
Abstract
Cranial irradiation-induced inflammation plays a critical role in the initiation and progression of radiation-induced brain injury (RIBI). Anti-inflammation treatment may provide therapeutic benefits. Corilagin (beta-1-O-galloyl-3, 6-(R)-hexahydroxydiphenoyl-D-glucose, C27H22O18) was a novel member of the tannin family with anti-inflammatory properties and is isolated from some medicinal plants, such as Phyllanthus amarus and Caesalpinia coriaria. In this study, the effect of Corilagin on RIBI was investigated and the underlying mechanisms were explored. Spatial learning and memory ability of mice were investigated by the Morris water maze test. Evans blue leakage and electron microscopy were used to assess the integrity of blood-brain barrier (BBB). The mRNA and protein expressions of inflammatory cytokines, TNF-α and IL-1β, were measured by using real-time PCR and Western blotting. The activation of microglial cells and expression of TNF-α were examined by immunofluorescence staining. Phosphorylated signal transducers and activators of transcription 3 (p-STAT3) and IκBα, and the translocation of p65 from cytoplasm to nucleus were detected by using Western blotting. Morris water maze test showed that Corilagin ameliorated the neurocognitive deficits in RIBI mice. Evans blue leakage and electron microscopy exhibited that Corilagin partially protected the BBB integrity from cranial irradiation-caused damage; immunofluorescence staining showed that Corilagin could inhibit microglial activation and TNF-α expression. Real-time PCR and Western blotting revealed that Corilagin downregulated the expression of TNF-α and IL-1β and inhibited the irradiation-induced activation of NF-κB pathways by upregulating p-STAT3 expression. In conclusion, Corilagin could attenuate RIBI through inhibiting microglial activation and the expressions of inflammatory cytokines. Corilagin might inhibit the activation of NF-κB pathway in a STAT3-associated manner, thereby downregulating the inflammatory cytokine expressions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past decades, the incidence of primary and secondary tumors of the brain, head and neck has been on rise, and cranial radiation therapy (CRT) has been extensively used for the treatment of the tumors [1]. Nonetheless, CRT is associated with a common and severe side effect: radiation-induced brain injury (RIBI). About 50 % of survivors may develop late-onset irradiation-induced injury of normal brain tissues resulting from the damage of adjacent brain tissues [2–5]. As RIBI can substantially affect the quality of life of the patients, it is imperative to find an effective anti-RIBI therapy. Many preclinical studies have demonstrated that inflammatory responses tend to occur after CRT [6–8], suggesting that inflammation responses might contribute to RIBI. Our previous study found that 10-Gy CRT could induce RIBI in developing mice, as verified by water maze test. And we also found that the pro-inflammatory cytokine was released and microglial cells were activated in the brain tissues of rats with RIBI, indicating that the neuro-inflammation plays a key role in the development of RIBI [9].
Microglial cells are believed to be a key mediator in neuro-inflammation [10–12]. In the resting state, microglial cells exhibit a quiescent phenotype, which forms the basis of normal neurogenesis [13]. A recent study also found that in vivo brain irradiation triggered a conspicuous increase in the number of activated microglial cells, resulting in a concomitant decrease in neurogenesis in the hippocampus and deficits of spatial memory retention [9]. Moreover, in vitro studies also showed that irradiation could induce microglial activation and release of a variety of pro-inflammatory cytokines, including interleukin-1β (IL-1β), IL-6, tumor necrosis factor α (TNF-α) and COX-2 [14, 15]. These findings suggest that irradiation-induced microglial activation may play an important part in RIBI.
Our previous study exhibited that irradiation-induced TNF-α and IL-1β release resulted from the activation of NF-κB pathway, a critical transcription factor involved in inflammatory processes [9]. Moreover, it was reported that, in some cells, such as microglial cells, the transcriptional regulation of NF-κB fine-tuned the expression of downstream target genes in neuro-inflammatory response and the tuning required the antagonistic action of signal transducers and activators of transcription 3 (STAT3), another key regulator of inflammation. In neuro-inflammation response, the phosphorylation of STAT3 was decreased and its antagonizing effect on NF-κB pathway diminished, thereby activating NF-κB pathway [16, 17].
The structure of Corilagin (beta-1-O-galloyl-3, 6-(R)-hexahydroxydiphenoyl-D-glucose, C27H22O18), being given in Fig. 1, is a member of the tannin family and is isolated from some medicinal plants, such as Phyllanthus amarus and Caesalpinia coriaria [18, 19]. The medicinal plants, Phyllanthus amarus and Caesalpinia coriaria, were clinically shown to possess antioxidative, thrombolytic, and antihypertensive abilities [20, 21]. Corilagin has antioxidative, antiatherogenic, and antihypertensive effects, as exhibited by experiments with different animal models [18, 19, 22, 23]. A preliminary in vitro study suggested that Corilagin exerted its anti-inflammatory action by inhibiting the activation of NF-κB pathway [24, 25]. Post-irradiation microglial activation and pro-inflammatory cytokine release are believed to play key roles in RIBI [9]. In this study, we tried to clarify whether Corilagin administration could attenuate RIBI and explored its possible mechanism.

The structure of Corilagin
Materials and Methods
Animals
Eight-week-old C57BL/6J female mice were obtained from the Tongji Medical College, Wuhan, China. The mice weighed about 25 g on average and were housed, with four to five animals per cage, in laminar flow hoods and pathogen-free rooms to minimize the risk of infection. All animal experimentations were approved by the Institutional Animal Care and Use Committee of Tongji Medical College, Wuhan, China. Effort was made to reduce the number of animals used and mitigate their suffering in the experiment.
Cell Cultures
The mouse microglial cell line, BV-2, was maintained in the Laboratory Center of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology. Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 culture medium supplemented with 10 % fetal bovine serum (FBS) at 37 °C in a 5 % CO2-humidified incubator. Cells at the logarithmic phase were used in the experiment.
Radiation Schedule
BV-2 cells were irradiated at a single dose of 16-Gy X-ray. All irradiations were performed at room temperature, and control cells received sham irradiation. After irradiation, the cells were cultured in a CO2 incubator and maintained at 37 °C in 5 % CO2/95 % air for 3, 6, 12, and 24 h post-irradiation.
The mice were anesthetized by intraperitoneal injection of 50 mg/kg tribromoethanol (Sigma, Stockholm, Sweden), and then subjected to 10-Gy CRT as described previously [26] with minor modifications. Radiation was delivered by using a 6-MV X-ray (SIEMENS, Germany). The dose to the midplane of the brain was administered in a single fraction via a posterior field. A plastic jig was utilized to mount the mice. Radiation was given at a dose rate of 2.0 Gy/min, a gantry angle of 180°, a source-surface distance (SSD) of 100 cm, with the radiation field size being 3 cm × 30 cm. Tissue-equivalent material (1 cm in thickness) was placed under the head of each animal to establish electronic equilibrium and to ensure that the prescribed dose was delivered uniformly.
Treated animals were analyzed 3, 6, 24, 48, and 72 h and 1, 2, 4, 8, 16, and 24 weeks after irradiation. Each in vivo experiment was conducted in triplicate, and three mice were examined at each time point. Sham-irradiated control animals received the same treatment.
Animals and Cell Treatments
Corilagin standard substance (purity 99 %) was obtained from the China National Institute for the Control of Pharmaceutical and Biological Products (Fig. 1). Corilagin was administered in vivo by intraperitoneal injection once a day at doses of 30 mg/kg (body weight) or 15 mg/kg for 2 weeks. Mice were randomly divided into six groups: group 1: control group, or control, in which animals did not receive any treatment; group 2: Corilagin-alone group (Cori-alone), in which the animals were only given the intraperitoneal injection of Corilagin at 30 mL/kg/day for 2 weeks; group 3: RT-alone group (RT-alone), in which the animals were given normal saline for 2 weeks after 10-Gy CRT; group 4: RT + Corilagin (15 mg/kg/day) group (RT + L-Cori), in which the animals were intraperitoneally given Corilagin at 15 mL/kg/day for 2 weeks after 10-Gy CRT; group 5: RT + Corilagin (30 mg/kg) group (RT + H-Cori), in which the animals received Corilagin at 30 mL/kg/day, by intraperitoneal injection, for 2 weeks after 10-Gy CRT; group 6: RT + dexamethasone (5 mg/kg) group (RT + Dex), the animals were treated with dexamethasone at 5 mg/kg/d, by intraperitoneal injection, for 2 weeks after 10-Gy CRT.
For in vitro researches, BV-2 cells were divided into control group, RT-alone group, RT + Corilagin group, and RT + PS1145 (a novel specific IKK inhibitor) group. BV-2 cells were pretreated with culture medium, Corilagin and PS1145, respectively, for 12 h, and then, the supernatant was replaced. The cells were irradiated under the aforementioned conditions.
MTT Assay
The cytotoxicity of Corilagin and PS1145 was detected by using MTT assay. Cells were plated into a 96-well plate (1 × 105/well), each well containing 100 μL of medium. Upon overnight incubation, the medium from each well was discarded and replaced with fresh medium containing Corilagin at concentrations ranging from 0.125 to 80 μg/mL, and PS1145 at concentrations from 0.01 to 20 μmol/L. BV-2 cells treated with medium alone were used as controls. After treatment with PS1145 or Corilagin for 12 h, 20 μL MTT (5 mg/mL, pH 4.7) was added to each well. After 4 h, the supernatant was removed, 100 μL/well DMSO was added, and samples were shaken for 15 min. The optical density (OD) at 570 nm was measured on a microplate reader (Bio-Rad, Richmond, CA, USA) against blank wells (containing no cells). All experiments were performed in triplicate.
General Observation and Body Weight
After the treatments, the mice were housed under the identical experimental conditions to observe their feeding and drinking behaviors, limb movement, and the local skin reaction to the irradiation, and their body weight was recorded at each time point.
Morris Water Maze
To assess the cognitive change after CRT, the Morris water maze task was conducted, as described previously, 6 weeks after CRT [27]. The test was done in a circular black pool (width 127 cm, height 60 cm) with a black platform. The pool was filled with milk diluted with water at room temperature (21.0 ± 1 °C). The platform was set approximately 1.5 cm below the water surface.
The mice were placed into the pool at four possible start locations facing the wall of the pool, and a camera was simultaneously activated. Each mouse was allowed up to 60 s to locate the platform. The trial was terminated when the mouse found the platform within 60 s. If a mouse failed to locate the platform within 60 s, it was guided to locate the platform by a researcher and allowed to stay for 2 to 3 s. Each mouse was conditioned three times per day for 2 days to permit them to adapt to the pool environment (visible platform training), and then tested three times per day for 5 days to find the hidden platform (hidden platform training). The latency (the time taken to find the platform in the water), distance, and swim speed were recorded by employing an automated video tracking software package (Noldus™ EthoVision 2.3.19).
For data analysis, the pool was divided into four quadrants. During the visible platform training, the platform was moved to a different quadrant for each session. During the hidden platform training, the platform location was identical for each mouse (in the center of the target quadrant). Behaviors of the mice were tracked by using Ethovision 3.0 and analyzed for escape latency. In each trial, the average latency time for each mouse was calculated and recorded.
Evans Blue Leakage and Electron Microscopic Assessment of BBB Integrity
The ultrastructural changes were examined by HRP assay, as described previously with minor modifications, 48 h after CRT [28]. After anesthesia, animals received an intravenous injection of horseradish peroxidase (HRP; type II, Sigma Chemical Co., St. Louis, MO; 200 mg/kg body weight in 0.2 mL saline) through the catheter to acquire electron microscopic evidence of BBB permeability. HRP was allowed to circulate for 30 min, and then, animals were perfused transcardially with saline (50 mL), and then with 0.1 M sodium phosphate buffer (pH 7.4) containing 4 % paraformaldehyde and 2.5 % glutaraldehyde. Afterward, the brains were post-fixed with 2 % osmium in phosphate buffer. Samples were dehydrated through a graded series of ethanol and embedded. Ultra-thin sections (60 nm) were examined under a transmission electron microscope (FEI Tecnai G2 20 TWIN, USA).
Then, 48 h after the treatment, the functional integrity of the BBB was examined by using Evans blue dye extravasation in accordance with a previously reported technique [7] with some modifications, and the structural integrity of the BBB was assessed by electron microscopy. Three milliliters per kilogram of 2 % Evans blue dye (Sigma-Aldrich solution in saline) was administered via the femoral vein into the anesthetized mice 48 h after CRT. A successful injection was indicated by conjunctivae and limbs turning blue several seconds after the injection. The mice were anesthetized and the brains were transcardially perfused with 200 mL of saline through the left ventricle until perfusion fluid from the right atrium turned colorless. After decapitation, the brains were harvested, weighed, and then incubated in formamide solution at 50 °C for 72 h. The optical attenuation of the Evans blue formamide solution was spectrophotometrically determined at the excitation (emission) wavelength of 635 nm, and BBB permeability was expressed as microgram of Evans blue per milligram of brain tissue (μg/mg).
Brain Histology and Immunohistochemical Staining
The brain sections were stained with hematoxylin-eosin (HE) for pathological examination of post-CRT changes. And, after dewaxing in xylene and rehydration in graded alcohols, tissue sections were boiled in citrate buffer. The tissue sections were preincubated with H2O2 and blocked with rabbit or goat serum (DAKO Retrieval puffer, Glostrup, Denmark). Afterward, sections were incubated with rabbit polyclonal anti-caspase-3 (1:300, proteintech, Chicago, IL, USA), and then with goat-anti-rabbit antibody (1:200, Invitrogen, Carlsbad, USA). Sections were labeled by avidin-biotinperoxidase complex (Dako, Glostrup, Denmark), which was followed by diaminobenzidine development (Sigma, USA). Finally, sections were counter-stained with hematoxylin and mounted in Entellan (Merck, Darmstadt, Germany).
Isolation of RNA and Real-Time Quantitative RT-PCR
RNA extraction, cDNA synthesis, and real-time RT-PCR were performed as previously described [29]. Total RNA was isolated from the brain tissues of mice in different groups by using RNase Mini Kit (Qiagen, Valencia, CA, USA) according to instructions. Primer sequences were designed by using Beacon Designer software package (Bio-Rad). The primers are shown as follows: TNF-α sense 5′-AGG CGG TGC CTA TGT CTCA-3′ and anti-sense 5′-GAG GCC ATT TGG GAA CTT CT-3′; IL-1β sense 5′-GAA ATG CCA CCT TTT GAC AGTG-3′ and anti-sense 5′-CTG GAT GCT CTC ATC AGG ACA-3′; GAPDH sense 5′-TCA CCA CCA TGG AGA AGGC-3′ and anti-sense 5′-GCT AAG CAG TTG GTG GTG CA-3′. All primers were synthesized by Invitrogen (Groningen, Netherlands). Real-time PCR for cDNA analysis was conducted at 60 to 95 °C for 45 cycles on a Sequence Detection System (ABI Prism 7000, Applied Biosystems, Darmstadt, Germany) by following instructions and using SYBR Green Reaction Master Mix (TaKaRa Biotechnology Co. Ltd., Dalian, China). For each sample, GAPDH served as the housekeeping gene. Fold-change expression was calculated from the threshold cycle (Ct) values. For calculation of relative changes, gene expression measured in sham-irradiated tissues was taken as baseline value.
Western Blotting
Total proteins were extracted from the brain tissues and BV-2 cells by using a protein extraction kit (Pierce Biotechnology Inc., IL, USA) in accordance with the manufacturer’s protocol. In addition, cytoplasm and nuclear protein were also extracted respectively. Protein extracts were first dissolved in 15 % sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE), and then transferred to a nitrocellulose membrane at 150 mA. After being blocked with 5 % nonfat skim milk, which was diluted with Tris-buffered saline containing 0.1 % Tween 20 (TBST) for 1 h at room temperature, the membrane containing the protein extracts was incubated overnight with primary antibody diluted with 2 % bovine serum albumin in Tris-buffered saline with 0.1 % Tween 20 at 4 °C. The primary antibodies were as follows: mTNF-α and sTNF-α (1:400, Sigma, USA), pro-IL-1β and active-IL-1β (1:1000, Santa Cruz, USA), p-STAT3 (1:1000, Cell Signaling technology, USA), p-IκBα (1:1000, Cell Signaling technology, USA), p65 (1:500, Santa Cruz, CA, USA), GAPDH (1:10000, Bioworld, USA), histone (1:1000, Bioworld, USA), and β-actin (1:2000, Santa Cruz, USA). On the next day, proteins were visualized by using the enhanced chemiluminescence detection system (Pierce, USA) after incubation with respective horseradish peroxidase-conjugated secondary antibodies (1:1000), and then exposed to medical X-ray film. The intensity of the blots was quantified by employing a gel-image analyzer (JS380; Peiqing Science and Technology, Shanghai, China).
Immunofluorescence Staining
At the indicated time points after irradiation, BV-2 cells were fixed, permeabilized, and blocked with goat serum. Cell samples were stained with primary antibody: rabbit polyclonal antibody of p65 (1:400,Santa Cruz, CA, USA). And then, cell samples were probed with AlexaFluor 488-conjugated secondary antibody (1:200, Invitrogen, Carlsbad, CA, USA). Cell nuclei were counter-stained with 4′,6-diamidino-2-phenylindole (DAPI, Roche, Switzerland). Fluorescence intensity was then measured by using a confocal scanning microscope (BX41F; Olympus, Tokyo, Japan).
The brain sections (15 μm) were fixed in 4 % paraformaldehyde for 3–4 h at room temperature, and rinsed with PBS. After washing with PBS, the nonspecific binding sites were blocked with 10 % goat serum (GTX27481, GeneTex) for 1 h at room temperature, and then for costaining, the samples were incubated at 4 °C overnight with two primary antibodies, rat monoclonal anti-F4/80 (1:50, ABD Serotec, Raleigh, NC, USA) and rabbit polyclonal anti-TNF-α (1:50, ab9739, Abcam, USA), simultaneously in 1 % goat serum. Sections were washed with PBS and incubated in the dark for 1 h with secondary antibodies: Alexa Flour® 568 goat anti-rabbit IgG (H + L) (1:200, A11011, Invitrogen) and Alexa Flour® 488 goat anti-mouse IgG (H + L) (1:200, A11006, Invitrogen). After washing with PBS three times for 5 min, the nuclei were stained with DAPI (S36939, Invitrogen) for 15 min, and the sections were then examined by using a confocal scanning microscope (BX41F; Olympus, Tokyo, Japan).
Statistical Analysis
All of the quantitative data were expressed as mean ± SD. For comparison between more than two groups, ANOVA was used for repeated measurements, with appropriate post hoc test employed. All tests were two-tailed. A P less than 0.05 was considered statistically significant, and the artworks for the statistical analysis were created by GraphPad Prism 5. Mouse survival curves were calculated by using the Kaplan-Meier method and subjected to the log-rank test for comparison by using the Statistical 6.0 software package (Statsoft, USA).
Results
Cytotoxicity of Corilagin and PS1145 on BV2 Cells
Since MTT assay showed that the pretreatment with Corilagin ranging from 0.125 to 80 μg/mL for 12 h inhibited the viability of BV-2 cells at a very low inhibition rate (Fig. 2a), the concentration of 2.50 μg/mL was used for the in vitro study. Moreover, the MTT assay also revealed that inhibition rates of PS1145 to BV-2 cells were low at the concentration range from 0.01 to 20 μmol/L (Fig. 2b). Therefore, the concentration of 5 μmol/L was chosen for the following experiments.

Cytotoxicity of Corilagin and PS1145 on BV2 Cells. a The MTT assay of BV2 cells viability after the treatment of Corilagin. b The MTT assay of BV2 cells viability after the treatment of PS1145
Effect of Corilagin on Survival and Physical Status After CRT
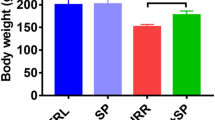
There was no significant difference in overall survival between control group and Cori-alone group (P > 0.05, Fig. 3a). But, the life span of mice was significantly reduced after CRT when RT-alone, RT + Dex, RT + H-Cori, and RT + L-Cori groups were compared with control and Cori-alone groups (P < 0.05), and the median survival in RT-only group lasted only 120 days. In contrast, mice in RT + Dex group had a longer median survival of 190 days as compared with RT-alone group (P < 0.05, log-rank), and mice in RT + L-Cori and RT + H-Cori had a median survival of 140 and 160 days. As compared to RT-alone group, the median survival of RT + H-Cori group was significantly longer (P < 0.05, log-rank). The data showed that 2-week Cori treatment could improve survival in RIBI mice.

Effect of Corilagin on survival and physical status after CRT. a Kaplan-Meier curves for overall survival. Mouse survival curves were calculated by using the Kaplan-Meier method and subjected to the log-rank test for comparison by using Statistical 6.0 software (Statsoft, USA). b The growth curves for body weight. Data shown are the mean ± SD (n = 3, significant difference from the respective values determined by ANOVA test)
After CRT, the mice in all radiation groups lost body weight as compared to the sham-irradiated groups (Fig. 3b), while Corilagin partially ameliorated this change and generally improved animal activity. Compared to controls, body weight dropped significantly in RT-alone and RT + L-Cori/H-Cori groups at the fourth, fifth, and sixth month (P < 0.05 vs. control group and Cori-alone group). H-Cori and Dex treatments relieved this irradiation-associated weight loss at the fifth and sixth month compared to RT-alone group (P < 0.05, RT + H-Cori group and RT + Dex group vs. RT-alone group).
The Effect of Corilagin on Irradiated-Associated Memory Deficits
To study whether Corilagin could attenuate the CRT-induced memory impairment, the Morris water maze test was performed (Fig. 4). The mean escape latency in the RT groups (RT-alone, RT + H-Cori, and RT + L-Cori groups) was increased when compared with sham-irradiated mice (P < 0.05 vs. control group and Cori-only group), while escape latency was shorter in H-Cori and Dex treatment groups than in RT-alone group after conditioning for 3 days (P < 0.05 vs. RT-alone group). On the sixth day, the platform was removed and the probe trial was conducted. The times of platform crossing were markedly greater in RT + H-Coril and RT + Dex groups than in RT-alone groups (P < 0.05 vs. RT-alone groups, Fig. 4b). The mice in RT-alone group spent significantly less time in the target quadrant than those in sham-irradiated group did (P < 0.05 vs. control group and Cori-alone group), while RT + H-Cori and RT + Dex groups spent more time in the target quadrant than RT-alone group (P < 0.05 vs. RT groups, Fig. 4c), and the percent distance of RT + H-Cori and RT + Dex groups in the target quadrant was also significantly increased compared with RT-only group (P < 0.05 vs. RT-alone groups, Fig. 4d). These results demonstrated that Corilagin treatment ameliorated memory deficits of CRT mice.

Effect of Corilagin on irradiated-associated memory deficits. a Escape latency for escape to a submerged platform in the training trials. b The times of crossing over the former location of the platform during the probe trail. c The time spent in the target quadrant during the probe trail. d The percent distance in the target quadrant during the probe trail (*P < 0.05 vs. control group; # P < 0.05 vs. RT-alone group, n = 8 per group, significant difference from the respective values determined by ANOVA test)
The Protective Effect of Corilagin on Irradiation-Induced BBB Damage
The ultrastructural and permeability changes of the BBB were assessed by HRP extravasation under a transmission electron microscope. No HRP extravasation into brain parenchyma was observed in control animals (Fig. 5a), while extensive HRP extravasation, both inside and outside BBB, was noted after CRT. Furthermore, mice in Cori and Dex treatment groups had less HRP extravasation after CRT, suggesting that Corilagin partially protected the permeability of BBB from CRT-induced damage. To further confirm the protective effect of Corilagin on the BBB after CRT, the amount of Evans blue dye extravasation was measured at the second day post-irradiation (n = 5 in each group). As shown in Fig. 5b, after 10-Gy CRT, Evans blue extravasation was obviously increased in brain tissues. On the other hand, H-Cori and Dex treatments significantly reduced the amount of Evans blue leakage after CRT, as compared to the RT-alone group (P < 0.05).

Effect of Corilagin on irradiation-induced blood-brain barrier damage. a The ultrastructure and permeability change of the BBB. b The amount of Evans blue dye extravasation 48 h after 10-Gy CRT (*P < 0.05 vs. control group; # P < 0.05 vs. RT-alone group, n = 5 in each group, significant difference from the respective values determined by ANOVA test)
The Effect of Corilagin on Irradiation-Induced Neuro-Inflammation and Apoptosis
In the control and Cori-alone groups, the neuronal nuclei in hippocampus were big, round, and transparent, and nucleoli were clear with a complete envelope. Twenty-four hours after CRT, the nuclear envelope disappeared, vacuole formed, neurons slightly swelled, and inflammatory cells infiltrated into the hippocampus in RT-alone group. Meanwhile, reticular interstitial hydropsy and neurophagia were seen. One week after CRT, the number of interstitial hydropsy and inflammatory cells were diminished, and some cells underwent apoptosis, losing nuclear envelop, with their nuclei being split (Fig. 6a). Furthermore, we found that Cori and Dex treatments could ameliorate these brain tissue damages, such as inflammatory cell infiltration and neuronal necrosis.

Effect of Corilagin on irradiated-induced neuroinflammation and apoptosis. a HE staning for pathological examination of post-CRT changes. White markers displayed the inflammatory cells infiltrating to the hippocampus zone. Black markers displayed that nuclear envelope disappeared, and vacuole formed. In the control and Cori-alone groups, the neuronal nuclei in hippocampus were big, round, and transparent, and nucleoli were clear with complete envelop. In RT-alone group, neurons were slightly swelling, and inflammatory cell infiltrated accompanying by reticular interstitium hydropsy and neuronophagia. Cori and Dex treatments attenuated these brain tissue damages. b Immunehistochemistry staining of caspase-3 in the hippocampus. Black markers were used to exhibit the caspased-3-positive cells, namely apoptotic cells. There was hardly any caspase-3-positive cell in the sham-irradiated groups (control and Cori-alone groups). The number of caspase-3-positive cells increased in RT-alone group. In the Cori and Dex treatment groups, the number of CRT-induced caspase-3-positive cells decreased, especially in H-Cori and Dex treatment groups
As previous study showed that ionizing radiation could induce caspase-3-dependent apoptosis in neural stem cells [30], we immunohistochemically detected the expression of caspase-3 in the hippocampus 24 h after CRT (Fig. 6b). There were hardly any caspase-3-positive cells in the sham-irradiated groups (control and Cori-alone groups), while, after CRT, the number of caspase-3-positive cells was increased significantly in RT-alone group. And, in the Cori and Dex treatment groups, after CRT, the number of caspase-3-positive cells was decreased, especially in H-Cori and Dex treatment groups. These data suggested that H-Cori and Dex treatments could ameliorate CRT-induced neuronal apoptosis.
The Effect of Corilagin on Irradiation-Induced Inflammatory Cytokines at mRNA and Protein Levels
We then examined the effect of Corilagin on the irradiation-induced expression of inflammatory cytokines in vivo in molecular term (Fig. 7). RNA was extracted from the brain tissues of mice from different groups at the given time points and was used for real-time PCR detection of inflammatory cytokines. Figure 7a, b shows that the levels of TNF-α and IL-1β in control microglial cells were relatively low, while in irradiated BV-2 cells, the level of these inflammatory cytokines was increased post-irradiation, peaking at the 6th hour (TNF-α, Fig. 7a) or the 72nd hour (IL-1β, Fig. 7b) (P < 0.05, vs. control). Additionally, Corilagin inhibited the expression of these cytokines, especially in RT + H-Cori and RT + Dex groups (P < 0.05).

Effect of Corilagin on irradiation-induced inflammatory factors in mRNA and protein lavels. a Real-time PCR for the mRNA levels of TNF-α. b Real-time PCR for the mRNA levels of IL-1β. c Western blotting for the protein levels of sTNF-α and mTNF-α. d Western blotting for the protein levels of active-IL-1β and pro-IL-1β. e Relative quantity analysis of western blotting for sTNF-α. f Relative quantity analysis of western blotting for active-IL-1β (*P < 0.05 vs. control group; # P < 0.05 vs. RT-alone group, n = 3 in each group, significant difference from the respective values determined by ANOVA test). g Western blotting for the protein level of β-actin
The protein expression of soluble TNF-α (sTNF-α, 17KD) was virtually negative in sham-irradiated group, but it was increased at the 3rd, 6th, 48th, and 72nd hour, and the 1st, 2nd, 4th, and 6th week after CRT (Fig. 7c). We further studied the protein level of pro-IL-1β (31 kDa) and active-IL-1β (17 kDa) by using Western blotting. It was found that 10-Gy CRT induced the expression of IL-1β protein (Fig. 7d). In sham-irradiated group, the expression of IL-1β was marginally detectable, but at the 3rd hour after CRT, the pro-IL-1β expression was induced and was increased steadily at the 6th, 24th, 48th, and 72nd hour, and the 1st, 2nd, and 6th week after CRT (Fig. 7d). Active IL-1β could be detected at the 3rd hour after CRT, and reached a peak at the 72nd hour (Fig. 7f). And, Corilagin pretreatment significantly inhibited the production of these cytokines, especially in RT + H-Cori and RT + Dex groups (P < 0.05, Fig. 7f).
The Effect of Corilagin on Microglial Activation
To determine the effect of Corilagin on the inflammation cytokine secretion and microglial activation after CRT, TNF-α and mouse EGF-like module-containing mucin-like hormone receptor-like 1 (F4/80) in the hippocampus of mice were detected by immunofluorescence staining (Fig. 8). The results indicated that the expression of TNF-α and F4/80 was increased after 10-Gy CRT. Moreover, TNF-α was mainly expressed in F4/80-positve cells. As F4/80 is a marker of activated microglial cells [31], we are led to infer that microglial cells were the main source of irradiation-induced TNF-α. While in the Cori or Dex treatment groups, after CRT, TNF-α expression was decreased, especially in RT + H-Cori and RT + Dex groups, suggesting that Corilagin could inhibit the microglial activation and the release of inflammatory cytokines in CRT mice.

Effect of Corilagin on microglial activation. Immunofluorescence staining detected TNF-α and F4/80 in the hippocampus of mice
Mechanisms of Corilagin Inhibiting Microglial Activation and Attenuating RIBI
We detected the activity of NF-κB and STAT3 pathways both in vitro and in vivo to explore the mechanism of Corilagin suppressing microglial activation and inflammation. Both immunofluorescence staining and Western blotting (Fig. 9) showed that, in vitro, the phosphorylated IκBα (p-IκBα) and intranuclear p65 was upregulated in RT groups, while in the irradiated BV-2 cells pretreated with 2.5 μg/mL Corilagin or 5.0 μmol/L PS1145, the level of p65 protein in nuclei and p-IκBα was significantly reduced 24 h post-irradiation (P < 0.05, Fig. 9d, e). Simultaneously, phosphorylated STAT3 (p-STAT3) was downregulated after irradiation, and the downregulation could be partially reversed by Corilagin (P < 0.05, Fig. 10a–c).

Effect of Corilagin on the activities of NF-κB pathways after RT in vitro. a The immunofluorescence stain for the translocation of p65 from cytoplasm to nucleus in BV2 cells 12 and 24 h after RT. b Western blotting analysis for p-IκBα, cytoplasm p65, and nuclei p65 24 h after RT. c Relative quantity analysis for western blotting of cytoplasm p65. d Relative quantity analysis for Western blotting of p-IκBα. e Relative quantity analysis for Western blotting of nuclei p65 (*P < 0.05 vs. control group; # P < 0.05 vs. RT-alone group, n = 3 in each group, significant difference from the respective values determined by ANOVA test)

Effect of Corilagin on the activities of STAT3 pathways after RT in vitro. a The immunofluorescence stain of p-STAT3 24 h after RT. b Western blotting analysis of p-STAT3 24 h after RT. c Relative quantity analysis for Western blotting of p-STAT3 (*P < 0.05 vs. control group; # P < 0.05 vs. RT-alone group, n = 3 in each group, significant difference from the respective values determined by ANOVA test)
In addition, the in vivo activity of NF-κB and STAT3 pathways was examined by Western blotting. Figure 11 shows that p-IκBα was upregulated in RT groups, while Corilagin treatment significantly decreased the protein level of p-IκBα after CRT (Fig. 11b). Additionally, the translocation of p65 from cytoplasm to nuclei induced by irradiation was inhibited by Cori (30 mg/kg) or Dex treatment (Fig. 11c, d). And, p-STAT3 was downregulated in RT groups, while Cori or Dex treatment significantly increased p-STAT3 (Fig. 11e). These data suggested that the Corilagin might work on RIBI by inhibiting the irradiation-induced activation of NF-κB pathway in a STAT3-associated manner.

Effect of Corilagin on the activities of NF-κB and STAT3 pathways after RT in vivo. a Western blotting analysis for p-IκBα, cytoplasm p65, nuclei p65, and p-STAT3. b Relative quantity analysis of western blotting for p-IκBα. c Relative quantity analysis of western blotting for cytoplasm p65. d Relative quantity analysis of Western blotting for nuclei p65. e Relative quantity analysis of Western blotting for p-STAT3 (*P < 0.05 vs. control group; # P < 0.05 vs. RT-alone group, n = 3 in each group, significant difference from the respective values determined by ANOVA test)
Discussion
Each year, practically 200,000 brain tumor patients are treated with either partial large field or whole brain irradiation in the USA [32, 33]. In fact, progressive impairment in cognitive ability, or RIBI, is identified in 40–50 % of brain tumor patients who survive more than 6 months after brain radiotherapy [34]. The impairment predominantly manifests as deficits in learning, memory, and spatial information processing [35]. Previous studies suggested that a series of biological processes are involved in RIBI, including alteration in the BBB [36, 37], a rapid induction of gene expression of the pro-inflammatory cytokines [38, 39], and the activation of microglia [40, 41]. The present study was designed to investigate the effect of Corilagin on RIBI and explore the underlying mechanisms. Our study demonstrated that the intraperitoneal injection of Corilagin after CRT significantly ameliorated irradiation-induced weight loss, the learning and memory deficits, partially protected BBB permeability from damage, suppressed inflammatory responses, and inhibited microglial activation in RIBI mice.
In the present study, the activities of daily living and weight of the experimental animals were observed and monitored. After CRT, the mice in all radiation groups lost body weight (Fig. 3b) and had less daily activities, which were observed in our previous study [42]. The irradiation-associated weight loss might be due to the most common side effects of CRT: nausea and loss of appetite. As we all know, these common side effects were caused by the irradiation-induced edema of brain tissue. This irradiation-associated weight loss could be partially alleviated by Corilagin. In addition, the reduced daily activities after CRT could also be generally improved by Corilagin. In previous researches, the body weight loss during the treatments was a poor prognostic factor for tumor patients [43, 44]. Therefore, Corilagin might improve the prognosis of the patients receiving CRT.
In this study, Morris water maze test showed that the RIBI model was successfully made in mice by giving 10-Gy CRT. Mice treated with Corilagin for 2 weeks after CRT found the platform more easily than those that received CRT alone on the days 4 and 5. These data indicated that the Corilagin treatment could alleviate neuro-cognitive deficits in learning and memory induced by 10-Gy CRT.
The cognitive decline in learning and memory has been suggested to be due to irradiation-induced impairment in the hippocampal neurogenesis [45]. In the present study, HE and caspase-3 immunohistochemical staining showed that CRT could induce inflammatory cell infiltration, neuronal necrosis, and apoptosis in the hippocampus, and Corilagin could attenuate these brain tissue damages.
Previous studies suggested that alteration in the BBB may be responsible for the injury of normal brain tissues after irradiation therapy [36, 37]. For example, irradiation disrupts the BBB by damaging the structural and functional integrity of the microvasculature of brain [46, 47]. In the present research, we used Evans blue dye extravasation to measure the vascular permeability in the CNS. After CRT, the BBB permeability was increased, while this increase could be partially reversed by the post-irradiation treatment with Corilagin. It was previously reported that BBB disruption had something do to the intricate network of cytokines and inflammatory mediators, such as TNF-α and IL-1β. These inflammatory cytokines were upregulated through the interaction among the signal transducer and activator of transcription (STAT) pathway, PI3K/Akt pathway, and NF-κB pathway [40, 47–52]. We postulate, on the basis of these findings, that Corilagin stabilizes the BBB after CRT, possibly by inhibiting the release of pro-inflammatory cytokines.
Although the etiology of RIBI is poorly understood, it is generally believed that inflammatory cytokines play an important part in RIBI [40, 53]. Irradiation has been reported to upregulate expression of pro-inflammatory cytokines and chemokines in the brain. A rapid upregulation of gene expressions of the pro-inflammatory cytokines, such as TNF-α and IL-1β, in response to radiation, has been reported to be implicated in the radiotherapy-associated damage to the brain [36, 39]. Interestingly, it was reported that cytokine expression was region-specific since the upregulated mRNA and protein expression of TNF-α and IL-1β was observed in hippocampal and cortical regions isolated from irradiated brains [40]. Moreover, TNF-α level was significantly elevated in cortex than in hippocampus and IL-1β level was elevated in hippocampus compared to cortical samples [40]. Furthermore, both in vitro and in vivo studies showed that whole brain irradiation-induced pro-inflammatory environment in the brain may be, at least in part, mediated by the activation of microglia, suggesting that, after radiation, specific type of cells might contribute to the overexpression of pro-inflammatory mediators in the brain [40, 41]. In this study, the in vivo test showed that the number of TNF-α-positive cells was increased after CRT. Moreover, TNF-α was principally expressed in F4/80-positve cells. Since F4/80 is a specific marker of microglial cells [31], we hypothesize that microglial cells are the main source of irradiation-induced TNF-α. What is more, this study exhibited that irradiation-induced expression of TNF-α was inhibited by the post-irradiation treatment with Corilagin, especially at the dose of 30 mg/kg. And, Western blotting examination further verified that Corilagin (30 mg/kg/day) significantly inhibited the sharp increase of the pro-inflammatory cytokines (TNF-α and IL-1β) caused by CRT, suggesting that Corilagin could inhibit the microglial activation and the expressions of inflammation cytokines. In addition, it was found that the expression of TNF-α in the lung tissue was biphasic: one peak was at approximately 6 h, and the second peak was at approximately 8 weeks [54]; our results also demonstrate a complex pattern of elevated TNF-α expression after cranial radiation therapy, pathologically suggesting a two-phase mechanism. An immediate upregulated expression of pro-inflammatory cytokines was observed 3 h after irradiation. After normalization of the values in 1 week, a second peak of TNF-α expression occurred 4 weeks later. In prior researches [54, 55], TNF-α was thought to play a prominent role in the initiation of the cytokine cascade and further promote the inflammatory process. Moreover, it was reported that the cellular source of TNF-α could be displayed via TNF-α immunoreactivity as well as histopathologic changes [56]. Therefore, the two expression peaks might be due to inflammatory cascade. Moreover, this study showed that Corilagin exerted an inhibiting effect on the two peaks of TNF-α expression.
Our previous study exhibited that irradiation-induced TNF-α and IL-1β were downstream cytokines of NF-κB pathway [9]. Transcriptional regulation of genes often requires the cooperative or antagonistic interaction among several factors, and this interaction fine-tunes the expression of downstream target genes in response to a variety of stimuli. Depending on the cell type and the nature of the inducing stimuli, NF-κB and STAT3 pathways (another key regulator of inflammation), functionally, can either promote or antagonize each other. In normal immune cells, STAT3 could directly interact with NF-κB and antagonize cytokine activation, and exert inhibitory effects on stimuli-induced IKK, an IkB kinase complex that induces IkB phosphorylation [57]. Moreover, it was reported that in microglial cells of some inflammation models, the transcriptional regulation of NF-κB fine-tuned the expression of downstream target genes requiring the antagonistic action of STAT3. During the neuro-inflammatory response, the phosphorylation of STAT3 was downregulated and its antagonizing effect on NF-κB pathway was suppressed, thereby allowing NF-κB pathway to be activated [16, 17].
STAT3 is abundantly expressed in brain and principally takes part in the regulation of genes involved in inflammation and tumorigenesis. In the course of neuroinflammation, cytokine signal transduction is predominantly mediated through the JAK/STAT signaling pathway [58]. Previous studies showed that STAT3 could downregulate the expression of some pro-inflammatory mediators, such as IL-12 [59], TNF-α [60], and iNOS [61], directly or indirectly. Moreover, STAT3 deletion in macrophages resulted in chronic inflammation, with an overrelease of pro-inflammatory cytokines [62]. Recently, it was found that during the neuroinflammatory response in a murine model of Alzheimer’s disease (AD) and in AD patients, the expression of p-STAT3 (the activated state of STAT3) in hippocampal neurons was decreased [63]. In addition, an anti-AD agent was found to suppress neuroinflammation by inhibiting the activation of NF-κB pathway in a STAT3-dependent fashion [17]. On the other hand, our previous in vitro study showed that Corilagin inhibited the double strand break-triggered (DSB)/NF-κB pathway in irradiated microglial cells [23]. From the aforementioned findings, we are led to postulate that Corilagin attenuated RIBI through inhibiting microglial activation and the release of inflammatory cytokines via NF-κB pathway in a STAT3-associated manner.
In this study, we demonstrated that after CRT, the irradiation-induced translocation of p65 (one protein of NF-κB dimers) from cytoplasm to nucleus and p-IκBα was inhibited by Corilagin both in vitro and in vivo. And, the phosphorylation of STAT3 was downregulated in RT groups, while Corilagin treatment significantly increased p-STAT3. These findings suggested that irradiation-induced neuroinflammation was mediated via the activation of NF-κB pathway, which was antagonized by activated STAT3, and Corilagin worked on RIBI via NF-κB pathways in a STAT3-associated manner.
Conclusion
In summary, this study demonstrated that treatment with Corilagin after CRT could attenuate structural and biochemical abnormalities of mouse RIBI model by inhibiting microglial activation and the expression of inflammatory cytokines. Corilagin might inhibit the activation of NF-κB pathway in a STAT3-associated manner, thereby downregulating the inflammatory cytokine release. Corilagin promises to be a new protecting agent for radiation-induced brain injury.
References
Khuntia D, Brown P, Li J, Mehta MP (2006) Whole-brain radiotherapy in the management of brain metastasis. J ClinOncol 24(8):1295–1304
Johannesen TB, Langmark F, Lote K (2003) Cause of death and long-term survival in patients with neuro-epithelial brain tumours: a population-based study. Eur J Cancer 39(16):2355–2363
Cole AM, Scherwath A, Ernst G, Lanfermann H, Bremer M, Steinmann D (2013) Self-reported cognitive outcomes in patients with brain metastases before and after radiation therapy. Int J Radiat Oncol Biol Phys 87(4):705–712
Beltran C, Naik M, Merchant TE (2010) Dosimetric effect of target expansion and setup uncertainty during radiation therapy in pediatric craniopharyngioma. Radiother Oncol 97(3):399–403
Li J, Bentzen SM, Li J, Renschler M, Mehta MP (2008) Relationship between neurocognitive function and quality of life after whole-brain radiotherapy in patients with brain metastasis. Int J Radiat Oncol Biol Phys 71(1):64–70
Schnegg CI, Kooshki M, Hsu FC, Sui G, Robbins ME (2012) PPARδ prevents radiation-induced proinflammatory responses in microglia via transrepression of NF-kappaB and inhibition of the PKCα/MEK1/2/ERK1/2/AP-1 pathway. Free RadicBiol Med 52(9):1734–1743
Liu JL, Tian DS, Li ZW, Qu WS, Zhan Y, Xie MJ, Yu ZY, Wang W et al (2010) Tamoxifen alleviates irradiation-induced brain injury by attenuating microglial inflammatory response in vitro and in vivo. Brain Res 1316:101–111
Zhao W, Robbins ME (2009) Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications. Curr Med Chem 16(2):130–143
Dong X, Luo M, Huang G, Zhang J, Tong F, Cheng Y, Cai Q, Dong J et al (2015) Relationship between irradiation-induced neuro-inflammatory environments and impaired cognitive function in the developing brain of mice. Int J Radiat Biol 91(3):224–239
Stoll G, Jander S (1999) The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol 58(3):233–247
Gebicke-Haerter PJ (2001) Microglia in neurodegeneration: molecular aspects. Microsc Res Tech 54(1):47–58
Pocock JM, Liddle AC (2001) Microglial signalling cascades in neurodegenerative disease. Prog Brain Res 132:555–565
Luo XG, Chen SD (2012) The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener 1(1):9
Xue J, Dong JH, Huang GD, Qu XF, Wu G, Dong XR (2014) NF-κB signaling modulates radiation-induced microglial activation. Oncol Rep 31(6):2555–60
Kyrkanides S, Moore AH, Olschowka JA, Daeschner JC, Williams JP, Hansen JT, Kerry OM (2002) Cyclooxygenase-2 modulates brain inflammation-related gene expression in central nervous system radiation injury. Brain Res Mol Brain Res 104(2):159–169
Hutchins AP, Poulain S, Miranda-Saavedra D (2012) Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood 119(13):e110–9
Zhang ZH, Yu LJ, Hui XC, Wu ZZ, Yin KL, Yang H, Xu Y (2014) Hydroxy-safflor yellow A attenuates Aβ1-42-induced inflammation by modulating the JAK2/STAT3/NF-KB pathway. Brain Res 1563:72–80
Shen ZQ, Dong ZJ, Peng H, Liu JK (2003) Modulation of PAI-1 and tPA activity and thrombolytic effects of corilagin. Planta Med 69(12):1109–1112
Duan W, Yu Y, Zhang L (2005) Antiatherogenic effects of phyllanthusemblica associated with corilagin and its analogue. YakugakuZasshi 125(7):587–591
Bharti S, Nidhi V, Juan PM (2014) Aparajita M (2014) An overview of important ethnomedicinal herbs of Phyllanthus species: present status and future prospects. Sci World J 2014:839172
Shibuya H, Kitagawa I (1996) Chemical study of Indonesian medicinal plants. J Pharm Soc Jpn 116(12):911–27
Kinoshita S, Inoue Y, Nakama S, Ichiba T, Aniya Y (2007) Antioxidant and hepatoprotective actions of medicinal herb Terminalia catappa L. from Okinawa Island and its tannin corilagin. Phytomedicine 14(11):755–762
Cheng JT, Lin TC, Hsu FL (1995) Antihypertensive effect of corilagin in the rat. Can J Physiol Pharmacol 73(10):1425–1429
Guo YJ, Luo T, Wu F, Liu H, Li HR, Mei YW, Zhang SL, Tao JY et al (2014) Corilagin protects against HSV1 encephalitis through inhibiting the TLR2 signaling pathways in vivo and in vitro. Mol Neurobiol. doi:10.1007/s12035-014-8947-7
Dong XR, Luo M, Fan L, Zhang T, Liu L, Dong JH, Wu G (2010) Corilagin inhibits the double strand break-triggered NF-kappaB pathway in irradiated microglial cells. Int J Mol Med 25(4):531–536
Schindler MK, Forbes ME, Robbins ME, Riddle DR (2008) Aging-dependent changes in the radiation response of the adult rat brain. Int J Radiat Oncol Biol Phys 70(3):826–834
Zhu L, Zhang L, Zhan L, Lu X, Peng J, Liang L, Liu Y, Zheng L et al (2014) The effects of Zibu Piyin Recipe components on scopolamine-induced learning and memory impairment in the mouse. J Ethnopharmacol 151(1):576–582
Cevik NG, Orhan N, Yilmaz CU, Arican N, Ahishali B, Kucuk M, Kaya M, Toklu AS (2013) The effects of hyperbaric air and hyperbaric oxygen on blood–brain barrier integrity in rats. Brain Res 1531:113–121
Overbergh L, Giulietti A, Valckx D, Decallonne R, Bouillon R, Mathieu C (2003) The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J Biomol Tech 14(1):33–43
Ivanov VN, Hei TK (2014) A role for TRAIL/TRAIL-R2 in radiation-induced apoptosis and radiation-induced bystander response of human neural stem cells. Apoptosis 19(3):399–413
Austyn JM, Gordon S (1981) F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol 11(10):805–15
Stone HB, Moulder JE, Coleman CN, Ang KK, Anscher MS, Barcellos-Hoff MH, Dynan WS, Fike JR et al (2004) Models for evaluating agents intended for the prophylaxis, mitigation and treatment of radiation injuries. Report of an NCI Workshop, December 3–4, 2003. Radiat Res 162(6):711–728
Moulder JE, Cohen EP (2007) Future strategies for mitigation and treatment of chronic radiation-induced normal tissue injury. Semin Radiat Oncol 17(2):141–8
Warrington JP, Csiszar A, Mitschelen M, Lee YW, Sonntag WE (2012) Whole brain radiation-induced impairments in learning and memory are time-sensitive and reversible by systemic hypoxia. PLoS One 7(1), e30444
Acharya MM, Christie LA, Lan ML, Donovan PJ, Cotman CW, Fike JR, Limoli CL (2009) Rescue of radiation-induced cognitive impairment through cranial transplantation of human embryonic stem cells. Proc Natl AcadSci USA 106(45):19150–19155
Diserbo M, Agin A, Lamproglou I, Mauris J, Staali F, Multon E, Amourette C (2002) Blood–brain barrier permeability after gamma whole-body irradiation: an in vivo microdialysis study. Can J Physiol Pharmacol 80(7):670–678
Nordal RA, Wong CS (2005) Molecular targets in radiation-induced blood–brain barrier disruption. Int J Radiat Oncol Biol Phys 62(1):279–287
Hong JH, Chiang CS, Campbell IL, Sun JR, Withers HR, McBride WH (1995) Induction of acute phase gene expression by brain irradiation. Int J Radiat Oncol. Biol Phys 33(3):619–626
Gaber MW, Sabek OM, Fukatsu K, Wilcox HG, Kiani MF, Merchant TE (2003) Differences in ICAM-1 and TNF-alpha expression between large single fraction and fractionated irradiation in mouse brain. Int J Radiat Biol 79(5):359–366
Lee WH, Sonntag WE, Mitschelen M, Yan H, Lee YW (2010) Irradiation induces regionally specific alterations in pro-inflammatory environments in rat brain. Int J Radiat Biol 86(2):132–144
Conner KR, Forbes ME, Lee WH, Lee YW, Riddle DR (2011) AT1 receptor antagonism does not influence early radiation-induced changes in microglial activation or neurogenesis in the normal rat brain. Radiat Res 176(1):71–83
Zhang J, Tong F, Cai Q, Chen L-j, Dong J-h, Wu G, Dong X-r (2015) Shenqi Fuzheng Injection attenuates irradiationinduced brain injury in mice via inhibition of the NF-κB signaling pathway and microglial activation. Acta Pharmacol Sin 36:1288–1299
Ross PJ, Ashley S, Norton A, Priest K, Waters JS, Eisen T, Smith IE, O’Brien MER (2004) Do patients with weight loss have a worse outcome when undergoing chemotherapy for lung cancers? Br J Cancer 90:1905–11
Langius JAE, Bakker S, Rietveld DHF, Kruizenga HM, Langendijk JA, Weijs PJM, Leemans CR (2013) Critical weight loss is a major prognostic indicator for disease-specific survival in patients with head and neck cancer receiving radiotherapy. Br J Cancer 109:1093–1099
Rola R, Raber J, Rizk A, Otsuka S, VandenBerg SR, Morhardt DR, Fike JR (2004) Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol 188(2):316–330
Baker DG, Krochak RJ (1989) The response of the microvascular system to radiation: a review. Cancer Invest 7(3):287–294
Rubin P, Gash DM, Hansen JT, Nelson DF, Williams JP (1994) Disruption of the blood–brain barrier as the primary effect of CNS irradiation. Radiother Oncol 31(1):51–60
Barichello T, Lemos JC, Generoso JS, Cipriano AL, Milioli GL, Marcelino DM, Vuolo F, Petronilho F et al (2011) Oxidative stress, cytokine/chemokine and disruption of blood–brain barrier in neonate rats after meningitis by Streptococcus agalactiae. Neurochem Res 36(10):1922–30
Takata F, Dohgu S, Matsumoto J, Takahashi H, Machida T, Wakigawa T, Harada E, Miyaji H et al (2011) Brain pericytes among cells constituting the blood–brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J Neuroinflammation 8:106
Liu T, Zhang T, Yu H, Shen H, Xia W (2014) Adjudin protects against cerebral ischemia reperfusion injury by inhibition of neuroinflammation and blood–brain barrier disruption. J Neuroinflammation 11:107
Li L, McBride DW, Doycheva D, Dixon BJ, Krafft PR, Zhang JH, Tang J (2015) G-CSF attenuates neuroinflammation and stabilizes the blood–brain barrier via the PI3K/Akt/GSK-3β signaling pathway following neonatal hypoxia-ischemia in rats. Exp Neurol pii: S0014–4886(15):00005–9
Lee YW, Hennig B, Toborek M (2003) Redox-regulated mechanisms of IL-4-induced MCP-1 expression in human vascular endothelial cells. Am J Physiol Heart Circ Physiol 284(1):H185–92
Greene-Schloesser D, Robbins ME (2012) Radiation-induced cognitive impairment from bench to bedside. Neuro Oncol 14(4):iv37–44
Rübe CE, Wilfert F, Palm J, König J, Burdak-Rothkamm S, Liu L, Schuck A, Willich N et al (2004) Irradiation induces a biphasic expression of pro-inflammatory cytokines in the lung. Strahlenther Onkol 180(7):442–8
Ardestani S, Deskins DL, Young PP (2013) Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species. J Mol Signal 8(1):12–12
Rübe CE, Wilfert F, Uthe D, Schmid KW, Knoop R, Willich N, Schuck A, Rübe C (2001) Modulation of radiation-induced tumor necrosis factor (TNF-α) expression in the lung tissue by pentoxifylline. Radiother Oncol 64:177–87
Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, Kano A, Iwamoto Y et al (2003) STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A 100(4):1879–1884
Gautron L, De Smedt-Peyrusse V, Layé S (2006) Characterization of STAT3-expressing cells in the postnatal rat brain. Brain Res 1098(1):26–32
Smith AM, Qualls JE, O’Brien K, Balouzian L, Johnson PF, Schultz-Cherry S, Smale ST, Murray PJ (2011) A distal enhancer in Il12b is the target of transcriptional repression by the STAT3 pathway and requires the basic leucine zipper (B-ZIP) protein NFIL3. J Biol Chem 286(26):23582–23590
Mandal P, Park PH, McMullen MR, Pratt BT, Nagy LE (2010) The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat Kupffer cells. Hepatology 51(4):1420–1429
Yu Z, Zhang W, Kone BC (2002) Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J 367(Pt 1):97–105
Fu XY (2006) STAT3 in immune responses and inflammatory bowel diseases. Cell Res 6(2):214–219
Chiba T, Yamada M, Sasabe J, Terashita K, Shimoda M, Matsuoka M, Aiso S (2009) Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol Psychiatry 14(2):206–222
Acknowledgments
This study is sponsored by the National Nature Science Foundation of China (nos. 81172595 and 81573090), Post-doctoral Program foundation of China (no. 20100480905), and a grant from the Foundation of Special Post-doctoral Program of China (201104440).
Conflict of Interest
No potential conflicts of interest were disclosed.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Fan Tong and Jian Zhang are considered co-first authors.
Fan Tong and Jian Zhang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Tong, F., Zhang, J., Liu, L. et al. Corilagin Attenuates Radiation-Induced Brain Injury in Mice. Mol Neurobiol 53, 6982–6996 (2016). https://doi.org/10.1007/s12035-015-9591-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9591-6