochondrial dysfunction (Gigante et al. 2011; Andreazza et al. 2010). In addition, several components of the electron transport chain have been reported to be reduced in the brains of bipolar patients (Cataldo et al. 2010; Rollins et al. 2009).
The Krebs cycle, or citric acid cycle, is the central point of oxidative metabolism, providing carbon for biosynthesis and reducing agents for generation of ATP. Inactivation of any Krebs cycle step can alter mitochondrial ATP production (Blass and Brown 2000). In addition, alterations in the Krebs cycle would greatly alter the rate of brain metabolism and the production of free radicals (Lyubarev and Kurganov 1989; Velot et al. 1997). Some of the enzymes that form part of the Krebs cycle are citrate synthase (CS), malate dehydrogenase (MDH) and succinate dehydrogenase (SDH). These enzymes are implicated in some abnormalities in the central nervous system, such as BD, depression, and schizophrenia (Hroudova and Fisar 2010; Bubber et al. 2011; Feier et al. 2013).
Citrate synthase catalyzes the first step within the cycle, the condensation of acetyl-coenzyme A with oxaloacetate to form citrate; it is the only enzyme in the cycle that can catalyze the formation of a carbon–carbon bond (Wiegand and Remington 1986). MDH catalyzes the conversion of oxaloacetate and malate, utilizing the NAD/NADH coenzyme system. This reaction plays a key part in the malate/aspartate transport across the mitochondrial membrane, and in the Krebs cycle within the mitochondrial matrix (Minard and McAlister-Henn 1991). SDH or mitochondrial complex II, is a multimeric enzyme that is bound to the inner membrane of the mitochondria and has a dual role, it serves both as a critical step of the Krebs cycle and as a member of the respiratory chain that transfers electrons directly to the ubiquinone pool (Oyedotun and Lemire 2004; Kantorovich and Pacak 2010).
Valproic acid (VPA), a drug used for treatment and prophylaxis of BD, has been characterized as a histone deacetylases (HDAC) inhibitor (Göttlicher et al. 2001). HDAC inhibitors promote transcriptional activation by relaxing the DNA conformation. The HDAC inhibitors, including VPA, phenylbutyrate, sodium butyrate (SB), and trichostatin A, cause chromatin remodeling through histone hyperacetylation, increasing the expression of proteins (Phiel et al. 2001). Several studies have investigated the use of HDAC inhibitors as a treatment for a variety of disorders (Peedicayil 2012). In previous studies, our laboratory found that amphetamine decreased the activity of mitochondrial respiratory-chain complexes in the prefrontal cortex, hippocampus, striatum, and amygdala of rats, and VPA or SB were able to reverse and prevent this impairment (Moretti et al. 2011; Valvassori et al. 2010).
The effects of stimulants, such as amphetamine, on behavior have been widely used as an animal model of mania because it induces psychomotor agitation, which is commonly observed during mania. Also, locomotor activity is easily measured in rats (Berggren et al. 1978; Davies et al. 1974). Thus, we examined the effects of SB on activities in mitochondrial enzymes of the Krebs cycle in the prefrontal, hippocampus, and striatum from rats submitted to an animal model of mania induced by d-AMPH.
Experimental methods
In the present study, we have extended the investigation of the effects of SB on d-AMPH induced neurochemical alterations in an animal model of mania by measuring the activities of mitochondrial enzymes of the Krebs cycle in prefrontal, hippocampal, and striatal samples that were kept frozen at −80 °C from one of our previous experiments (Moretti et al. 2011). The detailed description of the experiments has been published elsewhere (Moretti et al. 2011); therefore, we summarize here the treatment regimens and describe the subsequent steps performed for the present investigation.
Animals
The subjects were adult male Wistar rats (weighing 250–350 g) obtained from our breeding colony. With food and water available ad libitum and they were maintained on a 12-h light/dark cycle (lights on at 7:00 a.m.) at a temperature of 22 ± 1 °C. All experimental procedures were performed in accordance with, and with the approval of the local ethics committee in the use of animals at the Universidade do Extremo Sul Catarinense.
Drugs and pharmacological procedures
Rats received intraperitoneal (ip) injection of either d-AMPH (2 mg kg−1) or Sal once a day for a period of 14 days. From the 8th to the 14th day (treatment for 7 days), d-AMPH and Sal-treated animals also received Sal (ip,twice a day) or SB (0.6 g kg−1 ip,twice a day), totaling four experimental groups of 12 animals per group: Sal + Sal, Sal + SB, d-AMPH + Sal, and d-AMPH + SB. No behavioral assessment was performed between days 1 and 14. On the 15th day of treatment, the animals received a single injection of d-AMPH or Sal, and after 2 h , they were killed by decapitation and prefrontal cortex, hippocampus, and striatum were manually dissected on ice, rapidly frozen on dry ice and stored at −70 °C until assayed.
The range of doses of SB employed in this work was chosen based on our previous study (Moretti et al. 2011).
Activities of enzymes of Krebs cycle
Citrate synthase activity
Citrate synthase activity was assayed according to the method described by Shepherd and Garland (1969). The reaction mixture contained 100 mM Tris, pH 8.0, 100 mM acetyl CoA, 100 mM 5,5′-di-thiobis-(2-nitrobenzoic acid), 0.1 % triton X-100, and 2–4 μg supernatant protein and was initiated with 100 μM oxaloacetate and monitored at 412 nm for 3 min at 25 °C.
Malate dehydrogenase activity
Malate dehydrogenase was measured as described by Kitto (1969). Aliquots (20 mg protein) were transferred into a medium containing 10 mM rotenone, 0.2 % Triton X-100, 0.15 mM NADH, and 100 mM potassium phosphate buffer, pH 7.4, at 37 °C. The reaction was started by the addition of 0.33 mM oxaloacetate. Absorbance was monitored as described above.
Succinate dehydrogenase activity
Succinate dehydrogenase activity was determined according to the method of Fischer et al. (1985), and measured by following the decrease in absorbance due to the reduction of 2,6-di-chloro-indophenol (2,6-DCIP) at 600 nm with 700 nm as a reference wavelength (ε = 19.1 mM−1 cm−1) in the presence of phenazine methasulphate (PMS). The reaction mixture consisting of 40 mM potassium phosphate, pH 7.4, 16 mM succinate and 8 μM 2,6-DCIP was pre-incubated with 40–80 μg homogenate protein at 30 °C for 20 min. Subsequently, 4 mM sodium azide, 7 μM rotenone and 40 μM 2,6-DCIP were added and the reaction was initiated by the addition of 1 mM PMS and was monitored for 5 min.
Statistical analysis
Data were analyzed by two-way analysis of variance followed by the Tukey’s test, when F was significant and are expressed as mean ± SEM. All analyses were performed using the Statistical Package for the Social Science (SPSS; version 16.0) software.
Results
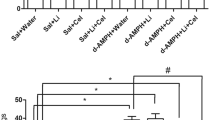
As depicted in Fig. 1a, CS activity was significantly decreased in all brain structures evaluated in d-AMPH administered rats, as compared to the control group. This reduction was significantly reversed by the treatment with SB.

Activities of enzymes of Krebs cycle. a Citrate synthase activity: in the prefrontal, hippocampus, and striatum. b Malate dehydrogenase activity: in the prefrontal, hippocampus and striatum. c Succinate dehydrogenase activity: in the prefrontal, hippocampus, and striatum. Bars represent mean ± error; *p < 0.05 vs. Sal + Sal group, according to two-way ANOVA followed by the Tukey’s test. # p < 0.05 vs. d-AMPH + Sal, according to two-way ANOVA followed by the Tukey’s test
Data from the two-way ANOVA for d-AMPH administration [prefrontal cortex: F(1.12) = 13.13, p = 0.03; hippocampus: F(1.12) = 2.72, p = 0.12; striatum: F(1.12) = 12.32, p = 0.04], treatment [prefrontal cortex: F(1.12) = 29.95, p < 0.001; hippocampus: F(1.12) = 22.35, p < 0.001; striatum: F(1.12) = 30.62, p < 0.001], and d-AMPH administration × treatment interaction [prefrontal cortex: F(1.12) = 14.95, p = 0.02; hippocampus: F(1.12) = 13.68, p = 0.03; striatum: F(1.12) = 8.89, p = 0.01].
As can be observed in Fig. 1b, MDH activity was significantly decreased in all brain structures evaluated of d-AMPH administered rats as compared to the control group. This decrease induced by d-AMPH administration was significantly reversed by the treatment with SB.
Data from the two-way ANOVA for d-AMPH administration [prefrontal cortex: F(1.12) = 26.11, p < 0.001; hippocampus: F(1.12) = 85.61, p < 0.001; striatum: F(1.12) = 16.93, p = 0.001], treatment [prefrontal cortex: F(1.12) = 16.82, p = 0.001; hippocampus: F(1.12) = 25.66, p < 0.001; striatum: F(1.12) = 14.49, p = 0.002], and d-AMPH administration × treatment interaction [prefrontal cortex: F(1.12) = 19.23, p < 0.001; hippocampus: F(1.12) = 73.47, p < 0.001; striatum: F(1.12) = 3.75, p = 0.07].
As shown in Fig. 1c, in the prefrontal, hippocampus, and striatum, d-AMPH administered rats displayed lower SDH activity than the control group. This reduction was significantly reversed by repeated treatment with SB.
Data from the two-way ANOVA for d-AMPH administration [prefrontal cortex: F(1.12) = 11.98, p = 0.004; hippocampus: F(1.12) = 2.08, p = 0.17; striatum: F(1.12) = 33.7, p < 0.001], treatment [prefrontal cortex: F(1.12) = 7, p = 0.02; hippocampus: F(1.12) = 15.39, p = 0.002; striatum: F(1.12) = 26.47, p < 0.001], and d-AMPH administration × treatment interaction [prefrontal cortex: F(1.12) = 12.6, p = 0.03; hippocampus: F(1.12) = 20.82, p < 0.001; striatum: F(1.12) = 14.75, p = 0.002].
Discussion
Mitochondrial dysfunction and oxidative stress may underlie the pathophysiology of BD (Gigante et al. 2011; Andreazza et al. 2010). Oxidative stress can lead to neurotransmitter abnormalities, DNA damage, protein inactivation, altered gene expression, and apoptotic events. Therefore, the link between oxidative mechanisms and neuronal plasticity may be an explanation to the pathophysiology of BD (Ben-Shachar 2002).
The effects of stimulants, such as amphetamine, on behavior have been widely used as an animal model of mania because it induces psychomotor agitation, which is commonly observed during mania. Also, locomotor activity is easily measured in rats (Berggren et al. 1978; Davies et al. 1974). Studies have suggested that alteration in the dopaminergic system is a predominant etiological factor for BD (Berk et al. 2007; Valvassori et al. 2010). Studies with this animal model show that manic-like hyperactivity induced by d-AMPH is associated with severe brain damage by an increased formation of lipid and protein oxidation products, and a decrease in the BDNF levels (Frey et al. 2006a, b).
Alterations in the Krebs cycle would profoundly alter the rate of brain metabolism and the production of free radicals. Inactivation of any step can disrupt mitochondrial bioenergetics (Blass and Brown 2000). In the present study, we found that the d-AMPH administration induced a marked reduction in the Krebs cycle enzymes (CS, MDH, and SDH) in all brain structures evaluated. In agreement with these findings, previous studies described that amphetamine administration inhibited the activity of Krebs cycle enzymes (CS, MDH, and SDH) and mitochondrial respiratory chain complexes (I, II, III, and IV) in the hippocampus, striatum, and prefrontal cortex of rats, and that mood stabilizers, lithium and VPA, exerted protective effects against the amphetamine-induced mitochondrial dysfunction (Feier et al. 2013; Valvassori et al. 2010). From this, we suggest that the inhibition of the Krebs cycle induced by amphetamine can alter the function of the mitochondrial respiratory chain complexes, and consequently, the rate of brain metabolism. Considering that mood stabilizers modulate Krebs cycle enzymes we can suggest that it is an important target to study in BD.
The Krebs cycle enzymes participate in energy production, neurotransmitter metabolism and metabolic interaction between mitochondria and cytoplasm. Previous studies have demonstrated that reduction in the activities of these enzymes can alter brain function, increasing the release of dopamine and decreasing the release of acetylcholine (Bubber et al. 2011; Gibson et al. 1989; Blass et al. 2002; Rose and O’Connell 1967). Since dopamine regulates and organizes numerous important behavioral processes including motor activity (Schultz 2007), we suggest that the Krebs cycle enzymes’ activity decrease may contribute for behavioral alterations induced by d-AMPH, such as hyperactivity. On the other hand, several studies have reported that amphetamine inhibits oxidative phosphorylation and the Krebs cycle, suggesting that it might be due to mitochondrial damage induced by dopaminergic toxicity (Sailasuta et al. 2010; Shima et al. 2011; Feier et al. 2013). It is well described in the literature that chronic amphetamine administration leads to long-term deficits in dopaminergic systems in the brain, resulting from dopamine generated reactive oxygen species (ROS) (LaVoie and Hastings 1999). Monoamine oxidase is located in the outer membrane of the mitochondria and could be a significant source of ROS production, mainly in dopaminergic neurons (Adam-Vizi 2005). Mitochondria are the main source of ROS, which are produced in the complexes of the electron transport chain (Mattiasson et al. 2003). Moreover, a shift in the antioxidant/pro-oxidant balance toward oxidative stress may inhibit Krebs cycle and electron transport chain complexes, leading to decreases in ATP production and cellular dysfunction (Calabrese et al. 2001).
Another significant finding in this study is that SB reverses the AMPH’s effects on Krebs cycle enzymes’ (CS, MDH, and SDH) activity. In a previous study, it was shown that SB was able to reverse and prevent the hyperactivity and impairment in the mitochondrial chain complexes induced by d-AMPH, suggesting that the mechanism of action in SB involves the improvement of mitochondrial function, parallel with behavioral changes (Moretti et al. 2011). Chen et al. (2010) have shown that the tubacin, a specific HDAC6 inhibitor, dramatically enhanced mitochondrial movement in hippocampal neurons, suggesting that HDAC plays an important role in the modulation of mitochondrial transport. In addition, a previous study showed that SB administration increases muscle mitochondrial respiration in mice (Gao et al. 2009), suggesting a relation between HDAC inhibition and changes in energy metabolism. A previous study has demonstrated that the activities of key enzymes that control the direction of glycolysis versus gluconeogenesis and the branching between citrate cycle and glyoxylate bypass were all regulated by acetylation. This modulation is mainly controlled by a pair of lysine acetyltransferase and deacetylase, whose expressions are coordinated with growth status (Wang et al. 2010). Therefore, we suggest that the protective effects of SB can be explained by the fact that this drug inhibits HDAC, which may be increasing the expression of the enzymes of the Krebs cycle, thereby providing substrates for the cycle.
In addition, Wu et al. (2008) have demonstrated that HDAC inhibitors, sodium SB and trichostatin A, up-regulate GDNF and BDNF expression in astrocytes, and protect DA neurons. Evidence suggests that oxidative stress may be increased in conditions where BDNF is described to be decreased in BD (Andreazza et al. 2007; Kapczinski et al. 2008). The oxidative stress may inhibit mitochondrial electron transport chain complexes, leading to a decrease in ATP production and cellular dysfunction (Calabrese et al. 2001). Furthermore, preclinical studies have shown that mood stabilizers, lithium and valproate, increase the brain-derived neurotrophic factor (BDNF) levels, and protect the rat brain against oxidative damage and mitochondrial impairment (Frey et al. 2006a, b). In the culture of neurons and glia, SB and other HDAC inhibitors upregulate gene transcription for the BDNF (Yasuda et al. 2009; Wu et al. 2008). In addition, treatment with SB increases brain protein levels of BDNF in rats (Kim et al. 2009). Evidence indicates that neurotrophic factors, such as BDNF, prevent neuronal damage caused by oxidative stress, promoting neuronal protection and the regulation of synaptic function in the central nervous system, via stimulating intracellular signaling cascades (Huang and Reichardt 2003; Numakawa et al. 2010).
In conclusion, the present study shows the inhibition of Krebs cycle enzymes in an animal model of mania induced by d-AMPH. Thus, we suggest an interaction between Krebs cycle enzymes’ inhibition and mitochondrial dysfunction induced by d-AMPH. We also have demonstrated that SB can reverse d-AMPH-induced mitochondrial dysfunction. These findings will enhance the understanding in the pathology of disorder, and suggest that HDAC can be a possible new therapeutic strategy to improve the mitochondrial deficit seen in BD.
References
Adam-Vizi V (2005) Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal 7:1140–1149
Andreazza AC, Cassini C, Rosa AR, Leite MC, De Almeida LM, Nardin P, Cunha AB, Cereser KM, Santin A, Gottfried C, Salvador M, Kapczinski F, Gonçalves CA (2007) Serum S100B and antioxidant enzymes in bipolar patients. J Psychiatr Res 41:523–529
Andreazza AC, Shao L, Wang JF, Young LT (2010) Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch General Psychiatry 67:360–368
Barnes SJ, Weitzman PD (1986) Organization of citric acid cycle enzymes into a multienzyme cluster. FEBS Lett 201:267–270
Ben-Shachar D (2002) Mitochondrial dysfunction in schizophrenia: a possible linkage to dopamine. J Neurochem 83:1241–1251
Berggren U, Tallstedt L, Ahlenius S, Engel J (1978) The effect of lithium on amphetamine-induced locomotor stimulation. Psychopharmacology 59:41–45
Berk M, Dodd S, Kauer-Sant’anna M, Malhi GS, Bourin M, Kapczinski F, Norman T (2007) Dopamine dysregulation syndrome: implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr Scand Suppl 434:41–49
Blass JP, Brown AM (2000) Lower activity of Krebs cycle enzymes than of electron transport in human brain: disease implications. Neurobiol Aging 21:81
Blass JP, Gibson GE, Hoyer S (2002) The role of the metabolic lesion in Alzheimer’s disease. J Alzheimers Dis. 4:225–232
Bubber P, Hartounian V, Gibson GE, Blass JP (2011) Abnormalities in the tricarboxylic acid (TCA) cycle in the brains of schizophrenia patients. Eur Neuropsychopharmacol 21:254–260
Calabrese V, Scapagnini G, Giuffrida Stella AM, Bates TE, Clark JB (2001) Mitochondrial involvement in brain function and dysfunction: relevance to aging, neurodegenerative disorders and longevity. Neurochem Res 26:739–764
Cataldo AM, McPhie DL, Lange NT, Punzell S, Elmiligy S, Ye NZ, Froimowitz MP, Hassinger LC, Menesale EB, Sargent LW, Logan DJ, Carpenter AE, Cohen BM (2010) Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol 177:575–578
Chen S, Owens GC, Makarenkova H, Edelman DB (2010) HDAC6 regulates mitochondrial transport in hippocampal neurons. Plos One 26:10848
Davies JA, Jackson B, Redfern PH (1974) The effect of amantadine, L-dopa, (plus)-amphetamine and apomorphine on the acquisition of the conditioned avoidance response. Neuropharmacology 13:199–204
Feier G, Valvassori SS, Varela RB, Resende WR, Bavaresco DV, Morais MO, Scaini G, Andersen ML, Streck EL, Quevedo J (2013) Lithium and valproate modulate energy metabolism in an animal model of mania induced by methamphetamine. Pharmacol Biochem Behav 103:589–596
Fischer JC, Ruitenbeek W, Berden JA, Trijbels JM, Veerkamp JH, Stadhouders AM, Sengers RC, Janssen AJ (1985) Differential investigation of the capacity of succinate oxidation in human skeletal muscle. Clin Chim Acta 153:23–26
Frey BN, Valvassori SS, Réus GZ, Martins MR, Petrolino FC, Bardini K, Dal-Pizzol F, Kapczinski F, Quevedo J (2006a) Effects os lithium and valproato on amphetamine-induced oxidative stress generation in an animal model of mania. J Psychiatry Neurosci 31:326–332
Frey BN, Andreazza AC, Cereser KM, Martins MR, Valvassori SS, Reus GZ, Quevedo J, Kapczinski F (2006b) Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci 79:281–286
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M (2009) Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58:1509–1517
Gibson GE, Manger T, Toral-Barza L, Freeman G (1989) Cytosolic-free calcium and neurotransmitter release with decreased availability of glucose or oxygen. Neurochem Res 14:437–443
Gigante AD, Andreazza AC, Lafer B, Yatham LN, Beasley CL, Young LT (2011) Decreased mRNA expression of uncoupling protein 2, a mitochondrial proton transporter, in post-mortem prefrontal cortex from patients with bipolar disorder and schizophrenia. Neurosci Lett 505:47–51
Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20:6969–6978
Hroudova J, Fisar Z (2010) Activities of respiratory chain complexes and citrate synthase influenced by pharmacologically different antidepressants and mood stabilizers. Neuro Endocrinol Lett 31:336–342
Huang EJ, Reichardt LF (2003) Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72:609–642
Kantorovich V, Pacak K (2010) Pheochromocytoma and paraganglioma. Prog Brain Res 182:343–373
Kapczinski F, Frey BN, Andreazza AC, Kauer-Sant’Anna M, Cunha AB, Post RM (2008) Increased oxidative stress as a mechanism for decreased BDNF levels in acute manic episodes. Rev Bras Psiquiatr 30:243–245
Kim HJ, Leeds P, Chuang DM (2009) The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J Neurochem 110:1226–1240
Kitto GB (1969) Intra- and extramitochondrial malate dehydrogenases from chicken and tuna heart. Methods Enzymol 23:106–116
LaVoie MJ, Hastings TG (1999) Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci 19:1484–1491
Lyubarev AE, Kurganov BI (1989) Supramolecular organization of tricarboxylic acid cycle enzymes. Biosystems 22:91–102
Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T (2003) Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med 9:1062–1068
Minard KI, McAlister-Henn L (1991) Isolation, nucleotide sequence analysis, and disruption of the MDH2 gene from Saccharomyces cerevisiae: evidence for three isozymes of yeast malate dehydrogenase. Mol Cell Biol 11:370–380
Moretti M, Valvassori SS, Varela RB, Ferreira CL, Rochi N, Benedet J, Scaini G, Kapczinski F, Streck EL, Zugno AI, Quevedo J (2011) Behavioral and neurochemical effects of sodium butyrate in an animal model of mania. Behav Pharmacol 22:766–772
Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H (2010) BDNF function and intracellular signaling in neurons. Histol Histopathol 25:237–258
Oyedotun KS, Lemire BD (2004) The quaternary structure of the Saccharomyces cerevisiae succinate dehydrogenase. Homology modeling, cofactor docking, and molecular dynamics simulation studies. J Biol Chem 279:9424–9431
Peedicayil J (2012) Role of epigenetics in pharmacotherapy, psychotherapy and nutritional management of mental disorders. J Clin Pharm Ther 37:499–501
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS (2001) Histone deacetylase is a direct target os valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276:6734–6741
Rollins B, Martin MV, Sequeira PA, Moon EA, Morgan LZ, Watson SJ, Schatzberg A, Akil H, Myers RM, Jones EG, Wallace DC, Bunney WE, Vawter MP (2009) Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. Plos One 4:4913
Rose IA, O’Connell EL (1967) Mechanism of aconitase action I. The hydrogen transfer reaction. J Biol Chem 242:1870–1879
Sailasuta N, Abulseoud O, Harris KC, Ross BD (2010) Glial dysfunction in abstinent methamphetamine abusers. J Cereb Blood Flow Metab 30(5):950–960
Schultz W (2007) Behavioral dopamine signals. Trends Neurosci 30:203–210
Shepherd D, Garland PB (1969) The kinetic properties of citrate synthase from rat liver mitochondria. Biochem J 114:597–610
Shima N, Miyawaki I, Bando K, Horie H, Zaitsu K, Katagi M, Bamba T, Tsuchihashi H, Fukusaki E (2011) Influences of methamphetamine-induced acute intoxication on urinary and plasma metabolic profiles in the rat. Toxicology 287:29–37
Valvassori SS, Rezin GT, Ferreira CL, Moretti M, Gonçalves CL, Cardoso MR, Streck EL, Kapczinski F, Quevedo J (2010) Effects of mood stabilizers on mitochondrial respiratory chain activity in brain of rats treated with d-amphetamine. J Psychiatr Res 44:903–909
Velot C, Mixon MB, Teige M, Srere PA (1997) Model of a quinary structure between Krebs TCA cycle enzymes: a model for the metabolon. Biochemistry 36:14271–14276
Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327:1004–1007
Wiegand G, Remington SJ (1986) Citrate synthase: structure, control, and mechanism. Annu Rev Biophys Biophys Chem 15:97–117
Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS (2008) Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol 11:1123–1134
Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM (2009) The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry 14:51–59
Zarate CA Jr, Manji HK (2006) Cellular plasticity cascades targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry 59:1006–1020
Acknowledgments
We thank CNPq, FAPESC, CAPES and UNESC for financial support.
Conflict of interest
None of the authors or funding sources has conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Valvassori, S.S., Calixto, K.V., Budni, J. et al. Sodium butyrate reverses the inhibition of Krebs cycle enzymes induced by amphetamine in the rat brain. J Neural Transm 120, 1737–1742 (2013). https://doi.org/10.1007/s00702-013-1056-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-013-1056-3