
Abstract
Adult neurons, which are terminally differentiated cells, demonstrate substantial radioresistance. In contrast, human neural stem cells (NSC), which have a significant proliferative capacity, are highly sensitive to ionizing radiation. Cranial irradiation that is widely used for treatment of brain tumors may induce death of NSC and further cause substantial cognitive deficits such as impairing learning and memory. The main goal of our study was to determine a mechanism of NSC radiosensitivity. We observed a constitutive high-level expression of TRAIL-R2 in human NSC. On the other hand, ionizing radiation through generation of reactive oxygen species targeted cell signaling pathways and dramatically changed the pattern of gene expression, including upregulation of TRAIL. A significant increase of endogenous expression and secretion of TRAIL could induce autocrine/paracrine stimulation of the TRAIL-R2-mediated signaling cascade with activation of caspase-3-driven apoptosis. Furthermore, paracrine stimulation could initiate bystander response of non-targeted NSC that is driven by death ligands produced by directly irradiated NSC. Experiments with media transfer from directly irradiated NSC to non-targeted (bystander) NSC confirmed a role of secreted TRAIL for induction of a death signaling cascade in non-targeted NSC. Subsequently, TRAIL production through elimination of bystander TRAIL-R-positive NSC might substantially restrict a final yield of differentiating young neurons. Radiation-induced TRAIL-mediated apoptosis could be partially suppressed by anti-TRAIL antibody added to the cell media. Interestingly, direct gamma-irradiation of SK-N-SH human neuroblastoma cells using clinical doses (2–5 Gy) resulted in low levels of apoptosis in cancer cells that was accompanied however by induction of a strong bystander response in non-targeted NSC. Numerous protective mechanisms were involved in the maintenance of radioresistance of neuroblastoma cells, including constitutive PI3K-AKT over-activation and endogenous synthesis of TGFβ1. Specific blockage of these survival pathways was accompanied by a dramatic increase in radiosensitivity of neuroblastoma cells. Intercellular communication between cancer cells and NSC could potentially be involved in amplification of cancer pathology in the brain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Radiation therapy alone or in combination with chemotherapy is a standard modality for treatment of many types of tumors, including cancers in the nervous system. Normal adult neurons, which are terminally differentiated cells, demonstrate a substantial radioresistance. In contrast, neural stem and progenitor cells (NSC/NPC) having significant proliferative capacities are highly sensitive to ionizing radiation. Cranial irradiation used for treatment of brain tumors may cause substantial cognitive deficits such as impairing learning, attention and memory, which are especially pronounced in children, due to inhibition of the proliferation and death of neural stem cells (NSC) [1–6].
Ionizing irradiation generates reactive oxygen species (ROS) that cause DNA damage and affect numerous cell signaling pathways and the corresponding gene expression followed by inhibition of cell proliferation, induction of the DNA repair mechanisms and, finally, either cell survival or cell death (via apoptosis, necrosis or mitotic catastrophe) depending on a balance between cell survival and cell killing mechanisms [7, 8]. Besides the primary induction of ROS in the cell as a result of ionizing radiation, there are numerous mechanisms that accelerate ROS/RNS production after irradiation. These are dependent on modulation of the mitochondrial functions or NADPH oxidase-1 activity [8, 9]. Consequently, directly irradiated cells can dramatically change the regulation of gene expression by induction of SOS programs, sharing multiple common features with a general response to oxidative stress. Induction of gene expression of proinflammatory cytokines (IL1β, TNFα, IL6, IL17 and IL33) and COX2 directed by activation of the transcription factors NF-κB, STAT3 and AP1 is a common feature of different types of stress response and, furthermore, a basis for the induction of a bystander response in non-targeted cells induced by signaling machinery of directly irradiated cells [10, 11]. Numerous investigations of the radiation-induced bystander response of non-targeted cells during the last two decades have dramatically changed the paradigm of radiobiology concerning general regulation of radiation response [9, 12, 13].
In the present study we investigate radiation-induced effects in directly irradiated NSC or in human neuroblastoma cells with the subsequent induction of intercellular communication and bystander response of non-targeted NSC that could ultimately result in the death or inhibition of differentiation of non-targeted cells.
Results
Radiation-induced death of human NSC
In human SOX2-positive, Nestin-positive neural stem/progenitor cells (NSC) exposed to graded doses of γ-irradiation (2.5–10 Gy), there was the early (6 h after treatment) pronounced activation of caspase-3 evaluated by downregulation of the latent form of procaspase-3 (p32) and induction of caspase3-dependent cleavage of PARP1 that was suppressed by pan-caspase inhibitor zVAD-fmk (40 μM), rather than by a necroptosis inhibitor Necrostatin-1 (40 μM) (Fig. 1a, b). Survival of irradiated NSC was substantially decreased (Fig. 1c), while apoptotic levels of irradiated NSC (determined by PI staining and FACS analysis as % cells in pre-G1 area) were gradually increased 24–72 h after treatment in dose-dependent manner (Fig. 1d). Proliferation and survival of human NSC is critically dependent on several growth factors, FGF2, EGF and IGF1 [14], which via the corresponding receptors induce the PI3K–AKT and MAPK signaling cascades. LY294002, a PI3K–AKT small molecule inhibitor, induced notable levels of apoptosis in NSC and dramatically upregulated radiation-induced apoptosis of NSC (Fig. 1e). Based on comparison of levels of apoptosis and total cell death, it was evident that the vast majority of irradiated NSC died via apoptotic pathways (Fig. 1e). In general, death of NSC was easily observed as early as 6 h after irradiation by pronounced flotation of dying NSC and decreased confluence of adherent cells.

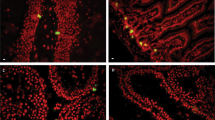
Radiation-induced apoptosis in human neural stem cells (NSC). a Confocal analysis of immunofluorescent images of NSC was performed using monoclonal antibody against Nestin, an early neuroprogenitor cell marker (red), and polyclonal antibody against Sox2, a pluripotency marker (green). b Western blot analysis of expression of pro-Caspase3 and PARP1 in human NSC 6 h after treatment by increasing doses of γ-irradiation. A universal caspase inhibitor zVAD-fmk (40 μM) and an inhibitor of necroptosis, Necrostatin (40 μM), were added 30 min before irradiation, as indicated. Beta-Actin was used as a loading control. c Effect of γ-irradiation on survival of human NSC. Numbers of live cells attached to fibronectin matrix were determined before irradiation at time point 0 and 48 h after treatment. Relative cell density (normalized to initial cell density at time point 0) is indicated. d, e Cell cycle-apoptosis analysis of human NSC after treatment by increasing doses of γ-radiation. A PI3K–AKT inhibitor LY294002 (40 μM) was added 30 min before irradiation. At indicated time points, cells were stained by propidium iodide and analyzed by the flow cytometry. Pooled results of four independent experiments are shown in d. Error bars represent mean ± SD (p < 0.05, Student’s t test); star indicates a significant difference. A typical experiment is shown in e. TD means levels of total cell death that were determined using Trypan blue exclusion assay
Radiation-induced apoptosis in NSC is mediated by the endogenous TRAIL/TRAIL-R2 expression
SOX2 and Nestin-positive NSC (Figs. 1a, 2a) were further characterized by constitutively high level of TRAIL-R2 (a synonym DR5) expression that was not further upregulated upon γ-irradiation (Fig. 2a); TRAIL-R1/DR4 expression was not detectable in NSC. On the other hand, total levels of p53-dependent TRAIL and Fas-Receptor [15, 16], but not Fas-Ligand (Fas-L), were notably upregulated after irradiation in parallel with an increase of p53 protein levels. These changes were in concert with a substantial downregulation of anti-apoptotic Survivin (Fig. 2a). Immunostaining and FACS analysis further confirmed upregulation of total TRAIL levels and the presence of constitutively high TRAIL-R2 levels after irradiation of NSC (Fig. 2b). Apoptotic commitment was evident 6 h after irradiation, due to downregulation of the latent pro-Caspase-8 that reflected activation of the TRAIL-R2-mediated death signaling cascade (Fig. 2a) followed by pro-Caspase-3 activation and PARP1 cleavage (see Fig. 1b).

Proapoptotic and prosurvival signaling during radiation-induced apoptosis in NSC. a Expression levels of indicated proteins were determined using Western blot analysis. Beta-Actin was used as a loading control. b Endogenous TRAIL and TRAIL-R2/DR5 expression was determined using specific immunostaining and FACS analysis. For detection of total protein expression in NSC, cells were fixed and permeabilized using FIX & PERM cell fixation and permeabilization reagents, while detection of surface expression was performed without permeabilization (not shown). Percentage of positively stained cells with the corresponding levels of median fluorescent intensity (MFI) are indicated. c NSC were γ-irradiated in the presence or in the absence (control with the vehicle solution, 0.1 % DMSO) of specific small molecule inhibitors of signaling pathways: U0126 (MEK-ERK; 10 μM), SP600125 (JNK; 20 μM) SB203580 (MAPK p38; 10 μM), IKK-NF-κB (BMS345541; 10 μM), LY294002 (PI3K-AKT; 50 μM) and Stat3-inhibitor-6 (25 μM), which were added to the media 30 min before irradiation. Cell cycle—apoptosis analysis was performed using PI staining and FACS analysis. Stars indicate a significant difference
Furthermore, IKK-NF-κB and AKT activities (determined as levels of the corresponding phosphoproteins) were substantially decreased after irradiation. Activities of two MAPK’s, JNK and especially p38, were also decreased, while ERK activity was relatively stable in irradiated cells. Stat3 total levels were also relatively stable (Fig. 2a). Combined treatment of NSC by irradiation in the presence of specific small molecule inhibitors of cell signaling pathways, U0126 (a MEK-ERK inhibitor), SP600125 (a JNK inhibitor), SB203580 (a MAPK p38 inhibitor), BMS345541 (an IKK-NF-κB inhibitor), LY294002 (a PI3K–AKT inhibitor), and Stat3-inhibitor-6 highlighted a prosurvival role for AKT and JNK activation during radiation-induced apoptotic commitment of NSC (Fig. 2a). Radiation-induced endogenous expression of TRAIL in TRAIL-R2/DR5-positive NSC and almost negligible levels of endogenous Fas-L expression in these cells allow us to suggest the TRAIL–TRAIL-R2 interaction as a possible driver of radiation-induced death of NSC.
To further address a question about the functional significance of TRAIL-R-mediated death signaling cascade in human NSC, we used the exogenous recombinant TRAIL (10–50 ng/ml). Even at high dose (50 ng/ml), TRAIL alone only modestly increased PARP1 cleavage and induced relatively low levels of apoptosis in NSC 24 after treatment (Fig. 3a–c). As expected, a combination of TRAIL (50 ng/ml) and CHX (1 μg/ml) efficiently cleaved PARP1 and increased levels of apoptosis 24 h after treatment (Fig. 3a–c). Caspase-3-mediated cleavage of PARP1 and TRAIL-induced apoptosis were substantially blocked by zVAD-fmk, a universal caspase inhibitor, rather than by Necrostatin-1 (40 μM), a necroptosis inhibitor (Fig. 3a). Interestingly, Necrostatin-1 also dramatically upregulated AKT activity and, thereafter, further stabilized the Nanog (an AKT target) protein level (Fig. 3a). It also provided an increase in general survival functions in NSC. In contrast, no significant effects of Necrostatin-1 were observed in the regulation of MAPK’s signaling pathways (Fig. 3a). Levels of RIP1, which is critical for the necroptotic signaling cascade [17], were somewhat modulated after treatment of NSC with TRAIL or Necrostatin-1 alone or in combinations (Fig. 3A).

Exogenous TRAIL/TRAIL-R2-induced apoptosis in NSC. Effects of combined treatment with TRAIL and cycloheximide (CHX) on the cell cycle and apoptosis of human NSC. a Western blot analysis of indicated proteins was performed 6 h after treatment with TRAIL (50 ng/ml), CHX (1 μg/ml), zVAD-fmk (40 μM), Necrostatin (40 μM) alone or in combination. PARP1 cleavage was used as a characteristic feature of Caspase3-mediated apoptosis. Beta-Actin was used as a loading control. b, c Cell cycle-apoptosis analysis was performed after treatment of human NSC with TRAIL, TNFα (50 ng/ml) and CHX alone or in combination in the presence or in the absence of pan-caspase inhibitor zVAD-fmk (40 μM). PI staining of DNA and FACS analysis was performed 24–48 h after indicated treatment. A typical experiment is presented in b; pooled results of four independent experiments are shown in c. Error bars represent mean ± SD (p < 0.05, Student’s t test); star indicates a significant difference
Similar to TRAIL and CHX treatment, a combination of the exogenous TNFα and CHX could induce well pronounced apoptosis in NSC (Fig. 3c). However, this combination was able to induce both apoptotic (zVAD-fmk-sensitive) and necrotic (Necrostatin-sensitive) death pathways, while TRAIL in combination with CHX induced mainly apoptosis that did not respond to Necrostatin-1 cotreatment (Suppl. Fig. 1A).
To further confirm a leading role of the endogenous TRAIL upregulation (Fig. 4a) in radiation-induced apoptosis, we used human TRAIL ELISA kit (R&D Systems) for detection of TRAIL levels in the media 16 h after γ-irradiation at 5 Gy (Fig. 4b). Two-fold increase in TRAIL concentration was detected. Basal level of TRAIL in non-treated media probably connected with excretion from dead cells. Next we added inhibitory anti-TRAIL antibody (50 μg/ml) to the culture media 30 min before irradiation. This allowed us to partially suppress the TRAIL–TRAIL-R2-mediated signaling cascade. Treatment with this antibody resulted in escape from PARP1 cleavage (Fig. 4c), increase of cell survival and inhibition of radiation-induced apoptosis (Fig. 4d, e). Taken together, these results highlight the key role for endogenously produced TRAIL in the radiation-induced apoptosis of human NSC in cell culture conditions. The exogenous Fas-L in combination with CHX was also able to induce modest levels of apoptosis in NSC (data not shown), due to low surface expression of Fas-receptor, which, however, notably increased after irradiation (Fig. 4f). The endogenous production of Fas-L was insignificant. The exogenous production of TNFα and Fas-L that potentially could target NSC in vivo is located in microglial and glial cells [18, 19].

Radiation-induced apoptosis in NSC is mediated by endogenous TRAIL/TRAIL-R2 expression. a Total expression levels of TRAIL-R2 (DR5) and TRAIL 16 h after γ-irradiation of NSC. Specific immunostaining and FACS analysis were performed. b TRAIL levels (pg/ml, normalized for 2 × 106 cells) were detected in the media 16 h after irradiation of the confluent NSC cultures using ELISA. Control was a fresh cell-free media. c Dose-dependent cleavage of PARP1 and its suppression by inhibitory anti-TRAIL Ab and control IgG that were added to the culture media 30 min before irradiation. d Effects of anti-TRAIL Ab (50 μg/ml) in the culture media on increased survival of NSC after irradiation. Cells were immunostained using anti-Nestin Ab. e Effects of anti-TRAIL Ab and control nonspecific IgG on levels of radiation-induced apoptosis 48 h after treatment. Pooled results of four independent experiments are shown. Error bars represent mean ± SD (p < 0.05, Student’s t test); star indicates a significant difference. f Radiation-induced changes in surface expression of Fas and total expression of Fas-L in NSC 16 h after irradiation (5 Gy). US mean unstained cells
Beside TRAIL, TNFα and Fas-L, we further tested several cytokines, growth factors and PGE2 as probable candidates for intercellular communication before and after γ-irradiation of NSC. We demonstrated that TGFβ1 could modestly increase basal apoptotic levels in human NSC 24 h after treatment (Suppl. Fig. 1B), reflecting the presence of the corresponding receptors in NSC. Furthermore, pretreatment with TGFβ1 (5 ng/ml) before irradiation further upregulated levels of radiation-induced apoptosis in NSC (Suppl. Fig. 1C). Surprisingly, we did not observe notable protective effects against radiation-induced apoptosis of NSC by IL1β and PGE2 (Suppl. Fig. 1C).
Radiation-induced bystander effects in NSC: analysis of cell cycle and apoptosis in directly irradiated and non-targeted cells
Media-transfer experiments from directly irradiated NSC with upregulated endogenous production and secretion of several cytokines, such as TRAIL, to non-irradiated native NSC (Suppl. Fig. 2) allow us to detect induction/upregulation of apoptosis in non-targeted cells. Relatively low, but a well pronounced apoptosis was detected 24–48 h after irradiated media transfer, demonstrating a twofold increase compared to control (non-irradiated) media transfer (Fig. 5a, b). Caspase-3-mediated cleavage of PARP1 in bystander cells was also evident in these conditions (Fig. 5c). In contrast, immortalized human fetal astrocytes (IHFA) exhibited a strong radioresistance after treatment with clinical doses of γ-radiation (Fig. 5d, e). These cells did not produce TRAIL or Fas-L upon irradiation (data not shown) and were resistant to the exogenous TRAIL/TRAIL-R- mediated apoptosis due to the absence of surface TRAIL-R expression [20]. Furthermore, as recently established, astrocytes lack functional DNA damage response signaling that tightly correlated with their radioresistance [21]. Thereafter, media transfer from irradiated IHFA did not notably upregulate apoptosis and did not induce PARP1 cleavage in bystander NSC (Fig. 5d–f).

Radiation-induced bystander effects in NSC: cell cycle and apoptosis analysis of directly irradiated and non-targeted cells. a, b NSC were irradiated at 5 Gy. 24 h after irradiation, the media from irradiated and control (non-irradiated) cells were transfer to naive NSC, which were cultured for additional 24 or 48 h. Cell cycle-apoptosis analysis was performed for directly irradiated cells 48 and 72 h after irradiation (the left part of a and b) and for non-targeted cells 24 and 48 h after media transfer (the right parts of a and b). c Analysis of PARP1 cleavage was performed for control and bystander cells 24 and 48 h after media transfer. d–f Similar experiments were performed with media transfer from control (non-irradiated) and directly irradiated immortalized human fetal astrocyte (IHFA) to NSC. Cell cycle apoptosis analysis is shown on d and e; Western blot analysis of PARP1 is shown on f
Bystander effects of irradiated media transfer on neuronal differentiation of NSC
In general, the early radiation-induced bystander response with the end-point of induction of apoptosis in NSC was reproducible and convincing, but exhibited a relatively low consequence at this time point. The next aim of our study was to determine delayed radiation-induced bystander effects that could affect neuronal differentiation of NSC in the cell culture conditions. We used again in our experiments pretreatment of the native NSC with media transferred from non-irradiated (control) cells and from directly irradiated NSC (24 h after treatment) with the subsequent induction of neural differentiation of NSC using the specific differentiation media (see Suppl. Fig. 2). Removing FGF2 and EGF from the NSC media induced neuronal differentiation of human NSC (Fig. 6; Control-1 cells) that was accompanied by high levels of precursor cell death during differentiation. The gradual disappearance of an early neuroprogenitor cell marker, Nestin (red), which was substituted with a neuronal marker, Doublecortin (green) was evident in surviving cells (Fig. 6). Gamma-irradiation before induction of differentiation significantly retarded this process with a strong decrease in a green/red ratio (a ratio of the number of Doublecortin-positive green cells to the number of Nestin-positive red cells that reflected the degree of neuronal differentiation) and in a relative cell survival. Relative cell survival after differentiation of control or irradiated cells was determined by direct counting cells that were attached to the laminin matrix in six microscopic areas 10 days after initiation of differentiation. Media transfer from irradiated NSC (24 h after irradiation) to naive bystander NSC affect cell survival, compared to non-irradiated media transfer to control-2 cells and decreased the ratio of green Doublecortin-positive young neurons among differentiating cells (Fig. 6d). Non-specific IgG did not change the dramatic effects of direct irradiation of NSC while a pretreatment with anti-TRAIL antibody (50 μg/ml) added to media before irradiation of NSC, partially reversed inhibition of survival and differentiation (Fig. 6c). Anti-TRAIL pretreatment notably, but still modestly increased the yield of young neurons among bystander cells (Fig. 6f).

Neuronal differentiation of NSC in culture: bystander effect as a result of irradiated media transfer. a–c NSC were non-treated (Control-1) or directly γ-irradiated at 5 Gy. d–f NSC were primed by 24 h-incubation with transferred media from non-irradiated cells (Control-2) or from irradiated NSC (Bystander cells). Neuronal differentiation of non-irradiated (Control-1), directly irradiated NSC and primed non-targeted NSC (Control-2 and Bystander) was initiated by differentiation media. Furthermore, non-specific IgG (b, e) or inhibitory anti-TRAIL Ab (50 μg/ml) (c, f) were added 30 min before irradiation to indicated cultures and were maintained for the next 24 h, before neuronal differentiation was initiated. Confocal analysis of immunofluorescent images was performed using monoclonal Ab against an early neuroprogenitor marker, Nestin (red), and polyclonal Ab against a neuronal marker, Doublecortin (green). A ratio of the number of green cells to the number of red cells and relative cell survival for irradiated cells 10 days after initiation of differentiation are indicated
On the other hand, direct pretreatment with exogenous recombinant TRAIL (10–50 ng/ml for 8 h) negatively affected the final yield of young neurons after differentiation in a dose-dependent manner (Fig. 7). The entire NSC population contained three sub-populations with higher, lower and negative surface expression of TRAIL-R2 (Fig. 7a). TRAIL-R-high NSC (with 110 MFI) could quickly die after TRAIL pretreatment during differentiation. However, at least some of the TRAIL-R-low or TRAIL-R-negative cells might overcome this treatment and perform neuronal differentiation (Fig. 7b). Taken together, these observations highlight a role for TRAIL in radiation-induced apoptosis of neural stem/progenitor cells. Inhibition of TRAIL significantly increased survival of differentiating cells, especially, for directly irradiated NSC and upregulated the degree of neuronal differentiation of both directly irradiated and bystander NSC.

Dose-dependent effects of the exogenous TRAIL on death and differentiation of NSC. a The whole population of NSC contained three subpopulations with negative (ns), lower (14 MFI) and higher (110 MFI) levels of surface TRAIL-R2 expression determined by immunostaining and FACS analysis. b, c NSC were pretreated for 8 h by increased doses of the exogenous TRAIL (10–50 ng/ml). Next, the neuronal differentiation of NSC was induced by changing to differentiation media. Three repeats of representative images of the neuronal differentiation of the control and TRAIL-treated cells are shown (after 10 days of culture in differentiation media) in b. The pooled results of three independent experiments are shown in c. Error bars represent mean ± SD (p < 0.05, Student’s t test); star indicates a significant difference
Radiation-induced signaling pathway and radiation-induced apoptosis in SK-N-SH human neuroblastoma cells
To further highlight the effects of intercellular communication after ionizing irradiation, we performed experiments with direct irradiation of cancer cells (human SK-N-SH neuroblastoma) trying to evaluate the induction of a bystander response in non-targeted NSC by signals from irradiated cancer cells. The primary neuroblastomas are characterized by a neural crest origin and extracranial location; however, highly metastatic neuroblastomas can go to the brain [22, 23] and potentially affect NSC. Even non-treated SK-N-SH neuroblastoma cells produce and secrete elevated levels of several cytokines, such as IL6, TNFα, TGFβ1, and COX-2-driven prostaglandin-E2 that could be found in cell cultures [24–27]. We therefor expected to detect potentially strong radiation-induced signal transmission from directly irradiated neuroblastoma cells to non-targeted NSC.
Dose-dependent effect of direct γ-irradiation on the main signaling pathways was really dramatic in SK-N-SH neuroblastoma cells (Fig. 8a, b), compared to NSC (see Fig. 2a). It includes stabilization of p53 protein levels and upregulation of p53-dependent BAX, TRAIL and DR5 levels, activation of NF-κB p65, MAPK p38, ERK and upregulation of JNK2 activity (at 2.5–5 Gy) in neuroblastoma cells (Fig. 8a, b). A modest downregulation of high constitutive levels of P-JNK and P-AKT could be detected in neuroblastoma cells after irradiation at 10 Gy. Endogenous TGFβ1 production was gradually decreased following irradiation, while TGFβ-R2 levels were upregulated (Fig. 8b). Caspase-8 activation was not detectable, a modest down-regulation of the latent pro-Caspase-3 was evident only 6 h after treatment at 10 Gy; however, PARP1-clevage was still very slight at this time point. Levels of anti-apoptotic Survivin were also decreased, while XIAP levels were stable after irradiation (Fig. 8b). A notable, but still modest increase in total expression of TRAIL and pronounced increase in surface expression of DR5 were also detected after irradiation of neuroblastoma cells using immunostaining and FACS analysis (Fig. 8a, c). However, radiation-induced apoptosis was not really detectable 16–24 h after irradiation of SK-N-SH cells, reflecting a strong prevalence of surviving functions.

Radiation-induced proapoptotic and antiapoptotic signaling pathways in SK-N-SH human neuroblastoma cells. a, b Western blot analysis of indicated proteins 6 h after treatment with increasing doses of γ-irradiation. c TRAIL-R2/D5 surface expression was determined in directly irradiated SK-N-SH cells by immunostaining and FACS analysis. d Effect of γ-irradiation (5 Gy) on TRAIL-induced apoptosis of SK-N-SH cells. Error bars represent mean ± SD (p < 0.05, Student’s t test); star indicates a significant difference. e Effects of inhibitory anti-TGFβ1 Ab on radiosensitivity of SK-N-SH cells. Vehicle solution, 0.1 % DMSO, and zVAD-fmk (40 μM) were used. Error bars represent mean ± SD (p < 0.05, Student’s t test); star indicates a significant difference
The exogenous TRAIL at high dose (50 ng/ml) alone or in combination CHX induced low levels of apoptosis 16 h after treatment (Fig. 8d). However, γ-radiation pretreatment in combination with the exogenous TRAIL induced higher levels of apoptosis, due to a notable upregulation of TRAIL-R2 surface expression in irradiated neuroblastoma cells (Fig. 8c). Taken together, these observations demonstrate a modest level of endogenous TRAIL expression and efficient anti-apoptotic protection that suppressed radiation-induced apoptosis in SK-N-SH cells at the early time points. Consequently, higher levels of radiation-induced apoptosis of neuroblastoma cells were observed only 72 h after treatment with 10 Gy (Suppl. Fig. 2A, B). On the other hand, the use of small molecule inhibitors of the protective signaling pathways, IKK-NF-κB, Stat3 or PI3K–AKT, in combination with radiation allowed us to dramatically increase induction of apoptosis, even at the early time points (Suppl. Fig. 2A). Additionally, suppression of endogenously produced TGF-β1 in cell media by inhibitory antibody also notably upregulated levels of radiation-induced apoptosis in neuroblastoma cells, highlighting anti-apoptotic function of TGFβ signaling in neuroblastoma cells (Fig. 8e), as it was recently observed for glioblastoma cells [28].
Radiation-induced effects of SK-N-SH neuroblastoma cells on cell cycle and apoptosis of non-targeted NSC
Even though, SH-N-SH cells did not develop apoptosis 16–24 after irradiation (Fig. 9a, c), media transfer experiments to non-targeted native NSC induced detectable levels of apoptosis in NSC that was clearly different from the basal levels. Additionally, PARP1 cleavage was observed in bystander NSC under these conditions (Fig. 9c, the right side). Media transfer from both control and irradiated neuroblastoma cells 48 h after treatment to non-targeted NSC induced pronounced apoptosis, although with a significant increase in apoptotic levels in NSC treated with irradiated media (Fig. 9b, c). This reflects the presence of death-inducing factors in the media of non-treated neuroblastoma cells that are increased after irradiation. Indeed, pretreatment with anti-TRAIL Ab of both control and irradiated media from SK-H-SH cells substantially (but not completely) decreased apoptotic levels in the non-targeted NSC (Fig. 9d). Taken together, these experiments indicate that high levels of anti-apoptotic protection in neuroblastoma cells [22] allowed to avoid the death of the vast majority of cancer cells after irradiation. However, non-targeted NSC could be quite efficiently killed by media transfer from control and, especially, from irradiated neuroblastoma cells.

Bystander effects of directly irradiated SK-N-SH cells on the neural differentiation of non-targeted NSC. a, b Media transfer from control (non-irradiated) and γ-irradiated SK-N-SH cells 24 and 48 h after irradiation to non-targeted naive NSC. Results of cell cycle-apoptosis analysis are shown 24–48 h for directly irradiated cells and 24 h after media transfer to non-targeted cells. c Western blot analysis of PARP1 cleavage in directly irradiated SK-N-SH cells (24 h after irradiation with two independent repeats—1 and 2) and in non-targeted NSC (24 after media transfer). d Both control and irradiated media were pretreated with anti-TRAIL Ab (50 μg/ml) before irradiation. Cell-cycle-apoptosis analysis was performed
Radiation-induced effects of SK-N-SH cells on the neuronal differentiation of non-targeted NSC
As expected, a strong cytotoxic activity of media from non-irradiated and, especially, from irradiated neuroblastoma cells that were used for pretreatment of non-targeted NSC before initiation of differentiation caused dramatic suppressive effects on neuronal differentiation. Irradiated media from neuroblastoma cells finally killed the vast majority of NSC and completely blocked neuronal differentiation (Fig. 10, the upper panel). The presence of anti-TRAIL antibody increased a survival of differentiating cells for both types of media transfer, although effect of anti-TRAIL on restoring neuronal differentiation was relatively modest (Fig. 10, the bottom panel). In summary, these observations indicated that TRAIL produced by cancer cells might be involved in modulating cell death and suppression of neuronal differentiation of non-targeted NSC. However, this death ligand appears to operate in a complex combination with other cytotoxic and protective factors, such as Fas-L, IL6, TNFα and TGFβ1 produced by cancer cells that also could affect NSC survival and differentiation.

Effects of media transfer from non-irradiated and irradiated (5 Gy) SK-N-SH cells on neuronal differentiation of NSC. NSC were treated for 8 h with media transferred from non-irradiated or irradiated SK-N-SH cells in the presence of nonspecific IgG or anti-TRAIL Ab (50 μg/ml). Next, the neuronal differentiation of NSC was induced by changing to differentiation media. Immunostaining with anti-Nestin and anti-Doublecortin Abs was performed after 12 days of differentiation. Relative cell survival for control and non-targeted differentiating cells 12 days after initiation of differentiation is indicated
Discussion
Results of our study demonstrated that radiation-induced apoptosis of NSC in cell culture is mainly mediated by TRAIL/TRAIL-R2 interaction through autocrine or paracrine stimulation. This finding is in contrast to the popular belief that this signaling mechanism preferentially kills cancer cells. Indeed, the vast majority of cancer cell lines express TRAIL-receptors and approximately 50 % of these lines can efficiently induce the TRAIL-R-death signaling cascade after treatment by exogenous TRAIL. In contrast, other lines require an additional co-treatment for the induction of TRAIL-dependent death, such as inhibition of PI3K–AKT or NF-κB signaling pathways [29, 30]. The exogenous TRAIL in vivo can be produced at low levels by many types of TRAIL-R-negative normal cells and at higher levels by some types of cells of the immune system. Recombinant TRAIL or anti-TRAIL-R agonistic antibodies were used with variable success to kill different types of cancer cells [31]. Radiation-induced stimulation of TRAIL production in TRAIL-R-positive NSC, however, could be an additional complication on the use of radiation therapy to specifically kill cancer cells. Paradoxically, human NSC with ectopic overexpression of the membrane form of TRAIL were successfully used as a vehicle for delivery of a death ligand to glioblastoma xenotransplant in animal experiments with the subsequent induction of apoptosis (in combination with bortezomib) [32]. We think, however, that effective protection of the endogenous human NSC against membrane TRAIL, which is delivered by vehicle NSC, should be considered in similar type of treatment of glioblastoma in human.
Human NSC were also sensitive to Fas-L- and, especially, to TNFα-induced cell death, due to the presence of the corresponding receptors. TNFα could be produced in vivo by microglial cells of the brain [18], targeting via TNFR1 both apoptotic and necrotic pathways in NSC. While the TNFR1-mediated apoptotic pathway might be blocked by caspase-3 inhibitors, the TNFR1-mediated necroptotic pathway was suppressed using RIP1 inhibitor, Necrostatin-1 [17]. Surprisingly, Necrostatin-1 was demonstrated in the present study as a strong inducer of a protective AKT activity in NSC (see Fig. 3a) that could additionally maintain the integrity of mitochondria and suppress development of oxidative stress. Necrostatin-1 treatment was previously used for efficient suppression of cell death during ischemic brain injury [33]. On the other hand, beside induction of cell death, TNFα may serve as a classical inducer of cell survival pathways via NF-κB activation. Such complicated functions of TNFα highlights its central role in balancing between life and death during different types of stress responses [34–36]. An additional possibility for negative regulation of NSC survival was linked with Fas-L production by cancer cells with the simultaneous radiation-induced upregulation of Fas-Receptor levels in NSC and down-regulation of antiapoptotic protein regulators. Similarly with TNFR-signaling, Fas-Receptor-mediated signaling cascades could be either proapoptotic, or prosurvival, due to numerous regulatory mechanisms, including NF-κB activation by this pathway [15]. Prosurvival features of Fas-Receptor pathway are probably well pronounced in mouse NSC/NPC [37].
In general, ionizing irradiation of cells targets numerous cell signaling pathways and induces changes in expression of several hundred genes [38]. However, radiation-induced gene activation shares some common features with other types of cell stress, including modulation of the pro-inflammatory and anti-inflammatory gene expression of specific sets of cytokines and their receptors, COX2 expression and COX2-directed production of PGE2, expression of death ligands and death receptors [9, 39] (see Suppl. Fig. 4). Production and secretion of cytokines and PGE2 after irradiation may serve as a basis for induction of propagating bystander response of non-targeted cells.
We and others previously observed radiation-induced induction/upregulation of COX2 gene expression in normal embryonic fibroblasts [40] and cancer cells, such as neuroblastoma, glioblastoma and melanoma [25, 41], and demonstrated a significant role of COX2-PGE2 signaling pathway in regulation and propagation of the bystander response [40, 42]. Surprisingly, we did not detect high levels of inducible COX2 expression in NSC. However, the bystander effect in these cells could be modulated by distinct signaling transmitters, including TRAIL (see Suppl. Fig. 4). We previously demonstrated that oxidative stress induced by sodium arsenite treatment of NSC resulted in activation of the inner mitochondrial death pathway accompanied by high levels of death of NSC and suppression of neuronal differentiation [43]. Interestingly, radiation-induced apoptosis of NSC, which was mediated mainly by the TRAIL/TRAIL-R2 apoptotic pathway in concert with additional regulators of cell survival, also suppressed neuronal differentiation. Furthermore, bystander suppression of NSC differentiation was especially strong in case of radiation-induced intercellular communication between irradiated cancer (neuroblastoma or glioblastoma) cells and NSC. We expect that further investigations of mechanisms of radiation-induced neurotoxicity at the level of human NSC may open new opportunities to protect and maintain neurogenesis after anti-cancer therapy, especially for patients with brain tumors.
Materials and methods
Materials
Fibronectin, laminin, polyornithine and prostaglandin E2 (PGE2) were obtained from Sigma-Aldrich (St. Louis, MO, USA). PI3K inhibitor LY294002, IKK inhibitor BMS-345541, STAT3 inhibitor-6 S3I-201 (also known as NSC 74859), PI3K inhibitor LY294002, MEK inhibitor U0126, MAPK p38 inhibitor SB203580, JNK inhibitor SP600125 and pan-caspase inhibitor zVAD-fmk was purchased from Calbiochem (La Jolla, CA, USA). Necrostatin-1, a RIP-1 inhibitor, was obtained from EMD Millipore (Billerica, MA, USA). Human soluble Killer-TRAIL (recombinant) and Fas-L (recombinant) were purchased from Alexis (San Diego, CA, USA); human TNFα, TGFβ1, IL1β and IL6 were obtained from R&D Systems (Minneapolis, MN, USA).
Human embryonic NSC in culture
Cryopreserved human embryonic NSC were obtained from Gibco/Life Technologies (Carlsbad, CA, USA) as a commercially available product (N7800-200). The cells were derived from NIH approved H9 (WA09) human embryonic stem cells. The cells were plated in 6-well culture plates coated with fibronectin and incubated at 37 °C in complete growth medium NSC/SFM, which contained serum-free DMEM/F12 supplemented with 2 mM GlutaMAX, bFGF (20 ng/ml), EGF (20 ng/ml) and StemPRO neural supplement (2 %). All reagents were obtained from from Gibco/Life Technologies (Carlsbad, CA, USA).
Neuronal differentiation of human NSC in culture
NSC were plated on polyornithine- and laminin-coated 6-well plates, which contained similarly coated cover slips, in complete NSC/SFM. After 2 days, neuronal differentiation was initiated by neuronal differentiation media, which contains Neurobasal medium, B-27 Serum-free supplement (2 %) and 2 mM GlutaMAX (Gibco/Life Technologies). Medium was changed every two days. A neuronal phenotype was confirmed using immunofluorescence detection 10 days after initiation of differentiation.
Cell lines
IHFA were kindly provided by Dr. M. Davidson (Columbia University, New York, NY, USA). Human neuroblastoma SK-N-SH (HTB-11, ATCC) and human glioblastoma (HTB-14, ATCC) cell lines were obtained from ATCC (Manassas, VA, USA).
Immunocytochemistry analysis
Cells were fixed with 4 % paraformaldehyde in PBS for 60 min. Immunochemical staining was performed using standard protocols. Cells were stained for the undifferentiated NSC marker, Nestin (using mAb from Millipore, Temecula, CA, USA) and for the neuronal marker, Doublecortin using Ab from Cell Signaling, (Danvers, MA, USA). Additional markers include Sox2 and phospho-JNK (using Abs from Cell Signaling). The secondary Abs were Alexa Fluor 594 goat anti-mouse IgG and Alexa Fluor 488 goat anti-rabbit IgG from Molecular Probes/Life Technologies (Carlsbad, CA, USA). A laser scanning confocal microscope (Nikon TE 2000 with EZ-C1 software, Tokyo, Japan) was used for immunofluorescence image analysis.
Irradiation procedures
To determine sensitivity to γ-rays, plated NSC, astrocytes, neuroblastoma and glioblastoma cells were exposed to radiation from a Gammacell 40 137Cs irradiator (dose rate, 0.82 Gy/min) at Columbia University. 6–72 h after irradiation, cells were stained with PI and analyzed by flow cytometry.
FACS analysis of TRAIL, TRAIL-R2/DR5, TRAIL-R1/DR4, Fas and Fas-L levels
Total and surface levels of TRAIL, TRAIL-R2/DR5, TRAIL-R1/DR4, Fas and Fas-L on human cell lines were determined by staining with a PE-conjugated anti-human-TRAIL, anti-human-DR5, anti-human-DR4, anti-human Fas and anti-human-Fas-L mAbs (R&D System, Minneapolis, MN, USA and eBioscience, San Diego, CA, USA) and subsequent flow cytometry. For detection of total levels of antigen proteins, cells were fixed and permeabilized using 0.5 % Triton X-100 in PBS. PE-conjugated nonspecific mouse IgG1 was used as an immunoglobulin isotype control. A FACS Calibur flow cytometer (Becton–Dickinson, Mountain View, CA, USA) and the CellQuest program were used to perform flow cytometric analysis. All experiments were independently repeated 3–5 times.
Apoptosis studies
For induction of apoptosis, cells were exposed to γ-irradiation (2–10 Gy) alone or in the presence of small molecule inhibitors of cell signaling pathways. Furthermore, apoptosis was induced TRAIL, TNFα, Fas-L, TGFβ and CHX alone or in combination. Apoptosis was then assessed by PI staining and quantifying the percentage of hypodiploid nuclei (pre-G1) using FACS analysis that was performed on a FACS Calibur flow cytometer (Becton–Dickinson) using the CellQuest program. Trypan blue exclusion test was used for determination of cell viability and total death levels.
Western blot analysis
Total cell lysates (50 μg protein) were resolved on SDS-PAGE, and processed according to standard protocols. The monoclonal antibodies used for Western blotting included: anti-β-Actin (Sigma, St. Louis, MO, USA); anti-caspase-8, anti-caspase-3 (Cell Signaling, Danvers, MA, USA); The polyclonal antibodies used included anti-phospho-p44/p42 MAP kinase (T202/Y204) and anti-p44/p42 MAP kinase; anti-phospho-JNK and anti-JNK1-3; anti-phospho-cJun (S73) and anti-cJun; anti-phospho-AKT (S473) and anti-AKT; anti-phospho-p65 (S536) NF-κB and anti-p65 NF-κB, anti-phospho-STAT3 (Y705) and anti-STAT3; anti-p53, anti-Bax, anti-β-Catenin, anti-Sox2, anti-Nanog, anti-TGF-β, anti-TGF-β-Receptor-2 and anti-PARP-1 (Cell Signaling, Danvers, MA, USA); anti-Fas, anti-Fas-L, anti-DR5/TRAIL-R2, anti-DR4/TRAIL-R1 and anti-TRAIL (Alexis, San Diego, CA, USA); anti-Survivin (R&D, USA) The secondary antibodies were conjugated to horseradish peroxidase; signals were detected using the ECL system (Thermo Scientific, Rockford, IL, USA).
ELISA for TRAIL detection in the media
Antibody pair used in sandwich ELISA for detection of human TRAIL was from R&D System (Minneapolis, MN, USA).
Statistical analysis
Data from four to five independent experiments were calculated as means and standard deviations. Comparisons of results between treated and control groups were made by the Student’s t tests. A p value of 0.05 or less between groups was considered significant.
Abbreviations
- FACS:
-
Fluorescence-activated cell sorter
- FGF2:
-
Fibroblast growth factor-2 (basic)
- DR5:
-
Death receptor-5 (synonym for TRAIL-R2)
- IκB:
-
Inhibitor of NF-κB
- IKK:
-
Inhibitor nuclear factor kappa B kinase
- JNK:
-
c-Jun N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
MAPK/ERK kinase
- MEF:
-
Median fluorescence intensity
- NF-κB:
-
Nuclear factor kappa B
- NSC:
-
Neural stem cells
- PARP1:
-
Poly (ADP-ribose) polymerase-1
- PI:
-
Propidium iodide
- STAT:
-
Signal transducers and activators of transcription
- TGFβ:
-
Transforming growth factor beta
- TGFβ-R:
-
TGFβ-receptor
- TNFα:
-
Tumor necrosis factor alpha
- TRAIL:
-
TNF-related apoptosis ligand
- TRAIL-R:
-
TRAIL-receptor
- zVAD:
-
Carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone
References
Monje ML, Mizumatsu S, Fike JR, Palmer TD (2002) Irradiation induces neural precursor-cell dysfunction. Nat Med 8:955–962
Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR (2003) Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Res 63:4021–4027
Acharya MM, Lan ML, Kan VH, Patel NH, Giedzinski E, Tseng BP, Limoli CL (2010) Consequences of ionizing radiation-induced damage in human neural stem cells. Free Radic Biol Med 49:1846–1855
Acharya MM, Christie LA, Lan ML, Giedzinski E, Fike JR, Rosi S, Limoli CL (2011) Human neural stem cell transplantation ameliorates radiation-induced cognitive dysfunction. Cancer Res 71:4834–4845
Hellstrom NA, Bjork-Eriksson T, Blomgren K, Kuhn HG (2009) Differential recovery of neural stem cells in the subventricular zone and dentate gyrus after ionizing radiation. Stem Cells 27:634–641
Greene-Schloesser D, Robbins ME, Peiffer AM, Shaw EG, Wheeler KT, Chan MD (2012) Radiation-induced brain injury: a review. Front Oncol 2:73
Okada H, Mak TW (2004) Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 4:592–603
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135:1311–1323
Hei TK, Zhou H, Chai Y, Ponnaiya B, Ivanov VN (2011) Radiation induced non-targeted response: mechanism and potential clinical implications. Curr Mol Pharmacol 4:96–105
Ivanov VN, Ghandhi SA, Zhou H, Huang SX, Chai Y, Amundson SA, Hei TK (2011) Radiation response and regulation of apoptosis induced by a combination of TRAIL and CHX in cells lacking mitochondrial DNA: a role for NF-kappaB-STAT3-directed gene expression. Exp Cell Res 317:1548–1566
Ivanov VN, Zhou H, Ghandhi SA, Karasic TB, Yaghoubian B, Amundson SA, Hei TK (2010) Radiation-induced bystander signaling pathways in human fibroblasts: a role for interleukin-33 in the signal transmission. Cell Signal 22:1076–1087
Prise KM, O’Sullivan JM (2009) Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer 9:351–360
Morgan WF (2003) Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro. Radiat Res 159:567–580
Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132:645–660
Allen JE, El-Deiry WS (2012) Regulation of the human TRAIL gene. Cancer Biol Ther 13:1143–1151
Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E et al (1995) Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol 15:3032–3040
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4:313–321
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Haase G, Pettmann B, Raoul C, Henderson CE (2008) Signaling by death receptors in the nervous system. Curr Opin Neurobiol 18:284–291
Song JH, Bellail A, Tse MC, Yong VW, Hao C (2006) Human astrocytes are resistant to Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. J Neurosci 26:3299–3308
Schneider L, Fumagalli M, d’Adda di Fagagna F (2012) Terminally differentiated astrocytes lack DNA damage response signaling and are radioresistant but retain DNA repair proficiency. Cell Death Differ 19:582–591
Cheung NK, Dyer MA (2013) Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer 13:397–411
Jaing TH, Yang CP, Hung IJ, Wang HS, Tseng CK, Hsueh C (2003) Brain metastases in children with neuroblastoma–a single-institution experience. Med Pediatr Oncol 41:570–571
Ivanov VN, Hei TK (2011) Regulation of apoptosis in human melanoma and neuroblastoma cells by statins, sodium arsenite and TRAIL: a role of combined treatment versus monotherapy. Apoptosis 16:1268–1284
Johnsen JI, Lindskog M, Ponthan F, Pettersen I, Elfman L, Orrego A, Sveinbjornsson B, Kogner P (2004) Cyclooxygenase-2 is expressed in neuroblastoma, and nonsteroidal anti-inflammatory drugs induce apoptosis and inhibit tumor growth in vivo. Cancer Res 64:7210–7215
Rasmuson A, Kock A, Fuskevag OM, Kruspig B, Simon-Santamaria J, Gogvadze V, Johnsen JI, Kogner P, Sveinbjornsson B (2012) Autocrine prostaglandin E2 signaling promotes tumor cell survival and proliferation in childhood neuroblastoma. PLoS One 7:e29331
Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, Corrias MV, Moretta A, Bottino C (2013) Neuroblastoma-derived TGF-beta1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol 190:5321–5328
Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, Barcellos-Hoff MH (2012) Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res 72:4119–4129
Ashkenazi A, Holland P, Eckhardt SG (2008) Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol 26:3621–3630
Finnberg N, Klein-Szanto AJ, El-Deiry WS (2008) TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest 118:111–123
Adams C, Totpal K, Lawrence D, Marsters S, Pitti R, Yee S, Ross S, Deforge L, Koeppen H, Sagolla M, Compaan D, Lowman H, Hymowitz S, Ashkenazi A (2008) Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death Differ 15:751–761
Balyasnikova IV, Ferguson SD, Han Y, Liu F, Lesniak MS (2011) Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett 310:148–159
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1:112–119
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714
Karin M (2009) NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 1:a000141
Walczak H (2013) Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol 5:a008698
Knight JC, Scharf EL, Mao-Draayer Y (2010) Fas activation increases neural progenitor cell survival. J Neurosci Res 88:746–757
Ghandhi SA, Ming L, Ivanov VN, Hei TK, Amundson SA (2010) Regulation of early signaling and gene expression in the alpha-particle and bystander response of IMR-90 human fibroblasts. BMC Med Genomics 3:31
Ivanov VN, Ghandhi SA, Zhou H, Huang SX, Chai Y, Amundson SA, Hei TK (2011) Radiation response and regulation of apoptosis induced by a combination of TRAIL and CHX in cells lacking mitochondrial DNA: a role for NF-[kappa]B-STAT3-directed gene expression. Exp Cell Res 317:1548–1566
Zhou H, Ivanov VN, Lien YC, Davidson M, Hei TK (2008) Mitochondrial function and nuclear factor-kappaB-mediated signaling in radiation-induced bystander effects. Cancer Res 68:2233–2240
Ivanov VN, Hei TK (2006) Dual treatment with COX-2 inhibitor and sodium arsenite leads to induction of surface Fas Ligand expression and Fas-Ligand-mediated apoptosis in human melanoma cells. Exp Cell Res 312:1401–1417
Zhou H, Ivanov VN, Gillespie J, Geard CR, Amundson SA, Brenner DJ, Yu Z, Lieberman HB, Hei TK (2005) Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc Natl Acad Sci USA 102:14641–14646
Ivanov VN, Hei TK (2013) Induction of apoptotic death and retardation of neuronal differentiation of human neural stem cells by sodium arsenite treatment. Exp Cell Res 319:875–887
Acknowledgments
We would like to thank Drs. Adayabalam Balajee, Mercy Davidson, Peter Grabham and Howard Lieberman for advice, critical reading of the manuscript and discussion. This work was supported by NIH Grant P01 CA049062 and Pilot Grant of the Department of Dermatology, Columbia University (P30AR044531-11, Project GG006336).
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ivanov, V.N., Hei, T.K. A role for TRAIL/TRAIL-R2 in radiation-induced apoptosis and radiation-induced bystander response of human neural stem cells. Apoptosis 19, 399–413 (2014). https://doi.org/10.1007/s10495-013-0925-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-013-0925-4