
Abstract
Brain-derived neurotrophic factor (BDNF), in addition to its neurotrophic action, also possesses antioxidant activities. However, the underlying mechanisms remain to be fully defined. Sestrin2 is a stress-responsive gene implicated in the cellular defense against oxidative stress. Currently, the potential functions of sestrin2 in nervous system, in particular its correlation with neurotrophic factors, have not been well established. In this study, we hypothesized that BDNF may enhance sestrin2 expression to confer neuronal resistance against oxidative stress induced by 3-nitropropionic acid (3-NP), an irreversible mitochondrial complex II inhibitor, and characterized the molecular mechanisms underlying BDNF induction of sestrin2 in primary rat cortical cultures. We found that BDNF-mediated sestrin2 expression in cortical neurons required formation of nitric oxide (NO) with subsequent production of 3′,5′-cyclic guanosine monophosphate (cGMP) and activation of cGMP-dependent protein kinase (PKG). BDNF induced localization of nuclear factor-kappaB (NF-κB) subunits p65 and p50 into neuronal nuclei that required PKG activities. Interestingly, BDNF exposure led to formation of a protein complex containing at least PKG-1 and p65/p50, which bound to sestrin2 promoter with resultant upregulation of its protein products. Finally, BDNF preconditioning mitigated production of reactive oxygen species (ROS) as a result of 3-NP exposure; this antioxidative effect of BDNF was dependent upon PKG activity, NF-κB, and sestrin2. Taken together, our results indicated that BDNF enhances sestrin2 expression to confer neuronal resistance against oxidative stress induced by 3-NP through attenuation of ROS formation; furthermore, BDNF induction of sestrin2 requires activation of a pathway involving NO/PKG/NF-κB.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondrial dysfunction has been implicated in the pathogenesis of a number of different neurodegenerative disorders [1]. 3-Nitropropionic acid (3-NP), an irreversible inhibitor of succinate dehydrogenase in the complex II of electron transport chain [2], induces ATP depletion and increases oxidative damage [3], leading eventually to mitochondrial dysfunction and neuronal death. In particular, 3-NP has been used in the context of Huntington’s disease (HD) because 3-NP administration in rodents and nonhuman primates results in behavioral and motor deficits recapitulating the HD phenotypes in humans [4, 5]. Previously, we have demonstrated that brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, exerts remarkable protective effects against 3-NP-induced toxicity in primary rat cortical cultures [6]. Apart from its well-established neurotrophic effects [7, 8], BDNF has also been proposed to possess antioxidant actions; these include increased expression levels of superoxide dismutases and glutathione reductase [9] as well as reduction in the extents of tyrosine nitration, an index for oxidative protein damage [10]. Recently, we have also reported that BDNF may induce the expression of an ATP-dependent antioxidant protein, namely sulfiredoxin, to counteract mitochondrial inhibition in cortical neurons [11]. In addition, BDNF has also been shown to restore the reduced mitochondrial electron coupling capacity and upregulate mitochondrial uncoupling protein 2 (UCP2), which acts as an antioxidant by reducing superoxide anion production, in rostral ventrolateral medulla of spontaneously hypertensive rats [12]. Thus, it is tempting to speculate that antioxidative mechanisms of BDNF may contribute significantly to its pro-survival effects in various experimental systems modeling neurodegeneration, including 3-NP-induced oxidative damages in the nervous systems.
Sestrin2, also known as hypoxia-inducible gene 95 (Hi95) that may be activated by prolonged hypoxia [13], is important for the maintenance of metabolic homeostasis. Sestrin2 is a stress-responsive protein that plays critical roles in hypoxia [14], DNA damage [15], and oxidative stress [16]. Previously, induction of sestrin2 has been proposed to carry cytoprotective activity against various stressors, such as hydrogen peroxide and ischemia, through regeneration of functional peroxiredoxins from the overoxidized ones under severe oxidative stress [13]. Recently, possible antioxidant functions of sestrin2 in the nervous systems begin to emerge. For example, a recent report revealed the protective effect of sestrin2 induction against neurotoxicity associated with MPP+, a mitochondrial complex I inhibitor, in neuroblastoma SH-SY5Y cells [17]. Sestrin2 also controls reactive oxygen species (ROS)-dependent neuropathic pain signaling after peripheral nerve injury [18]. Together, these findings point to the neuroprotective potential of sestrin2, at least in part via attenuation of oxidative stress, in the experimental models mimicking neurodegeneration.
Despite the identified mediators downstream of BDNF, such as sulfiredoxin [11], the multi-faceted defensive mechanisms of BDNF, especially those relating to antioxidant action, remain obscure. The potential link of sestrin2 to neurotrophic factors, in particular neurotrophins like BDNF, has never been established. In the present study, we tested the hypothesis that BDNF may enhance sestrin2 expression to confer neuronal resistance against oxidative stress induced by 3-NP and characterized the molecular mechanisms underlying BDNF induction of sestrin2 in primary rat cortical cultures.
Materials and Methods
Chemicals and Reagents
BDNF and 3-NP were prepared as described in our previous reports [6, 19]. The stock solutions of L-NG-Nitroarginine methyl ester (L-NAME; 10 mM; Cat. No. N5751, Sigma; St. Louis, MO, USA), 8-bromoguanosine 3′,5′-cyclic monophosphate (8-Br-cGMP; 10 mM; Cat. No. 17110, Sigma), and SN50 (1 mM; Cat. No. P-600, Enzo Life Sciences; Farmingdale, NY, USA) were all prepared in sterile ddH2O. The stock solutions of KT5823 (100 μM; Cat. No. K1388, Sigma), 1H-[1, 2, 4] Oxadiazolo [4,3-a] quinoxalin-1-one (ODQ; 10 mM; Cat. No. 0880, Tocris Bioscience; Minneapolis, MN, USA), pifithrin-α (PFTα; 272 mM; Cat. No. 506132, Millipore, Billerica, MA, USA), and 2-methooxyestradiol (2ME2; 10 mM; Cat. No. M6383; Sigma) were dissolved in dimethyl sulfoxide (DMSO). The anti-TrkB IgG (Cat. No. AF1494; R&D Systems Inc.; Minneapolis, MN, USA) used for blocking TrkB receptor was dissolved in sterile PBS at 0.2 mg/ml as a stock solution.
Primary Fetal Rat Cortical Culture
All the procedures for animal care and preparation of fetal rat cortical cultures were reviewed and approved by the “Institutional Animal Care and Use Committee (IACUC) of National Yang-Ming University” under the protocol title of “Roles of sestrin2 in BDNF neuroprotection against mitochondrial dysfunction” with the IACUC approval number of 1011225. All the experiments involving animals were performed humanely in accordance with the guidelines described in the “User Manual of Laboratory Animal Center at National Yang-Ming University”. Primary neuronal cultures were prepared from cortices of Sprague-Dawley fetal rat brains at embryonic day 18 as previously described [20]. The rat cortical cultures were a neuron-enriched co-culture system as evidenced by more cells immunostained with antibodies recognizing MAP-2 (approximately 85 %) than with GFAP (approximately 15 %), the respective cellular marker for neurons and astrocytes [20].
Transfection of siRNA
The detailed protocols for transfection of siRNAs into primary cortical cultures have been described in our earlier publication [11]. All the siRNAs, including the negative control (NC) siRNA with scrambled sequences, were purchased from GE Dharmacon Inc. (Lafayette, CO, USA). The target sequences for the Accell SMARTpool siRNA mixtures are as follows: GUUUUGAGCUGGAGAAGUC (A-052642-13), CCAUCAAUGUGAAAGUUGG (A-052642-14), GCAGCCUGUUCUUUGGUUA (A-052642-15), UCUUUGGCAUCAGAUACGA (A-052642-16) for sestrin2; CCGUCUGGUUGUAGGAAUA (A-089158-13), CUCUCAACUAUGUAUGUAC (A-089158-14), CCAUUUGUAAUAAAGUAUA (A-089158-15), GCGCUGUGAUCGUUUAUAA (A-089158-16) for c-jun; GCAUUAAUUCGAGAUAUAC (A-080047-13), CACAUUUCCUUCAUGGUUU (A-080047-14); UCAGCAUGUUACGUGAUGA (A-080047-15); UGUGUAAGUGUAAAUACUA (A-080047-16) for nuclear factor (erythroid-derived 2)-like 2 (Nrf2); CCGUGAGGCUGUUUGGUUU (A-080033-13), GCCUUAUAUCCAGUGUCUU (A-080033-14), CUUUUAUCAGCAAUAUCUC (A-080033-15), UUGCCAGACACAGACGAUC (A-080033-16) for p65; CCGCUGUCAUUAAGGUAUC (A-089855-13), CCCAUACCUUCAAAUACUA (A-089855-14), CCAUUGUCUUCAAAACACC (A-089855-15), CCUGCAAAGGUUAUCGUUC (A-089855-16) for p50. A non-targeting Accell siRNA (D-001910-01) was used as a negative control in all siRNA transfection experiments. Cortical cultures grown on 6-well or 24-well plates were transfected with the target siRNA or the NC siRNA, 1 μM each, for 3 days (days in vitro 4-7; DIV4-7) in neurobasal medium supplemented with B27 (NB/B27; Cat. No. 21130349 for NB and Cat. No. 17504044 for B27; Gibco; Grand Island, NY, USA).
Hoechst Staining for Cell Survival
Hoechst staining to assess cell survival was performed as described in our previous publication; the definitions of “cell survival (%)” in Hoechst staining was also described in details therein [6].
Nitric Oxide Fluorescence Assay
Nitric oxide (NO) fluorescence assay was performed as described in our previous publication [21]. After BDNF treatment, cortical cultures were loaded with 10 μM diaminodifluorofluorescein diacetate (DAF-DA; Cat. No. D-23842, Molecular Probes; Carlsbad, CA, USA) in 1× PBS for 60 min at room temperature; this was followed by one wash in 1 × PBS for 10 min to remove the excessive probes. For those cortical cultures grown on coverslips, the enhanced fluorescence signals indicative of NO production were detected under a confocal microscope (excitation/emission, 495 nm/515 nm; Zeiss LSM700, Jena, Germany).
Determination of Cellular 3′,5′-Cyclic Guanosine Monophosphate Contents
Cellular cGMP contents were determined using the Direct cGMP Enzyme Immunoassay Kit (Catalog No. 900-014, Assay Designs, Ann Arbor, MI, USA). In brief, cells were washed once with 1× PBS and lysed in 0.1 M HCl for 20 min at 4 °C. Lysates were centrifuged at 20,000×g for 20 min to pellet the debris. The cellular cGMP contents in the supernatant were determined according to the manufacturer’s instructions.
Preparation of Nuclear and Cytoplasmic Extracts
Nuclear and cytoplasmic extracts were fractionated using ProteoJET cytoplasmic and nuclear protein extraction kit (Cat. No. K0311; Fermentas International Inc., Burlington, Canada). Cortical cultures were seeded at a density of 1.3 × 106 cells per dish in 6-cm culture plates. Following experimental manipulations, cells were scraped off from the plates and collected by centrifugation at 250×g for 7 min. Fractionations for nuclear and cytosolic extracts were performed according to the manufacturer’s instructions.
Western Blotting
The rabbit antibodies against p50 (Cat. No. 04-234; Millipore) was diluted at 1:1000 in signal enhancer HIKARI solution 1 (Cat. No. NT08044-71R; Nacalai Tesque, Inc., Kyoto, Japan). The rabbit antibodies against sestrin2 (1:1000; Cat. No. 10795-1-AP; Proteintech Group Inc., Chicago, IL, USA), p65 (1:1000; Cat. No. sc-372; Santa Cruz BioTechnology, Inc., Santa Cruz, CA, USA), histone 3 (1:10000; Cat. No. 9715; Cell Signaling Technology), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1000; Cat. No. 2118; Cell Signaling Technology), and PKG-1 (1:1000; Cat. No. sc-25429; Santa Cruz BioTechnology, Inc.) were all diluted in 5 % non-fat milk in TTBS buffer (0.05 % Tween-20, 0.2 M NaCl in 20 mM Tris-HCl, pH 7.5). The mouse antibody against β-actin (1:7000; Cat. No. MAB1501; Millipore) was diluted in 5 % non-fat milk in TTBS. The conditions for membrane wash, detection of immunoreactive signals, and quantification of signal intensities on the blots were performed as described in our previous publication [19]. The β-actin was included in all the Western blotting experiments to serve as an internal control for equal loading of total cellular proteins in each lane. Where necessary, histone 3 and GAPDH were included as the respective internal reference for equal loading of nuclear and cytosolic proteins in each lane.
Immunocytochemistry by Confocal Microscopy
The following primary antibodies were diluted in blocking buffer (2 % normal goat serum, 0.3 % Triton X-100 in 1× PBS) at 1:100 for immunocytochemistry: the same rabbit antibodies used to detect sestrin2, p50, and PKG-1 for Western blotting, the mouse monoclonal antibodies against p65 (Cat. No. 610868; BD Biosciences) and MAP-2 (Cat. No. MAB378, Chemicon International, Inc.). The Dylight 594-conjugated goat anti-mouse IgG secondary antibodies (1:100; Cat. No. GTX76719; GeneTex, Hsinchu City, Taiwan) and Hylite 488-conjugated goat anti-rabbit IgG (1:100; Cat. No. 61056-H488; Anaspec, Fremont, CA, USA) were applied to respectively recognize the mouse and the rabbit IgGs. The coverslips were examined under a laser-scanning confocal microscope (Zeiss LSM700) equipped with filter sets to detect Dylight 594 (exCitation/emission, 595 nm/615 nm) and Hylite 488 (exCitation/emission, 494 nm/518 nm) fluorescence signals.
Co-immunoprecipitation
Nuclear extracts were fractionated using NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Cat. No. 78833; Thermo Scientific). Cortical cultures were scrapped off from the plates in CER I buffer supplied in this kit. After vigorous vortex at the highest speed for 15 s, the samples were incubated on ice for 10 min. Following addition of ice-cold CER II buffer supplied in the kit, the samples were then incubated on ice for additional 1 min and then centrifuged at 16,000×g for 5 min. The pellets were subjected to vortex at the highest speed for 15 s in the ice-cold NER buffer; this was followed by incubation for 40 min on ice with a 15-s vortex every 10 min. The nuclear extracts were collected by centrifugation at 16,000×g for 10 min. The same p65 and PKG-1 antibodies for Western blotting were used in the co-immunoprecipitation assay. Briefly, 50 μl magnetic beads were pre-incubated with 5 μg primary antibody against p65, PKG-1, or normal rabbit IgG (Cat. No. 12-370; Millipore) at room temperature for 4 h with gentle agitation at 4 rpm in 500 μl dilution buffer (Cat. No. CS200624; Millipore). The protein G magnetic beads (Cat. No. LSKMAGG10; Millipore) were washed twice in washing buffer (0.05 % Tween-20, 0.2 M NaCl in 20 mM Tris-HCl, pH 7.5). The immunoprecipitation reaction was conducted in the 500-μl mix containing the washed magnetic beads conjugated with the primary antibody and 70 μg nuclear proteins in dilution buffer at 4 °C overnight with gentle agitation at 4 rpm. At the completion of incubation, the magnetic beads conjugated with the primary antibody and its interacting proteins were washed three times in washing buffer and incubated in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 0.25 % Na-deoxycholate and 1 % NP-40) containing sample buffer at 95 °C for 10 min. The antibody and its interacting proteins detached from the magnetic beads were then subjected to Western blotting.
Real-Time Reverse Transcription-Coupled Polymerase Chain Reaction
Extraction of total RNA from cortical cultures, reverse transcription for cDNA construction, and real-time polymerase chain reaction (PCR) were performed as previously described [19]. The primers used in current study were: 5′- TACCTTAGCAGCTTCTGGCG-3′ (forward) and 5′- AGGTAAGAACACTGGTGGCG-3′ (reverse) for sestrin2; 5′-AGAGACAGCCGCATCTTCTTG-3′ (forward) and 5′-CGACCTTCACCATCTTGTCTATGA-3′ (reverse) for GAPDH. GAPDH was included as an internal control for PCR reaction.
Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation (ChIP) assay was performed using the Magna ChIPTM G (Cat. No. 17-611; Millipore) according to manufacturer’s protocols. Briefly, cortical cultures were washed with 1× PBS and then crosslinked with 1 % paraformaldehyde at room temperature for 15 min; this was followed by addition of the glycine supplied in kit for 5 min. Cultures were washed twice with 1× PBS and then collected in 1× PBS with protease inhibitor supplied in the kit. The samples were centrifuged at 425×g for 4 min and, after removing the supernatants, the pellets were re-suspended in 0.5 ml cell lysis buffer with protease inhibitor for 15 min on ice, vortexing briefly every 5 min. Thereafter, the samples were centrifuged at 425×g for 10 min and the pellets re-suspended in 0.5 ml nuclear lysis buffer with protease inhibitor. Following extraction of the nuclei, chromatin DNA was subjected to sonication (Sonicator S-4000; Qsonica, LLC., Newtown, CT, USA) under the condition of 2-s “pulse-on” and 1-s “pulse-off”, processed for a total of 2 min on ice. Equal amounts (100 μg) of nuclear proteins were incubated with 20 μl beads and 1 μg primary antibody against p65 (Cat. No. 610868; BD Biosciences), p50 (Cat. No. 04-234; Millipore), or normal rabbit IgG at 4 °C overnight with rotation at 4 rpm. Thereafter, the samples were washed sequentially, 5 min each, in low-salt buffer, high-salt buffer, LiCl buffer, and TE buffer on ice with shaking before addition of elution buffer with protease K. Samples were incubated at 62 °C for 2 h with shaking followed by incubation at 95 °C for 10 min. The immunoprecipitated DNA was purified through the Fast-Spin column provided in the kit and then eluted in 50 μl elution buffer supplied in the kit. The sequences of the primers used for ChIP assay were 5′-TGTAGCAACGAGGATTGCGA-3′ (forward) and 5′-TCCTGGAGGGACTGGTTAGG-3′ (reverse), with a predicted amplicon size of 236 bp. The PCR reactions were performed in a 10-μl reaction mixture that contained equal volume of DNA samples, forward and reverse primers (final concentration at 500 nM each), and 2× Taq DNA Polymerase Master Mix Red containing 1.5 mM MgCl2 (Cat. No. 180303, Ampliqon A/S, Denmark) and were subjected to the following thermocycle program: 95 °C for 3 min followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 57 °C for 30 s, and extension at 72 °C for 1 min. The PCR products were then analyzed by gel electrophoresis through 2 % agarose gels (Cat. No. UR-AGA001, UniRegion Bio-Tech, Taiwan) in 0.5× TBE buffer (Cat. No. UR-TBE-001-1L, UniRegion Bio-Tech).
Determination of ROS Production
Prior to BDNF preconditioning at DIV-7, cortical neurons were subjected to one medium exchange in neurobasal without B27 for 1 h in order for the cells to accommodate the B27-free culture condition. After BDNF pretreatment and subsequent 3-NP exposure, cortical cultures were stained with 5 μM CellROXTM Green Reagent (Cat. No. C10444; Molecular Probe) diluted in neurobasal medium without B27 at 37 °C for 30 min. The coverslips were washed three times in 1× PBS and then immediately observed under a laser-scanning confocal microscope (Zeiss LSM700) equipped with filter sets to detect ROS fluorescence signals (excitation/emission, 485 nm/520 nm). Quantitative analyses of the fluorescence signal intensities on the micrographs were conducted using the MetaMorph software.
Statistical Analysis
Results are expressed as mean ± SEM from the sample numbers (N). For counting of Hoechst-stained surviving cells, the N represents data collected from one independent experiment, which contained two to three coverslips for every experimental condition, using one cortical culture. On each coverslip, three vision fields were randomly selected for counting to obtain the average numbers of surviving cells in each vision field. The mean numbers of surviving cells for each independent experiment were therefore derived from a total of six to nine vision fields out of two to three coverslips for the same experimental condition. For Western blotting, real-time reverse transcription-coupled polymerase chain reaction (RT-PCR), and ChIP assays showing quantitative results, each N represents data collected from one experiment using one culture; combined data from at least three independent using three different cultures are shown. For all the double immunofluorescence staining, representative images from three independent experiments using three different cultures are shown. Multiple groups were analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test; two groups were analyzed by unpaired, two-tailed Student’s t test. P values of less than 0.05 were considered significant.
Results
Sestrin2 Induction Mediates BDNF-Dependent 3-NP Resistance in Cortical Neurons
We first examined the BDNF effects on the expression level of sestrin2 in primary rat cortical cultures. Results indicated that BDNF enhanced sestrin2 expression beginning at 4 h and the heightened sestrin2 level sustained until 8 h (Fig. 1a). Confocal microscopy revealed that BDNF-induced sestrin2 co-localized to MAP-2, indicating neuronal induction of this protein upon BDNF exposure (merged micrographs of MAP-2 and sestrin2; Fig. 1b). The cortical cultures used in the present study were a neuron-enriched culture system containing approximately 85 % of neurons and 15 % of astrocytes [20]; we therefore also examined the BDNF effects on sestrin2 expression in astrocytes using GFAP as a marker protein. The results indicated that basal sestrin2 expression can also be co-localized to GFAP-positive cells in both control and BDNF-treated cultures; however, BDNF exposure did not lead to significant changes in sestrin2 expression in the GFAP-positive cells (data not shown). BDNF binds to TrkB receptor on the surface of neuronal cells to trigger its downstream signaling pathways [22]. We therefore tested whether BDNF induction of sestrin2 in neurons requires TrkB receptor. The results indicated that BDNF-induced sestrin2 expression was inhibited by the anti-TrkB neutralizing antibody (Fig. 1c) capable of blocking TrkB functions, indicating that sestrin2 inducion by BDNF requires biological function of TrkB receptor. In the absence of BDNF, however, sestrin2 levels appeared unaffected by the anti-TrkB IgG, suggesting that basal expression of sestrin2 does not require BDNF-TrkB signaling (Fig. 1c). Given the co-localization of BDNF-induced sestrin2 in MAP-2-positive neurons (Fig. 1b) and the requirement of TrkB function, which is specific to neurons, for BDNF-mediated sestrin2 induction in cultures (Fig. 1c), we believed that sestrin2 induction mediated by BDNF occurs mainly, if not exclusively, in neurons of the mixed primary cultures. In addition to the neurotrophin BDNF, recently, we have reported that preconditioning of cortical cultures with oncostatin M, a cytokine of the interleukin-6 (IL-6) family, also protects neurons against 3-NP toxicity via induction of Mcl-1 with improvements of mitochondrial bioenergetics [23]. It is therefore interesting to test whether the observed sestrin2 induction is specific to BDNF. Results shown in Fig. 1d clearly demonstrated that oncostatin M, at the dosage of 100 ng/ml capable of conferring 3-NP resistance to cortical neuorns [23], failed to increase cellular sestrin2 contents, suggesting that induction of sestrin2 expression is specific to the neurotrophin BDNF.

Sestrin2 induction mediates BDNF-dependent 3-NP resistance in cortical neurons. a Cells were exposed to BDNF (100 ng/ml) at indicated times before detection of sestrin2 by Western blotting. N = 3. b Confocal micrographs showing BDNF-induced sestrin2 expression in MAP-2-positive neurons. Cortical cultures were treated with BDNF (100 ng/ml) for 4 h before immunostaining with the antibodies against MAP-2 (red) and sestrin2 (green). Hoechst 33258 served as counterstaining for nuclei (blue). Scale bar = 10 μm. Note heightened sestrin2 expression that is co-localized to MAP-2 signal upon BDNF exposure. c Cultures were exposed to BDNF (100 ng/ml) with normal mouse IgG (5 μg/ml) or anti-TrkB IgG (5 μg/ml) for 4 h before detection of sestrin2. N = 3. d Cells were exposed to oncostatin M (100 ng/ml) at indicated times before detection of sestrin2 by Western blotting. N = 3. Note that sestrin2 expression was not induced by oncostatin M in cortical neurons within the 8-h period of exposure time. e Cortical cultures were transfected with sestrin2 siRNA (1 μM) or negative control (NC) siRNA (1 μM) for 72 h before detection of sestrin2. N = 3. f The experimental condition was the same as described in e except that, at the end of transfection, cortical cultures were exposed to BDNF (100 ng/ml) for 4 h before sestrin2 detection. N = 4. g Cortical cultures were transfected with sestrin2 siRNA (1 μM) or NC siRNA (1 μM) for 72 h followed by exposure to BDNF (100 ng/ml) for 8 h and then 3-NP (2.5 mM) for additional 24 h. Cells were then subjected to Hoechst staining to determine the extents of cell survival. N = 6–15. Mean ± SEM. *, #, and § all denote P < 0.05. In (a), * denotes P < 0.05 as compared to control cultures without BDNF treatment
To further establish the causative relationship between BDNF-induced sestrin2 and its neuroprotective effects, siRNA was transfected into cortical neurons to knock down sestrin2 expression. We found that the sestrin2 siRNA at 1 μM effectively suppressed expression of both the endogenous (Fig. 1e) as well as the BDNF-induced (Fig. 1f) expression of sestrin2 proteins. Quantitative analyses revealed approximately 40 % reduction of endogenous sestrin2 contents compared to the cultures transfected with 1 μM NC siRNA in the absence of BDNF (Fig. 1e). Upon BDNF exposure, sestrin2 siRNA suppressed its protein expression down to the basal levels (Fig. 1f). Consistently, sestrin2 siRNA, but not the NC siRNA with scrambled sequences, abolished 3-NP resistance associated with BDNF preconditioning (Fig. 1g); however, sestrin2 siRNA alone did not affect cell survival in the control or 3-NP-treated cultures without prior BDNF exposure (Fig. 1g). Thus, sestrin2 induction plays a critical role in BDNF-mediated neuroprotection against 3-NP toxicity in cortical neurons.
NO/PKG-1 Pathway Is Involved in BDNF-Mediated Sestrin2 Induction
Robust evidence has shown that NO signaling cascades exert antioxidative action with cytoprotective effects in various experimental models [24–26]. Further, we have previously reported that BDNF-mediated neuroprotection against 3-NP involves nitric oxide synthase (NOS) activity [6]. We therefore first determined whether BDNF may affect NO production in cortical cultures. Confocal microscopy revealed BDNF (100 ng/ml) induced NO formation at 1 h (Fig. 2a). NO is known to activate soluble guanylate cyclase (sGC) to increase cellular cGMP levels [27]. In addition, we have demonstrated that S-nitrosoglutathione (GSNO)-dependent neuroprotective mechanism against amyloid beta-peptide (Aβ) toxicity was associated with activation of sGC/PKG pathway, in which cGMP is a well-known signaling mediator downstream of NO [28]. Therefore, we also determined whether BDNF may stimulate the production of cGMP in cortical cultures. Results indicated that exposure of cortical cultures to BDNF enhanced cellular cGMP contents at 1 and 2 h (Fig. 2b); at 2 h, the BDNF-induced cGMP production was abolished by the non-selective NOS inhibitor L-NAME (Fig. 2c). In addition to increasing cGMP production, we also detected a transient increase in the expression level of PKG-1 proteins at 1 and 2 h following BDNF exposure (Fig. 2d). Thus, BDNF may enhance PKG activity via two distinct pathways, namely increasing the production of cGMP through the NO-dependent mechanism and directly stimulating PKG-1 expression. Because BDNF-induced production of NO at 1 h (Fig. 2a) and cGMP contents at 1–2 h (Fig. 2b) preceded sestrin2 expression at 4–8 h (Fig. 1a), we hypothesized that BDNF-dependent sestrin2 induction may require prior activation of NO/cGMP/PKG-1. Consistent with this notion, BDNF-induced sestrin2 was abolished by L-NAME (Fig. 2e). Further, BDNF-induced sestrin2 was abrogated by ODQ and KT5823 (Fig. 2f), the respective inhibitor of sGC and PKG. Previously, we have reported that the membrane-permeable cGMP analogue 8-Br-cGMP at 20 μM protects cortical cultures against 3-NP toxicity [6]. In the scurrent study, we found that 8-Br-cGMP at the same dosage alone was sufficient to induce sestrin2 expression at 6–8 h (Fig. 2g). These findings thus indicated that BDNF-mediated neuroprotective effects involve production of NO with resultant induction of cGMP and enhancement of PKG-1 expression, together leading to PKG activation and ultimately sestrin2 expression.

Involvements of NO/cGMP/PKG in BDNF-dependent sestrin2 induction. a Cortical cultures were exposed to BDNF (100 ng/ml) for 1 h and then stained with DAF-DA before image acquisition under a confocal microscope. Scale bar = 100 μm. The arrows indicate NO-positive cells. b Cortical cultures were exposed to BDNF (100 ng/ml) for 1 and 2 h before the cellular cGMP contents were determined. N = 4. c Cortical cultures were exposed to BDNF (100 ng/ml) with or without L-NAME (100 μM) for 2 h before measurements of the cellular cGMP contents. N = 3. d Cultures were exposed to BDNF (100 ng/ml) at indicated times before detection of PKG-1. N = 4. e Cultures were exposed to BDNF (100 ng/ml) with or without the NOS inhibitor L-NAME (100 μM) for 4 h before detection of sestrin2. N = 3. f Cortical cultures were exposed to BDNF (100 ng/ml), with or without KT5823 (2 μM) or ODQ (10 μM), for 4 h before detection of sestrin2. N = 4. g Cultures were exposed to 8-Br-cGMP (20 μM) at indicated times before detection of sestrin2. N = 3. Mean ± SEM. * and # denote P < 0.05. In (d) and (g), * denotes P < 0.05 as compared to the respective control cultures without BDNF or 8-Br-cGMP treatments
Nuclear Factor-kappaB Mediates BDNF-Dependent Sestrin2 Induction and Neuroprotection
Having established the BDNF-NO-cGMP-PKG pathway mediating sestrin2 induction in cortical cultures, the transcription factor responsible for this BDNF effect remained unknown. The p53 [13], hypoxia-inducible factor-1 (HIF-1) [14], c-Jun [29], and Nrf2 [16] have all been reported to induce sestrin2 in various model systems. However, studies using the p53 inhibitor PFTα (Fig. 3a) and HIF-1α inhibitor 2ME2 (Fig. 3b) excluded potential involvements of these two transcription factors. Negative results were also obtained by using c-jun siRNA (Fig. 3c), which knocks down the expression of c-Jun protein effectively [11]. Further, BDNF-mediated sestrin2 was unaffected by nrf2 siRNA (Fig. 3e), despite its efficacy in suppressing Nrf2 protein expression (Fig. 3d). Together results shown in Fig. 3 indicated that sestrin2 induction by BDNF was not mediated by either of these four transcription factors. Next, we performed on-line search with the software (TRsearch) and identified the potential binding site for nuclear factor-kappaB (NF-κB) in the promoter region of sestrin2 gene. Interestingly, it has previously been demonstrated that PKG can enhance the transcriptional activity of NF-κB containing p65 and p50 subunits [30]. In this work, we found that BDNF induced a transient increase in the nuclear contents of p65 and p50 subunits at 2 h (Fig. 4a); immunocytochemical staining also confirmed BDNF-induced colocalization of p65 and p50 in the nucleus (merged micrographs of p65 + p50; Fig. 4b). Further, co-immunoprecipitation assay revealed that BDNF treatment increased nuclear contents of the immunoprecipitable p65, along with the associated p50 (Fig. 4c). Together, these results demonstrated that BDNF increases the nuclear p65/p50 contents and enhances their physical association to form the heterodimeric NF-κB complex.

Transcription factors p53, HIF-1, c-Jun, and Nrf2 are not involved in BDNF-mediated sestrin2 induction. a, b Cultures were exposed to BDNF (100 ng/ml) with or without PFTα (200 nM) in (a) or 2ME2 (20 μM) in (b) for 4 h before detection of sestrin2. N = 4 in (a) and N = 3 in (b). c Cortical cultures were transfected with c-jun siRNA (1 μM) or NC siRNA (1 μM) for 72 h. At the end of transfection, cells were exposed to BDNF (100 ng/ml) for 4 h before detection of sestrin2. N = 3. d Cortical cultures were transfected with nrf2 siRNA (1 μM) or NC siRNA for 72 h before detection of Nrf2. e Cortical cultures were transfected with nrf2 siRNA (1 μM) or NC siRNA (1 μM) for 72 h. At the end of transfection, cells were exposed to BDNF (100 ng/ml) for 4 h before detection of sestrin2. N = 3. Mean ± SEM. * denotes P < 0.05. The ns denotes “not significant”

Essential roles of NF-κB in BDNF-mediated sestrin2 induction and neuroprotection. a Cultures were exposed to BDNF (100 ng/ml) at indicated times before extraction of nuclear and cytoplasmic proteins for detection of p65 and p50; histone 3 and GAPDH served as the nuclear and cytoplasmic marker protein, respectively. N = 5. b Confocal micrographs showing nuclear co-localization of p65 and p50 induced by BDNF in cortical cells. Cultures were treated with BDNF (100 ng/ml) for 2 h before immunostaining with the antibodies against p65 (red) and p50 (green). Hoechst 33258 served as counterstaining for nuclei (blue). Scale bar = 10 μm. The arrows in the control cultures indicate relatively weaker nuclear signals of p65 and p50; the arrow heads indicate co-localization of p65 and p50 in the nuclei of the BDNF-treated cultures. c Cultures were treated with BDNF for 2 h before preparations of nuclear extracts. Equal amounts of nuclear proteins were subjected to immunoprecipitation with an antibody against p65; the immunoprecipitated samples were then immunoblotted by an antibody against p50. Representative blots from three independent experiments using three different cultures are shown. d, e Cortical cultures were transfected with p65 siRNA (1 μM), p50 siRNA (1 μM), or NC siRNA (1 μM) for 72 h. This was followed by exposure to BDNF (100 ng/ml) for 4 h before detection of p65 and p50 in (d) as well as sestrin2 in (e). N = 4. f The conditions for siRNA transfection were the same as described in e. At the end of transfection, cortical cultures were exposed to BDNF (100 ng/ml) for 8 h followed by incubation with 3-NP (2.5 mM) for additional 24 h before Hoechst staining to determine cell survival. N = 6. g Cortical cultures were exposed to BDNF (100 ng/ml) with or without SN50 (5 μM) for 4 h before detection of sestrin2. N = 4. h Cortical cultures were exposed to BDNF (100 ng/ml) with or without SN50 (5 μM) for 8 h followed by incubation with 3-NP (2.5 mM) or empty medium for additional 24 h before Hoechst staining. N = 9. Mean ± SEM. *, #, and § all denote P < 0.05. In (a), * denotes P < 0.05 as compared to the respective control cultures without BDNF treatment. In (d), * and # denote P < 0.05 as compared to the respective cultures transfected with NC siRNA
To test whether NF-κB subunits p65 and p50 play a critical role in BDNF-mediated sestrin2 induction and neuroprotection, we first tested the knockdown efficacies of p65 and p50 siRNAs. Results shown in Fig. 4d indicated that p65 siRNA at 1 μM significantly suppressed p65 expression; interestingly, expression of p50 was concomitantly downregulated by p65 siRNA. Quantitative analyses revealed approximately 40 and 20 % reduction of p65 and p50, respectively, as compared with the cultures transfected with 1 μM NC siRNA upon BDNF exposure. Reciprocally, transfection of p50 siRNA at 1 μM also resulted in 40 % inhibition of p50 and 30 % reduction of p65 as compared to the cortical cultures transfected with NC siRNA upon BDNF exposure (Fig. 4d). Importantly, BDNF-mediated sestrin2 induction was abrogated by both p65 and p50 siRNA at 1 μM (Fig. 4e); the same manipulations also abolished BDNF-dependent neuroprotection against 3-NP toxicity (Fig. 4f). In addition to siRNAs, the NF-κB peptide inhibitor SN50 also suppressed BDNF-mediated sestrin2 induction (Fig. 4g) and neuroprotection (Fig. 4h). Together, our results indicated that the NF-κB containing p65 and p50 subunits is a critical, if not the only, mediator in BDNF-stimulated sestrin2 expression and BDNF protection aganist 3-NP toxicity in cortical cultures.
NO/PKG Pathway Participates in BDNF-Induced Increases in Nuclear Contents of p65/p50 and Affects Their Binding Affinity to Sestrin2 Promoter
To further address the roles of NO/cGMP/PKG signaling in BDNF-induced nuclear translocation of p65/p50, cortical cultures were exposed to BDNF along with L-NAME, ODQ, or KT5823. We found that the increased nuclear contents of p65 and p50 as a result of BDNF exposure were inhibited by L-NAME (Fig. 5a) as well as KT5823 and ODQ (Fig. 5b). However, neither BDNF alone nor its combination with L-NAME significantly altered the total cellular contents of p65 and p50 proteins (Fig. 5c). Similar results were observed in the cultures exposed to BDNF with or without KT5823 and ODQ (Fig. 5d). Thus, BDNF-mediated activation of the NO/cGMP/PKG pathway enhanced nuclear contents of p65 and p50 without significantly altering their total cellular protein levels.

NO and PKG activity are important for BDNF-mediated nuclear translocation of p65 and p50. a, b Cortical cultures were exposed to BDNF (100 ng/ml) with or without L-NAME (100 μM) in (a) or KT5823 (2 μM) or ODQ (10 μM) in (b) for 2 h before detection of p65 and p50 in the nuclear fractions. N = 3. c, d The experimental conditions in (c) and (d) were the same as described in (a) and (b), respectively, except that total cellular contents of p65 and p50 were quantitatively determined. Mean ± SEM. *, #, and § all denote P < 0.05. Note that BDNF alone did not significantly increase the total cellular levels of p65 and p50; addition of L-NAME in (c) or KT5823 or ODQ in (d) was also without significant effects
To determine possible involvements of NF-κB in sestrin2 expression, we first tested whether sestrin2 induction by BDNF may be regulated at transcriptional level. Real-time RT-PCR shown in Fig. 6a demonstrated that BDNF significantly increased sestrin2 mRNA at 2–8 h, with the maximal induction observed at 4 h. We then conducted ChIP assays to demonstrate direct binding of NF-κB complex to the promoter of sestrin2 gene. Figure 6b is a schemic diagram to show the predicted NF-κB binding sites in the sestrin2 promoter as well as the positions of the PCR primers used in ChIP assays. Results indicated that BDNF enhanced binding of both p65 and p50 to the sestrin2 promoter, which was suppressed by the NOS inhibitor L-NAME (Fig. 6c) and the sGC inhibitor ODQ (Fig. 6d). Together, the results shown in Figs. 5 and 6 suggested that BDNF may induce nuclear translocation of NF-κB subunits p65 and p50 via activation of NO/cGMP/PKG pathway to enhance their binding to the sestrin2 promoter, resulting in upregulation of sestrin2 gene at mRNA level.

NO- and PKG-dependent enhancement of the p65/p50 binding affinity to the sestrin2 promoter induced by BDNF. a Cells were exposed to BDNF (100 ng/ml) at indicated times before total RNA isolation for amplification of sestrin2 mRNA by real-time RT-PCR. GAPDH was amplified as an internal standard. N = 3. b A diagram depicting the predicted NF-κB binding site in the sestrin2 promoter and the positions of the PCR primers used in ChIP assays. c, d Cultures were exposed to BDNF (100 ng/ml), alone or with L-NAME (100 μM) in (c) or ODQ (10 μM) in (d), for 2 h before ChIP assays using the antibodies against p65 and p50. The same amounts (1 μg) of normal rabbit IgG were also included in the ChIP assay to serve as the negative control (NC) for immunoprecipitation. N = 3. Mean ± SEM. * and # denote P < 0.05. In (a), * denotes P < 0.05 as compared to the control cultures without BDNF treatment
PKG-1 Is Involved in BDNF-Enhanced Sestrin2 Induction
Because nuclear localization signal (NLS) has been identified in PKG-1 sequence, we further explored possible nuclear translocation of PKG-1 in cortical cultures exposed to BDNF. Immunocytochemistry demonstrated BDNF-induced enhancement of PKG-1 immunofluorence in the MAP-2-positive cells, indicating its neuronal expression; more importantly, the enhanced PKG-1 signals induced by BDNF were detected in the nuclei (Fig. 7a). Fractionation of cytosolic and nuclear proteins followed by immunoblotting confirmed an increased nuclear content of PKG-1 in cultures exposed to BDNF for 2 h; indeed, basal level of PKG-1 was still detectable in the nuclear fractions even without BDNF treatment (Fig. 7b). Enhanced nuclear content of PKG-1 stimulated by BDNF required sGC activity as it was suppressed by ODQ (Fig. 7c). Because BDNF increased the nuclear contents of p65 and p50 (Fig. 4a) as well as PKG-1 (Fig. 7b), both at 2 h, we speculated that PKG-1 may interact with p65/p50 to form a protein complex. As expected, we found that the immunoprecipitated nuclear PKG-1 interacted, directly or indirectly, with p65 and p50 to a higher extent upon BDNF stimulation; further, a basal level of PKG-1 binding to p65 and p50 was observed in the nuclear extracts of control cultures even without BDNF treatment (Fig. 7d). Co-immunoprecipitation assay further revealed that association of PKG-1 with the NF-κB subunits p65 and p50 was abolished by the sGC inhibitor ODQ (Fig. 7e). These results indicated that increased nuclear PKG-1 as well as its interaction with p65/p50, both induced by BDNF, required sGC activity. Together, results shown in Fig. 7a–e suggested the possibility that BDNF may promote formation of a protein complex containing PKG-1, p65, and p50 that together binds to the sestrin2 promoter to stimulate its mRNA expression. Under this condition, binding of PKG-1 to the sestrin2 promoter can be expected. ChIP assay using the PKG-1 antibody confirmed this contention and revealed BDNF-induced binding of PKG-1, directly or indirectly, to the sestrin2 promoter containing the NF-κB binding motif (Fig. 7f).

BDNF-mediated nuclear localization of PKG-1, its association with NF-κB, and its binding to sestrin2 promoter. a Confocal micrographs showing BDNF-mediated increases in nuclear content of PKG-1. Cortical cultures were treated with BDNF (100 ng/ml) for 2 h before immunostaining with the antibodies against PKG-1 (green) and MAP-2 (red). Hoechst 33258 served as counterstaining for nuclei (blue). Scale bar = 10 μm. Note enhanced PKG-1 signal in the nuclei of the cortical cultures upon BDNF exposure, as indicated by the arrows. b Cultures were exposed to BDNF (100 ng/ml) for indicated times before extraction of nuclear and cytoplasmic proteins for the detection of PKG-1; histone 3 and GAPDH served as the respective marker for nuclear and cytoplasmic proteins. N = 3. c Cortical cultures were exposed to BDNF (100 ng/ml) with or without ODQ (10 μM) for 2 h before detection of nuclear contents of PKG-1. N = 3. d BDNF-induced physical association of PKG-1 with p65 and p50. Cortical cells were treated with BDNF (100 ng/ml) for 2 h before extraction of nuclear proteins. Equal amounts of nuclear proteins were immunoprecipitated with an antibody against PKG-1 or normal IgG, the latter served as the negative controls. The immunoprecipitated samples were then subjected to immunoblotting for detection of p50 and p65. e The experimental conditions were the same as described in (d) except addition of ODQ (10 μM) or the same amounts of solvent (DMSO) for 2 h. In both (d) and (e), representative blots from three independent experiments using three different cultures are shown. f Cultures were exposed to BDNF (100 ng/ml) for 2 h before ChIP assays using the PKG-1 antibody and the same set of PCR primers for demonstrating NF-κB binding to the sestrin2 promoter. N = 3. Mean ± SEM. * and # denote P < 0.05. In (a), * denotes P < 0.05 as compared to the control cultures without BDNF treatment
BDNF Attenuates 3-NP-Induced ROS Production
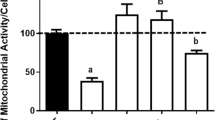
Having delineated the molecular mechanisms underlying BDNF-mediated sestrin2 induction, how sestrin2 may confer 3-NP resistance to cortical neurons still remains unknown. We have previously reported that N-acetyl-L-cysteine (NAC), a glutathione precursor capable of functioning as an antioxidant, attenuated 3-NP-induced cell death [20], suggesting that ROS production and oxidative stress may underlie 3-NP-mediated neuronal death. Sestrin2 has been shown to negatively regulate stress-induced ROS formation [31, 32]. To investigate the antioxidative properties of sestrin2 in cortical cultures under 3-NP-induced oxidative stress, the extents of ROS generation were determined by CellROXTM, a fluorescence probe for the detection of intracellular free radicals. Using confocal microscopy, we observed that 3-NP increased the cellular contents of ROS in cortical cultures, which was attenuated by BDNF preconditioning (Fig. 8). Notably, cotreatment with the NF-κB inhibitor SN50 or the PKG inhibitor KT5823 during the preconditioning phase reversed this antioxidant effect of BDNF (Fig. 8a). Further, sestrin2 siRNA, but not the NC siRNA, showed similar effects in increasing ROS formation despite the presence of BDNF (Fig. 8b); however, sestrin2 knockdown alone had no significant impact on the production of ROS in the absence of BDNF preconditioning (Fig. 8b). Together, these findings support the antioxidant property of BDNF mediated by NF-κB/PKG-dependent sestrin2 induction that together contributes to suppression of ROS generation and neurotoxicity resulted from 3-NP exposure in cortical neurons.

PKG/NF-κB/sestrin2 mediates BDNF-dependent attenuation of ROS production by 3-NP. a Cultures were exposed to BDNF (100 ng/ml) with or without SN50 (5 μM) or KT5823 (2 μM) for 8 h followed by incubation with 3-NP (2.5 mM) or empty medium for additional 10 h. b Cultures were transfected with sestrin2 siRNA (1 μM) or NC siRNA (1 μM) for 72 h. This was followed by exposure to BDNF (100 ng/ml) for 8 h and then incubation with 3-NP (2.5 mM) or empty medium for additional 10 h. In both (a) and (b), confocal microscopy was conducted to examine the extents of ROS production. Scale bar = 20 μm. The arrows indicate the cells with stronger ROS fluorescence signals. N = 3 in (a) and N = 3–6 in (b). Mean ± SEM. *, #, and § all denote P < 0.05. The ns denotes “not significant”
Discussion
In this work, we provided experimental evidence supporting a novel signaling cascade of “BDNF/TrkB→NO/cGMP/PKG→NF-κB→sestrin2→antioxidation→3-NP resistance” in primary cultures of fetal rat cortical neurons. Several novel findings are presented in the current study. First, to our best knowledge, the correlation of this stress-responsive protein, sestrin2, with neurotrophic factors and its contribution to the antioxidant effects of BDNF have never been revealed before. Second, unlike those previously reported transcriptional factors such as p53 [13], HIF-1 [14], c-Jun [29], and Nrf2 [16], we found in neuronal cells that NF-κB is the critical, if not the only, regulator controlling BDNF-dependent sestrin2 induction. Third, upstream of NF-κB is the activation of NO/cGMP/PKG pathway that enhances binding of a protein complex containing at least p65, p50, and PKG-1 to the sestrin2 promoter in the neuronal nuclei. In addition to PKG activation by production of cGMP, BDNF also stimulates PKG-1 expression and increases its nuclear levels. Together, these BDNF effects result in heightened nuclear contents of NF-κB subunits p65 and p50 without affecting their total cellular protein levels, leading ultimately to sestrin2 induction that contributes to the observed antioxidant effects of BDNF. Despite these novel findings supporting the crucial role of NF-κB-dependent sestrin2 induction mediating BDNF-induced antioxidant effects, whether overexpression of sestrin2 alone is sufficient to confer 3-NP resistance remains to be determined. However, it should be noted that sestrin2 may also promote autophagy in addition to antioxidant effects [33]. Although autophagy may help to eliminate the damaged intracellular organelles such as mitochondria, known as mitophagy [34], prolonged and excessive autophagy may result in autophagic cell death, thereby leading to neuronal demise [35]. Thus, it is speculated that properly regulated sestrin2 expression may be beneficial to neurons under oxidative stress while its overexpression may, on the contrary, induce autophagic cell death in the cortical neurons challenged with 3-NP. Further investigation is required to confirm this speculation.
NO is known to activate sGC to increase cellular cGMP levels [27]. Not surprisingly, we found that BDNF stimulated the formation of NO (Fig. 2a) and production of cGMP (Fig. 2b) and, as expected, BDNF-enhanced cellular cGMP contents were abolished by the non-selective NOS inhibitor L-NAME (Fig. 2c). Combined with our previous study demonstrating that BDNF-dependent neuroprotection is suppressed by L-NAME and KT5823 [6], these findings suggested the involvements of NO/cGMP pathway in BDNF neuroprotection against 3-NP toxicity. However, detailed targets downstream of this pathway that contribute to BDNF-dependent neuroprotective effects were not fully identified in that study. In this work, we demonstrated that L-NAME (Fig. 2e), KT5823 (Fig. 2f), and ODQ (Fig. 2f) all suppressed BDNF-induced sestrin2, suggesting that sestrin2 is one of the targets downstream of NO/cGMP/PKG signaling cascade. Consistent with the important roles of NO in sestrin2 expression, it has been demonstrated that the NO donor 2,2′-(hydroxynitrosohydrazono)bis-ethanimine (DETA-NO) may increase sestrin2 expression in macrophages [14]. In addition to NO, herein we also showed that the membrane-permeable cGMP analogue 8-Br-cGMP was sufficient to induce sestrin2 expression (Fig. 2g), consistent with a neuroprotective effect of 8-Br-cGMP against 3-NP toxicity in cortical neurons [6].
Downstream of NO/sGC/PKG, NF-κB is a previously unidentified transcription factor responsible for sestrin2 induction. During knockdown experiments, interestingly, we found that p65 siRNA significantly suppressed expression of p50 in addition to its designed target p65; reciprocally, transfection of p50 siRNA also resulted in inhibition of p65 expression (Fig. 4d). Thus, p65 and p50 may regulate the expression of each other, either directly or indirectly. Given this effect, it is not surprising that siRNA targeting at p65 or p50 alone was sufficient to efficaciously knock down the expression of BDNF-induced sestrin2 (Fig. 4e), despite the partial (30–40 %) inhibitory effects over their respective target gene when individually applied (Fig. 4d). Similarly, BDNF-dependent neuroprotection against 3-NP toxicity was also completely abrogated by either p65 siRNA or p50 siRNA (Fig. 4f).
Another interesting finding is the molecular mechanisms of PKG-1 mediating sestrin2 induction by BDNF in cortical neurons. Previously, it has been reported that a NLS sequence was identified in the PKG-1 protein, allowing it to be detected in both cytosol and nucleus [36]. In the present study, we also noted that, upon BDNF stimulation, PKG-1 was indeed translocated into the neuronal nuclei (Fig. 7a, b) in a process depending on sGC activity (Fig. 7c). Intriguingly, immunoprecipitation out of the nuclear extracts revealed that, upon BDNF exposure, PKG-1 formed a protein complex with p65 and p50 (Fig. 7d), in a sGC-dependent manner (Fig. 7e), that together bound to the sestrin2 promoter (Fig. 7f) for transcriptional activation. However, it should be noted that our results derived from co-immunoprecipitation assays cannot exclude possible involvements of additional, yet unidentified proteins mediating the interaction between PKG-1 and the NF-κB subunits p65/p50; in other words, this interaction may also be indirect. Apart from association with a transcription factor, such as NF-κB shown here, PKG-1 as a kinase is expected to regulate its targets via phosphorylation. For example, PKG activation stimulated by cGMP phosphorylates p65 at Ser-276/Thr-305 and p105 at Ser-335/Ser-940 in 293T cells, although the subcellular localization of the NF-κB subunits is not affected by PKG-mediated phosphorylation in these cells [30]. In our experimental paradigm, PKG activity was required for BDNF-induced nuclear translocation of p65 and p50 (Fig. 5b); PKG-1 also formed a protein complex with NF-κB subunits in the nucleus (Fig. 7d). However, whether PKG-1 phosphorylates NF-κB subunits p65 or p50 and, if so, the exact amino acid residues to be phosphorylated, all require further investigation.
Expression of sestrin2 may be regulated at multiple levels. We noted that, at protein level, sestrin2 expression induced by BDNF treatment was about 2-folds as compared to control cultures (Fig. 1a), whereas the mRNA increase of sestrin2 by BDNF was 4–6-folds as shown in Fig. 6a. The rationale behind such a discrepancy between sestrin2 expression at mRNA and protein levels induced by BDNF is still unknown. Our results indicated that BDNF enhanced NF-κB-dependent transcriptional activation of sestrin2 mRNA. However, as shown respectively in Fig. 6c, d, ChIP assay revealed only 1.7–2.5-folds induction of p65 and p50 binding to the sestrin2 promoter, which were less than the 4–6-folds increases in sestrin2 mRNA levels. Similarly, in Fig. 7b, BDNF exposure only resulted in a 3-fold increase in the nuclear contents of PKG-1. At present, we cannot rule out the possible involvements of additional transcription factors mediating the observed BDNF induction of sestrin2. More importantly, sestrin2 mRNA can also be regulated post-transcriptionally, such as via alterations of its mRNA stability. Similar to its mRNA, the protein product of sestrin2 gene is also subjected to regulation at multiple levels that may include translational efficiency and protein stability. We did not explore the possible effects of BDNF on translational efficiency of sestrin2 mRNA. However, we did test the effects of MG132 and bafilomycin A1, the respective inhibitor of proteasomal degradation and autophagy, on the expression of sestrin2. Our preliminary results indicated that both basal sestrin2 levels as well as those inducible by BDNF were increased by these two compounds (Wu and Yang, unpublished observations), suggesting that degradation of sestrin2, either endogenous or that induced by BDNF, depends on both proteasomes and autophagy. Further experiments are required to clarify the detailed regulatory mechanisms controlling the expression of sestrin2 at mRNA and protein levels.
In this study, we showed that sestrin2 contributes to the antioxidant function of BDNF in attenuating 3-NP-induced ROS formation (Fig. 8). While the detailed antioxidative mechanisms were not studied here, sestrin2 has previously been shown to defend cells against oxidative stress [13] via catalyzing the reduction of sulfinic acid in an ATP-dependent manner, thus maintaining peroxiredoxin levels in an active state [14, 31]. Alternatively, sestrin2 may also induce degradation of Kelch-like ECH-associated protein 1 (Keap1), a repressor of Nrf2, by autophagy to upregulate Nrf2 that exerts antioxidant functions against food deprivation-induced oxidative damage [37]. In primary cortical cultures, we have revealed that BDNF-dependent 3-NP resistance also involves c-Jun-dependent induction of sulfiredoxin [11], another reductase known to reduce the hyperoxidized peroxiredoxin back to the catalytically active thiol in an ATP-dependent manner [38, 39]. Thus, in the cortical neurons challenged with 3-NP to induce oxidative stress, BDNF may enhance sestrin2 and sulfiredoxin to confer 3-NP resistance via NF-κB and c-Jun, respectively.
Although our studies were conducted in primary cortical neurons in vitro, several key mediators in this proposed signaling cascade, such as cGMP/PKG and NF-κB, have been shown to attenuate brain injuries resulted from 3-NP exposure in vivo. For example, rats treated with sildenafil or vardenafil, two phosphodiesterase-5 (PDE5) inhibitors that are expected to increase cellular cGMP levels, showed improved neurologic scores with reduction in striatal lesion volumes following 3-NP administration; importantly, striatal levels of the phosphorylated cAMP response element-binding (CREB) protein along with the expression of BDNF were significantly increased in sildenafil-treated rats [40]. In contrast to sildenafil-induced BDNF by increasing cGMP contents in rat striatum, herein, we demonstrated an opposite pathway in which BDNF increases cGMP production with enhanced PKG activity in rat cortical neurons. Downstream of cGMP/PKG, we observed that the redox-sensitive NF-κB contributed to the antioxidant effects of BDNF against 3-NP toxicity, consistent with the previous in vivo observation demonstrating that mice lacking the p50 subunit of NF-κB suffer increased neuronal damage with aggravated motor dysfunction after 3-NP administration [41]. Indeed, pharmacological activation of NF-κB by natrium diethyldithiocarbamate trihydrate (NDDCT) in rats is sufficient to attenuate 3-NP-induced damages including loss of body weight, impairments in locomotor activity and grip strength, as well as deficits in learning and memory [42]. Together, our findings appear to suggest that the neuroprotective mediators reported in the current study may also carry in vivo significance in conferring resistance against mitochondrial dysfunction that is often implicated in neurodegeneration.
At present, direct evidence linking sestrin2 to mitigation of 3-NP-induced brain damage in vivo is still lacking. Nevertheless, potential involvements of sestrin2 in other experimental paradigms, like peripheral nerve injury, have been noted. Sestrin2 homozygous knockout mice exhibited increased late-phase neuropathic pain associated with elevated ROS levels in sensory neurons, which can be reversed by a free radical scavenger [18]. Interestingly, the PKG-1α activated by oxidants, but not by cGMP, mediates neuropathic pain after peripheral nerve injury [43]. While a direct connection between sestrin2 and PKG-1α is not established therein, these two in vivo studies nevertheless suggest an interesting possibility that higher oxidant levels in sestrin2 knockout mice may enhance PKG-1α activity, independently of cGMP, to mediate neuropathic pain after nerve injuries. In our studies, however, cGMP-dependent PKG activation induced by BDNF increases sestrin2 expression, thereby attenuating 3-NP-induced ROS production in cortical neurons in vitro. Thus, depending on its activators, PKG-1 may function differentially in various experimental systems.
Overall, our results show that NF-κB is a previously unidentified transcription factor for induction of sestrin2, a stress-responsive protein critically involved in antioxidation, upon BDNF exposure in neurons. Interaction of PKG-1 with p65 and p50 to facilitate nuclear translocation of this protein complex requires NO and PKG activities for BDNF-dependent sestrin2 expression. Further, sestrin2 contributes to BDNF-mediated suppression of ROS production caused by mitochondrial inhibition. Our findings thus provide new insights into this novel antioxidative mechanism of BDNF in addition to its well-established neurotrophic effects.
References
Quintanilla RA, Johnson GV (2009) Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res Bull 80(4-5):242–247
Alston TA, Mela L, Bright HJ (1977) 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc Natl Acad Sci U S A 74(9):3767–3771
Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E (2009) Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ 16(6):899–909
Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R et al (1993) Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 13(10):4181–4192
Brouillet E, Jacquard C, Bizat N, Blum D (2005) 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem 95(6):1521–1540
Wu CL, Hwang CS, Yang DI (2009) Protective effects of brain-derived neurotrophic factor against neurotoxicity of 3-nitropropionic acid in rat cortical neurons. Neurotoxicology 30(4):718–726
Baydyuk M, Xu B (2014) BDNF signaling and survival of striatal neurons. Front Cell Neurosci 8:254
Bothwell M (2014) NGF, BDNF, NT3, and NT4. Handb Exp Pharmacol 220:3–15
Mattson MP, Lovell MA, Furukawa K, Markesbery WR (1995) Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem 65(4):1740–1751
Lee B, Cao R, Choi YS, Cho HY, Rhee AD, Hah CK, Hoyt KR, Obrietan K (2009) The CREB/CRE transcriptional pathway: protection against oxidative stress-mediated neuronal cell death. J Neurochem 108(5):1251–1265
Wu CL, Yin JH, Hwang CS, Chen SD, Yang DY, Yang DI (2012) c-Jun-dependent sulfiredoxin induction mediates BDNF protection against mitochondrial inhibition in rat cortical neurons. Neurobiol Dis 46(2):450–462
Chan SH, Wu CW, Chang AY, Hsu KS, Chan JY (2010) Transcriptional upregulation of brain-derived neurotrophic factor in rostral ventrolateral medulla by angiotensin II: significance in superoxide homeostasis and neural regulation of arterial pressure. Circ Res 107(9):1127–1139
Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A et al (2002) Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 21(39):6017–6031
Essler S, Dehne N, Brune B (2009) Role of sestrin2 in peroxide signaling in macrophages. FEBS Lett 583(21):3531–3535
Sanli T, Linher-Melville K, Tsakiridis T, Singh G (2012) Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One 7(2):e32035
Shin BY, Jin SH, Cho IJ, Ki SH (2012) Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free Radic Biol Med 53(4):834–841
Zhou D, Zhan C, Zhong Q, Li S (2013) Upregulation of sestrin-2 expression via P53 protects against 1-methyl-4-phenylpyridinium (MPP+) neurotoxicity. J Mol Neurosci 51(3):967–975
Kallenborn-Gerhardt W, Lu R, Syhr KM, Heidler J, von Melchner H, Geisslinger G, Bangsow T, Schmidtko A (2013) Antioxidant activity of sestrin 2 controls neuropathic pain after peripheral nerve injury. Antioxid Redox Signal 19(17):2013–2023
Wu CL, Chen SD, Yin JH, Hwang CS, Yang DI (2010) Erythropoietin and sonic hedgehog mediate the neuroprotective effects of brain-derived neurotrophic factor against mitochondrial inhibition. Neurobiol Dis 40(1):146–154
Ju TC, Yang YT, Yang DI (2004) Protective effects of S-nitrosoglutathione against neurotoxicity of 3-nitropropionic acid in rat. Neurosci Lett 362(3):226–231
Huang CY, Yang HI, Chen SD, Shaw FZ, Yang DI (2008) Protective effects of lipopolysaccharide preconditioning against nitric oxide neurotoxicity. J Neurosci Res 86(6):1277–1289
Patapoutian A, Reichardt LF (2001) Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11(3):272–280
Chang SH, Hwang CS, Yin JH, Chen SD, Yang DI (2015) Oncostatin M-dependent Mcl-1 induction mediated by JAK1/2-STAT1/3 and CREB contributes to bioenergetic improvements and protective effects against mitochondrial dysfunction in cortical neurons. Biochim Biophys Acta-Mol Cell Res (in press)
Tang CM, Hwang CS, Chen SD, Yang DI (2011) Neuroprotective mechanisms of minocycline against sphingomyelinase/ceramide toxicity: roles of Bcl-2 and thioredoxin. Free Radic Biol Med 50(6):710–721
Kurauchi Y, Hisatsune A, Isohama Y, Sawa T, Akaike T, Katsuki H (2013) Nitric oxide/soluble guanylyl cyclase signaling mediates depolarization-induced protection of rat mesencephalic dopaminergic neurons from MPP(+) cytotoxicity. Neuroscience 231:206–215
Astort F, Mercau M, Giordanino E, Degese MS, Caldareri L, Coso O, Cymeryng CB (2014) Nitric oxide sets off an antioxidant response in adrenal cells: involvement of sGC and Nrf2 in HO-1 induction. Nitric Oxide 37(1-10
Collier J, Vallance P (1989) Second messenger role for NO widens to nervous and immune systems. Trends Pharmacol Sci 10(11):427–431
Ju TC, Chen SD, Liu CC, Yang DI (2005) Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic Biol Med 38(7):938–949
Zhang XY, Wu XQ, Deng R, Sun T, Feng GK, Zhu XF (2013) Upregulation of sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cell Signal 25(1):150–158
He B, Weber GF (2003) Phosphorylation of NF-kappaB proteins by cyclic GMP-dependent kinase. A noncanonical pathway to NF-kappaB activation. Eur J Biochem 270(10):2174–2185
Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM (2004) Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 304(5670):596–600
Eid AA, Lee DY, Roman LJ, Khazim K, Gorin Y (2013) Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol Cell Biol 33(17):3439–3460
Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G (2009) Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 8(10):1571–1576
Lee J, Giordano S, Zhang J (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 441(2):523–540
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132(1):27–42
Gudi T, Lohmann SM, Pilz RB (1997) Regulation of gene expression by cyclic GMP-dependent protein kinase requires nuclear translocation of the kinase: identification of a nuclear localization signal. Mol Cell Biol 17(9):5244–5254
Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D et al (2013) Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab 17(1):73–84
Biteau B, Labarre J, Toledano MB (2003) ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425(6961):980–984
Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG (2004) Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem 279(49):50994–51001
Puerta E, Hervias I, Barros-Minones L, Jordan J, Ricobaraza A, Cuadrado-Tejedor M, Garcia-Osta A, Aguirre N (2010) Sildenafil protects against 3-nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol Dis 38(2):237–245
Yu Z, Zhou D, Cheng G, Mattson MP (2000) Neuroprotective role for the p50 subunit of NF-kappaB in an experimental model of Huntington’s disease. J Mol Neurosci 15(1):31–44
Gupta S, Sharma B (2014) Pharmacological benefit of I(1)-imidazoline receptors activation and nuclear factor kappa-B (NF-kappaB) modulation in experimental Huntington’s disease. Brain Res Bull 102:57–68
Lorenz JE, Kallenborn-Gerhardt W, Lu R, Syhr KM, Eaton P, Geisslinger G, Schmidtko A (2014) Oxidant-induced activation of cGMP-dependent protein kinase I alpha mediates neuropathic pain after peripheral nerve injury. Antioxid Redox Signal 21(10):1504–1515
Acknowledgments
This study was supported by the National Science Council/Ministry of Science and Technology in Taiwan (NSC 101-2314-B-010-042MY2 and MOST 103-2314-B-010-013MY3 to Ding-I Yang), Ministry of Education in Taiwan-Aim for the Top University Plan (104AC-B5 to Ding-I Yang), Department of Health in Taipei City Government (10301-62-003 and 10401-62-022 to Ding-I Yang and Chi-Shin Hwang), and Cheng Hsin General Hospital (103F003C16 and CY10417 to Ding-I Yang and Jiu-Haw Yin).
Conflict of Interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Supplementary Figures: The entire Western blots shown in the present study, except β-actin, GAPDH, and histone3 that served as the respective loading controls for total proteins, cytosolic proteins, and nuclear proteins, are demonstrated in the corresponding supplementary figures. The numbers on the left side of each blot indicate the molecular weight markers in kDa. Because no immunoblots are presented in Fig. 6, there is no Suppl. Fig. 6.
Suppl. Fig. 1 and 2
(GIF 252 kb)
Suppl. Fig. 3 and 4
(GIF 273 kb)
Suppl. Fig. 5
(GIF 226 kb)
Suppl. Fig. 7
(GIF 229 kb)
Rights and permissions
About this article

Cite this article
Wu, CL., Chen, SD., Yin, JH. et al. Nuclear Factor-kappaB-Dependent Sestrin2 Induction Mediates the Antioxidant Effects of BDNF Against Mitochondrial Inhibition in Rat Cortical Neurons. Mol Neurobiol 53, 4126–4142 (2016). https://doi.org/10.1007/s12035-015-9357-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9357-1