
Abstract
Epidemiological studies have suggested a differential response, males versus female, in stroke incidence and prognosis. These divergences in brain response after damage are based mostly on hormonal differences. To date, estradiol and progesterone administered independently have demonstrated neuroprotection after ischemia in animal models. Nonetheless, contradictory results were revealed using a combined administration. In order to evaluate the effects of combinatorial treatment administered after ischemia induction, we used two different approaches: in vivo and in vitro models. Male rats which underwent permanent middle cerebral artery occlusion were treated with a combination of estradiol/progesterone at 6, 24 and 48 h after injury and sacrificed at 54 h post-ischemia. The rat brains were evaluated for reactive gliosis, NeuN-positive neurons, levels of synapse-associated proteins and activity levels of PI3K/Akt/GSK3/β-catenin survival pathway. Also, primary cortical neurons were subjected to oxygen and glucose deprivation for 17 h and returned to a normal environment in the presence of estradiol or estradiol/progesterone. Cell viability was evaluated, and activity levels of the PI3K/Akt/GSK3/β-catenin pathway. Our results indicate that some beneficial effects of estradiol were abolished in the presence of progesterone, particularly in the cerebral cortex (core). However, the combinatorial treatment showed positive effects in the hippocampus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke, a complex neurodegenerative disease, is the leading cause of disability and the second cause of death in humans worldwide [1]. Brain injury following an ischemic insult involves a complex succession of events that cause varying degrees of cell damage depending, not only on the characteristics of the initial insult, but also on the duration of the occlusion [2]. It has been accepted that the cell death in the ischemic core, the most affected area, is triggered by necrosis, whilst in the peri-infarct region (penumbra) is caused mainly by apoptosis [3, 2, 4]. Until now, there is no pharmacological therapy for this disorder except tissue plasminogen activator, but its use is limited. The middle cerebral artery occlusion (MCAO) is the in vivo model most commonly used to mimic and analyze the ischemic stroke damage and also the potential treatments [5]. Epidemiological studies suggest that the stroke risk is lower and the prognosis of patients is better in premenopausal women than in men. Nonetheless, the incidence of cerebrovascular diseases increases in women after menopause, being more elevated than in men [1]. On the other hand, a gender-differential response after ischemic injury has been demonstrated in animal models of stroke [6, 7].
17β-estradiol (E) and progesterone (P), the main female sex hormones, play a role in a multitude of physiological nervous processes acting both on neurons and in glial cells [8, 9]. They can cross the blood-brain barrier (BBB) and diffuse throughout the brain. However, they are considered as neurosteroids because they can be synthesized by the nervous system [10]. Nowadays, the neuroprotective effects of both hormones in different neurodegenerative diseases are well established [8, 9, 11]. Growing evidence strongly indicates that estradiol protects against ischemic stroke damage. In fact, estradiol administration before, during or after ischemia induction reduced the damage area in rodent models [12, 9]. Several mechanisms have been proposed to be involved in the estradiol neuroprotection against ischemia-induced damage, such as a reduction in pro-inflammatory events, glutamate-mediated excitotoxicity, oxidative damage and apoptosis [13–15]. However, only a few studies have examined the potential neuroprotective effect of progesterone, probably because the majority of the studies have focused on estradiol as the main source of neuroprotection seen in females. The neuroprotective effect of progesterone against ischemic brain damage includes decreasing infarct area, inflammation, BBB permeability and edema in animal models [16].
The detailed molecular mechanisms involved in neuroprotection by estradiol and progesterone are not completely understood. It has been described that both hormones exert their effects not only at the genomic level through the classical nuclear receptors, ‘slow responses’, but also by modulating cytosolic signalling cascades by interacting with membrane-localized and/or cytoplasmic receptors, ‘rapid response’ [11, 8]. These ‘cytoplasmic’ responses may play an important role in neural cells, especially in neuroprotection.
Moreover, the role of estradiol and progesterone has been described in synaptic plasticity [17], an important process in the recovery of brain function after damage [18, 19]. It has been reported that estradiol alters, through membrane receptors, the phosphorylation levels of synapsin I [20], a presynaptic protein that has a central role in neurotransmitter release, synaptic formation and maturation [19]. In addition, recent reports suggest an increase in postsynaptic density protein 95 (PSD95) expression, a postsynaptic protein located mainly at the glutamatergic synapses, mediated by estradiol [21].
To date, limited studies using a combination of estradiol and progesterone have been reported and the results obtained have been contradictory. Some point to the antagonistic effects between both hormones [22, 23, 17] whilst others report enhanced effects [24, 25].
In this study, we have analyzed the effect of post-ischemia estradiol and progesterone (E/P) treatment at physiological doses 54 h after injury. We performed two different experimental approaches using both in vivo (permanent middle cerebral artery occlusion, pMCAO) and in vitro (oxygen-glucose deprivation (OGD) in cortical neuronal primary culture) models. We have analyzed the effect of E/P post-ischemic treatments on reactive gliosis and NeuN-positive neurons. We have also evaluated the impact of this combined hormone therapy on the PI3K/Akt/GSK3/β-catenin survival pathway and on specific proteins implicated in synaptic plasticity and functionality (synapsin I, PSD95 and α-N-catenin). Our findings indicate that the addition of progesterone could abolish some of the beneficial effects observed previously by estradiol administration after pMCAO.
Materials and Methods
Animals
Adult male Wistar rats (8–12 weeks old, 250–310 g) obtained from the vivarium at Centro de Biologia Molecular “Severo Ochoa” (CBM-SO) were maintained under 12-h light/12-h dark cycle and with access to food and water ad libitum. All the animal care protocols were conformed to the appropriate national legislations (RD 53/2013) and the guidelines of the European Commission for the accommodation and care of laboratory animals (revised in Appendix A of the Council of Europe Convention ETS123). All procedures have the approval of the Bio-Ethical committee of CBM-SO.
Surgery and pMCAO Induction
pMCAO was performed by right middle cerebral artery occlusion using an intraluminal suture as we previously described [13]. Anaesthesia was induced by intraperitoneal injection (ip) of a mixture of medetomidine (250 μg/kg) and ketamine (75 mg/kg), accompanied by a subcutaneous injection of atropine (100 μg/kg).
Hormonal Administration
A total of 32 male rats were randomly distributed into the following experimental groups: sham-vehicle (SV, n = 8), ischemia-vehicle (IV, n = 8), sham-estradiol/progesterone (SEP, n = 8) and ischemia estradiol/progesterone (IEP, n = 8). The animals received three doses of 0.04 mg/kg β-estradiol (Sigma, St. Louis, MO, USA, ref E8875) as we previously described [13], combined with 4 mg/kg progesterone (Sigma, St. Louis, MO, USA, ref P7556) [26, 27] in a total volume of 500 μl. Intraperitoneal injections were administrated at 6, 24 and 48 h after pMCAO induction. Groups IV and SV received 500 μl of vehicle (1 % ethanol in saline). All animals were sacrificed 6 h after the last treatment.
Neurological Deficit Score
The neurological deficit score of each animal was measured before surgery, and at 6, 24, 48, 54 h after pMCAO induction, using the method described by Yrjänheikki [28]. A 7-point scale of neuromotor function was ranked as follows: grade 0, no spontaneous motion; grade 1, spontaneous circling towards the paretic side; grade 2, circling towards the paretic side when pulled by the tail; grade 3, circling towards the paretic side when pulled and lifted by the tail; grade 4, consistently reduced resistance to a lateral push towards the paretic side; grade 5, consistent flexion of the forelimb contralateral to the injured hemisphere; and grade 6, normal extension of both forelimbs towards the floor when lifted. Other neurological anomalies were also recorded, such as alterations in balance, sensorial perception (acoustic, visual localization) and reflex responses (corneal, palpebral, postural correction). In this study, we have excluded all the ischemic rats that did not reach an ischemic damage index (IDI) of 2–3 at 6 h post-MCAO.
Western Blotting
Tissue samples were homogenized and analyzed by Western blots as we previously described [13]. Membranes were incubated overnight at 4 °C with the following primary antibodies: Akt (1:1000; Cell Signaling no. 9272), phospho-AktSer473 (1:500; Cell Signaling no. 9271), phospho-AktThr308 (1:1000; clone C31E5E, Cell Signaling no. 2965), β-catenin (1:800; BD Transduction Laboratories no. 610,153), β-tubulin (1:1000; clone TUB 2.1, Sigma no. T4026), β-actin (1:1000; clone AC-15, Sigma no. A5441), GSK3 (1:1000; Biosource no. 44–610), phospho-GSK3Ser21/9 (1:1000; Cell Signaling no. 9331), synapsin (1:500; Cell Signaling no. 2312), phospho-synapsinSer9 (1:500; Cell Signaling no. 2311), PSD95 (1:1000; Cell Signaling no. 3450) and α-N-catenin (1:1000; clone C12G4; Cell Signaling no. 2163). Membranes were rinsed with PBS-Tween and incubated with appropriate secondary antibodies: anti-rabbit or anti-mouse IgG-HRP (Santa Cruz Biotechnology). The density of the immunoreactive bands was analyzed using Quantity One software (Bio-Rad; U-Max PowerLook 2100XL scanner) and the values were normalized to β-tubulin or β-actin levels detected in the same membrane and compared with control values (SV). The graphs represent mean ± standard error of mean (±SEM) of 3–5 independent experiments.
Tissue Collection
For immunoblotting analysis, animals were sacrificed by exposure to CO2 at 54 h after onset of pMCAO. The right cerebral cortex and hippocampus was dissected out and frozen at −80 °C for subsequent analysis.
For immunohistochemistry, the deeply anaesthetised animals using a mixture of medetomidine (375 μg/kg) and ketamine (112.5 mg/kg) ip, were transcardially perfused with ice-cold 0.1 M phosphate-buffered saline (PBS) followed by 4 % paraformaldehyde in 0.1 M PBS, pH 7.4. After perfusion, the brains were removed and post-fixed overnight at 4 °C followed by cryoprotection using 30 % sucrose in PBS at 4 °C for 48–72 h. Finally, cryoprotected brains were embedded in Tissue-Tek medium (Sakura, Zoeterwoude, Netherlands). Coronal cryostat sections (25-μm thick) were collected onto Superfrost Ultra Plus slides (Thermo Scientific, Braunschweig, Germany), air dried and stored at −20 °C. Sections located from +2 to −4 relative to bregma [29] were selected for immunohistochemistry/immunofluorescence.
Immunohistochemistry and Immunofluorescence
Immunohistochemical and immunofluorescence analyses were performed in a humidified chamber. The sections were permeabilized in PBS −0.25 % Triton X-100. After the tissue was incubated in blocking solution containing PBS −0.1 % Triton X-100 with 1 % horse serum. The slices were incubated overnight at 4 °C with the appropriate primary antibody in blocking solution: monoclonal mouse anti-glial fibrillar acidic protein (GFAP) (1:1000; Clon 4A11; BD Pharmingen no. 55632); rabbit anti-Iba1 (1:500; Wako no. 019–19741); monoclonal mouse anti-NeuN (1:100; Clon A60; Millipore MAB377). The sections were then washed with PBS −0.1 % Triton X-100 and incubated in PBS −1 % hydrogen peroxide. The slices were subsequently incubated at room temperature with the appropriate secondary antibody: biotinylated anti-mouse IgG or anti-rabbit IgG (1:200 in PBS −1 % horse serum or goat serum, respectively). After, the sections were incubated for 30 min with avidin-biotin-peroxidase complex from the Vector ABC Elite kit (VECTASTAIN, Vector laboratories Inc., Burligame, CA, USA), followed by 15-min incubation with FAST 3ʹ3 diaminobenzidine tablets (Sigma, D4168). After dehydration, the sections were cleared with xylol and mounted using Entellan New mounting medium (Electron Microscopy Sciences, Hatfield, PA, USA).
For immunofluorescence experiments, following incubation with the primary antibody, the sections were incubated with anti-mouse Alexa-555 (1:500; Invitrogen). The slices were mounted using Fluoromount-G (Southern Biotech). The immunostained slides were observed under an Olympus BX61 microscope and images captured using an Olympus DP50 camera.
Images Quantification
The intensity of immunostaining was quantified using Fiji image processing software [30]. For DAB-processed images (stained with GFAP and Iba-1), ×4-magnification images was used. After conversion of 16-bit, images was normalized subtracting background (rolling = 500 light). The threshold was automatically selected (AutoThreshold) using the Moments method. To determine the size of the stained selected area, we analyzed particles (selecting size = 100–5000 and circularity = 0.10–1.00). The values were normalized to the total area of the tissue including in the picture that was measured in each image. For NeuN quantification, ×10-magnification images were used. A grey-level threshold representing the boundary between background and positive staining was automatically selected by the software and then applied to measure the area of positive staining in each image. Prior to quantification, images was corrected using a variance filter (radius = 1 pixel). To determine whether the immunostaining area changed in any experimental group, we quantified the positive staining area in NeuN images and these values were normalized to the total area of the photographed sections.
Cortical Primary Neurons Culture and Oxygen-Glucose Deprivation
Primary mouse neurons were prepared from 16-day-old Swiss mice embryos. A pool of cerebral cortices were dissected and enzymatically digested with 0.05 % trypsin-EDTA (Invitrogen) and 10 mg/ml DNase (Roche Applied Science) for 15 min at 37 °C, followed by trituration with fire-polished glass pipettes. The cells were plated on 24-well plates for viability and 6-well plates for Western blot analysis at a density of 2.5 × 105 cells/ml. The plates were previously pre-coated with 0.5 mg/ml poly-l-lysine (Sigma-Aldrich) in borate buffer. Neurons were cultured with neurobasal medium (NB) supplemented with 2 % B27 and 2 mM glutamine (Invitrogen), and maintained at 37 °C in a humidified 95 % air/5 % CO2 atmosphere incubator. Five-day-old cultures were used for all experiments. The cultures contain almost 90–95 % of neurons and were negative to GFAP by western blot.
For oxygen-glucose deprivation (OGD), culture neurons were incubated in glucose-free Dulbecco’s modified Eagle’s medium (DMEM) and in a near-anaerobic incubator containing an atmosphere of 0.2 % O2, 5 % CO2 and 94.8 % N2 at 37 °C. As control (non-OGD condition), DMEM media and normal incubator were used. After 17-h OGD, the cultures were returned to a normal environment; glucose-free media was replaced with NB/B27 media and cultures were returned to a 95 % air/5 % CO2 incubator. Experimental parameters were assayed at 24 h following re-oxygenation/regeneration. Neuronal cultures were treated after OGD with β-estradiol (200 nM) and progesterone (20 μM) during 24 h.
MTT Cell Viability Assay
Cell viability was examined by MTT assay that depends on the reduction of the tetrazolium salt MTT to formazan by living cells. Mouse primary neurons were cultured in 24-well plates. After treatment, a MTT solution (0.3 mg/ml) made in phenol red-free NB was added to the cells. After 1-h incubation at 37 °C, the MTT solution was aspirated and DMSO was added to the cells. Aliquots were transferred to a 96-well plate and absorbance measured at 540 nm in a plate reader spectrophotometer (Opsys MR, Dynex Technologies). Results were expressed as percentages of the values obtained from the controls (non-OGD condition).
Statistical Analyses
GraphPad Prism software was used for all statistical analyses and all the data are presented as mean ± standard error of mean (SEM). Statistical significance was estimated using a two-tailed Student’s t test to compare differences between groups (SV vs IV; IV vs IEP). The neurological score was analyzed by two-way analysis of variance (ANOVA) with three groups (sham, IV and IEP), followed by Tukey’s post hoc test for group comparisons between groups (SV vs IV and IV vs IEP). A value of p ≤ 0.05 was considered statistically significant.
Results
Post-ischemia Administration of Estradiol/Progesterone Accelerates the Recovery of Neurological Status in Male Rats After pMCAO Induction
To investigate the effect of a combined hormone therapy administered after ischemic stroke, we treated pMCAO-induced rats at 6, 24 and 48 h with a mixture of estradiol/progesterone and monitored the behavioural scores. As Fig. 1 shows, all the animals attained the maximal ischemic damage index (IDI) before surgery was carried out. At 6 h post-surgery, immediately before the first treatment, the ischemic rats (IV and IEP groups) showed a neurological score significantly lower than the sham-operated animals (2.8 ± 0.374 vs 5.2 ± 0.374; ***p = 0.0002). This showed that pMCAO induction had a significant and consistent effect on the neurological status of the rats. At 24 h, the ischemic rats treated with the vehicle (IV) maintained significant lower values on IDI (4.6 ± 0.6; ***p = 0.0002). However, a unique injection of E/P induced a non-significant improvement on the neurological scores (5.1 ± 0.47). At 48-h post-pMCAO induction, the IV group conserved a reduced IDI of 5 ± 0.63 (*p = 0.031) whilst the IEP group recovered the maximum score index. Finally, all the experimental groups achieved the maximum IDI before sacrifice.

Post-ischemic estradiol/progesterone combined treatment accelerates the recovery of maximum ischemic damage index (IDI). The IDI was measured using a 7-point scale neurological test. Zero corresponds to the lowest and 6 to the highest degree of neurological recovery. Graphs represent the neurological score of all animals analyzed at 6, 24, 48 and 54 h after ischemic onset. Data represent the mean ± standard error of mean (SEM), the data were statistically significant at 6 h ***p = 0.0005 (SV vs IV and SV vs IEP), at 24 h ***p = 0.0005 (SV vs IV), At 48 h *p = 0.03 (SV vs IV) according to two-way ANOVA followed by Tukey’s test. SV, Sham-vehicle; IV, pMCAO-vehicle; IEP, pMCAO-estradiol/progesterone
The Estradiol/Progesterone Treatment After pMCAO Reduces the Reactive Gliosis Induced by Ischemia
After MCAO induction reactive gliosis has been reported [31, 13]. It is characterized by the activation and proliferation of microglia and astrocytes especially in regions near to the ischemic core and exerts both beneficial and detrimental effects on brain damage after injury. To further assess the effect of E/P treatment on reactive gliosis after pMCAO, we examined this phenomenon in coronal sections immunostained with astrocyte-specific and microglia-specific antibodies, GFAP and Iba-1, respectively (Fig. 2). We focused on the parietal and piriform cortex where we observed a larger proliferation of these cells after pMCAO induction.

The reactive gliosis is reduced after E/P treatment in the ischemic area. The reactive gliosis induced after 54-h pMCAO was measured in coronal brain sections by immunohistochemistry using GFAP and Iba1, markers of astrocytes and microglia, respectively. (a) Representative coronal sections corresponding to the injured area (bregma 0.48) of sham-vehicle (SV), pMCAO-vehicle (IV) and pMCAO-estradiol/progesterone (IEP) rats are shown. GFAP (a upper panel) and Iba1 (b bottom panel). b The graphs represent the percentage of marked area expressed as the percentage of labelled tissue with respect to the total area of the photographed sections. Staining was quantified in at least six serial coronal sections of each animal (from bregma 2.04 to −0.48). c A Nissl-stained section showing the affected areas selected for GFAP/Iba quantification, as indicated by rectangles. *p ≤ 0.05; scale bar = 100 μm
As we have already described [13], the GFAP-positive marked area in IV group was significantly higher than in the same area of SV group (4.81 ± 0.91 vs 1.67 ± 0.27 %; p ≤ 0.01; Fig. 2a). Additionally, a similar pattern was found when we examined the Iba1-positive stained area (10.62 ± 1.40 vs 5.99 ± 1.17 %; p ≤ 0.05; Fig. 2b). As expected, these findings indicate a marked accumulation of astrocytes and microglia in particular, near to the damaged area 54 h after ischemia onset. The administration of three doses of E/P after pMCAO induced a decrease in the percentage of GFAP-positive and Iba1-positive areas in the ischemic territory (Fig. 2a, b; right panels). The percentage of Iba1 marked surface decreased significantly to basal levels (6.09 ± 0.5 %; p ≤ 0.05), but the percentage of GFAP-marked surface decreased without reaching the levels of SV group (2.38 ± 0.8 %; p ≤ 0.05). This result suggests that post-pMCAO E/P treatment could reduce the reactive gliosis triggered by ischemia with a slightly greater effect on microglia than in astrocytes.
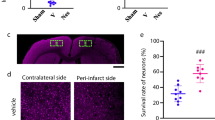
Estradiol/Progesterone Treatment Increases the Amount of NeuN-Positive Cells in CA2-CA3 but Not in the Cortex
After cerebral blood flow deprivation, some neurons, especially those located in the ischemic core, initiate a detrimental process that could induce cell death. The next step was to evaluate the impact of this combined hormone treatment on neuronal cells after pMCAO induction. It was revealed that the percentage of NeuN-positive marked area in IV group was significantly lower compared with the SV group (12.66 ± 0.24 vs 16.53 ± 1.2 %; p ≤ 0.05, Fig. 3) when the somatosensory cortex (ischemic core) was analysed by immunofluorescence. In addition, post-pMCAO treatments with a mixture of E/P did not cause any effect on the percentage of NeuN-positive marked area, since we detected similar levels in the IV group (13.62 ± 0.73).

The amount of NeuN-positive cells in CA2-CA3 increases after post-ischemic treatment with estradiol/progesterone. a Representative coronal sections corresponding to ischemic core (somatosensory cortex; bregma 0.12), and hippocampus (CA1 and CA2-CA3; bregma −3.36) of sham-vehicle (SV), pMCAO-vehicle (IV) and pMCAO-estradiol/progesterone (IEP) rats. b The bar graph represents the percentage of marked area normalized to the total area of the photographed sections. NeuN staining was quantified in at least six serial coronal sections of each animal, from bregma 2.04 to −0.48 (cortex) and bregma −2.76 to −3.84 (hippocampus). Arrows show NeuN-positive neurons in stratum lucidum/radiatum. c Nissl-stained sections showing the selected areas for NeuN quantification, as indicated by rectangles. Data represent mean ± standard error of mean (SEM). *p ≤ 0.05; scale bar = 100 μm
NeuN staining in the hippocampus CA1, and CA2-CA3 region, showed non-significant differences between SV (CA1 = 408 ± 47.3; CA2-CA3 = 451 ± 54) and IV groups (CA1 = 360.7 ± 16.8; CA2-CA3 = 314.6 ± 82; Fig. 3). These findings confirmed that the hippocampus did not suffer the same degree of injury as the somatosensory cortex 54 h after pMCAO induction. Remarkably, E/P treatments induced a significant increase in the percentage of NeuN-staining area in internal region around CA2-CA3 (584.5 ± 9.9; p ≤ 0.01) but this effect was not found in CA1 region (482.2 ± 56.9). This rise in NeuN staining was observed especially in stratum lucidum and radiatum of the hippocampus, just under the pyramidal cell layer (Fig. 3 arrows).
Effect of Estradiol/Progesterone on Synapse-associated Proteins
To evaluate more specifically this combined treatment on neurons, we analyzed some proteins directly associated with synaptic structure and function, highly sensitive to ischemia [32]. We focused on a member of the catenin family (α-N-catenin) [32], a member of postsynaptic density protein (PSD) family, PSD95; and a presynaptic protein (synapsin).
To demonstrate further the impact of E/P treatment on neurons, the levels of these proteins were checked 54 h after pMCAO induction (Fig. 4). The total levels of α-N-catenin did not show any significant differences between the experimental groups neither in the cortex nor the hippocampus (Fig. 4a, e). The PSD95 levels exhibited a slight increase in IV group in the cerebral cortex but not in the hippocampus (Fig. 4b, f). The administration of both hormones caused a non-significant reduction of the pMCAO-increased level (Fig. 4b, f). The total levels of synapsin were not altered after ischemia induction (Fig. 4c, g) in both regions studied. However, after 54-h pMCAO, the p-synapsinSer9 levels were significantly increased in both the cortex and the hippocampus when compared with the SV group, 130.12 ± 19 and 117.8 ± 4.6 %, respectively. The E/P treatment induced a reduction in these pMCAO-increased levels, but this was only statistically significant in the hippocampus (Fig. 4d, h).

Effect of post-pMCAO-estradiol/progesterone treatment on synapse-associated proteins. The total levels of αN-catenin (a, e), PSD95 (b, f) and synapsin (c, g) as well as the phosphorylated levels of p-synapsinSer9 were measured by Western blots of the ipsilateral cerebral cortex (a–d) and hippocampus (e-h) homogenates. The bar graphs represent the levels of these proteins normalized to β-actin/β-tubulin. A representative blot is shown above each graph. The bars represent the mean of 3–5 independent experiments. The total levels of αN-catenin both in the cortex and hippocampus (a, e, respectively) did not show significant variations between the different experimental groups. The pMCAO (IV group) induced a significant increase in PSD95 levels in the cortex but not in the hippocampus (b and f, respectively). The estradiol/progesterone treatment did not significantly alter the pMCAO-induced levels. In both the cortex and hippocampus, the total synapsin levels (c, g, respectively) did not change in any experimental group. But, the p-synapsinSer9 levels increased after pMCAO induction (IV group) in both the cortex (d) and hippocampus (h). Estradiol/progesterone treatment (IEP group) reduced p-synapsinSer9 down to control levels only in the hippocampus (h). Data were expressed as the mean ± SEM *p ≤ 0.05; n = 5 animals per group. SV, Sham-vehicle; SEP, Sham-estradiol/progesterone; IV: pMCAO-vehicle; IEP, pMCAO-estradiol/progesterone; r.u., relative units
Estradiol/Progesterone Treatment Induces a Recovery in PI3K/Akt/GSK3/β-catenin Survival Pathway in the Hippocampus but not in the Cortex
Although the detailed molecular mechanisms by which both estradiol and progesterone enhance neuronal survival are not clearly understood, it has been established that both hormones activate multiple intracellular signalling cascades. The PI3K/Akt/GSK3/β-catenin survival pathway is one of the well-described mechanisms, implicated in the neuroprotection triggered by estradiol and progesterone [33, 34]. Recently, we reported that estradiol treatments reverted the pMCAO-induced downregulation of the PI3K/Akt/GSK3/β-catenin pathway in the parietal cortex, reflecting an important neuroprotective effect in this ischemic area [13]. Therefore, in this study, we investigate whether the combination of estradiol and progesterone could modify the neuroprotective effect identified with estradiol.
The effect of E/P treatment on Akt and GSK3 activities was examined by Western blotting using phospho-specific antibodies against the active/inactive forms of theses kinases. The Akt activation was measured using pAktSer473 and pAktThr308 specific antibodies, the pGSK3Ser21/9 antibody can reflect the degree of inactivation of GSK3. Activate Akt can phosphorylate GSK3 in Ser21/9 residues reducing its kinase activity. We also checked the total amount of Akt and GSK3 using specific antibodies against these proteins. The effect of E/P treatment on β-catenin levels was analyzed using a specific antibody.
Firstly, we checked the activation status of this pathway in the parietal cortex. Unaltered levels of total Akt, GSK3 and β-catenin were found in all the experimental groups (Fig. 5) indicating no effect of the treatment on the total levels of these proteins. As we found in our previous study, the levels of pAktSer473, pAktThr308 and pGSKSer21/9 decreased significantly in cortical areas at 54-h pMCAO induction; indicating a downregulation of this survival pathway (Fig. 5b, c and e). The degree of reduction in the pAkt epitopes was different, with pAktSer473 affected more (40 ± 15 % of SV levels; p ≤0 .01) than pAktThr308 (60 ± 5.7 % of SV levels; p ≤ 0.01). The treatment using E/P did not significantly recover the pAktSer473 levels, but enhanced the levels of pAktThr308 which reached 85 ± 5.3 % of the SV levels (p ≤ 0.01; Fig. 5c). The levels of pGSK3Ser21/9, showed a reduction of 59 ± 9.7 % vs SV levels (Fig. 5e; p ≤ 0.05). In this case, E/P administration was not able to recover the activity levels of this kinase.

The estradiol/progesterone treatment did not recover the downregulation of the PI3K/Akt/GSK3/β-catenin pathway by ischemia in the parietal cortex. The activity levels of the PI3K/Akt/GSK3/β-catenin pathway in the cortex was examined in homogenates from the ipsilateral cerebral cortex using phospho-specific antibodies against active/inactive forms of Akt/GSK3 kinases, respectively. The bar graphs represent the total levels of Akt (a), pAktSer473 (b), pAktThr308 (c), total GSK3 (d), pGSK3Ser21/9 (e) and β-catenin (f) normalized to β-actin/β-tubulin. A representative blot is shown above each graph . The data represent the means of 3–4 independent experiments. No changes in the total Akt, GSK3 and β-catenin levels were observed in any of the experimental groups (a, d and f). The pMCAO (IV group) induced a significant reduction in pAktSer473 (b), pAktThr308 (c) and pGSK3Ser21/9 (e). The estradiol/progesterone treatment (IEP group) did not induce any significant changes compared with the vehicle-treated group (IV) in pAktSer473 (b) and pGSK3Ser21/9 (e). However, estradiol/progesterone treatment partially recovered the pAktThr308 (c) levels. The data are expressed as the mean ± SEM; *p ≤ 0.05 **p ≤ 0.01; n = 5 rats per group. SV, Sham-vehicle; SEP, Sham-estradiol/progesterone; IV: pMCAO-vehicle; IEP, pMCAO-estradiol/progesterone; r.u., relative units
On the other hand, analysis from the hippocampus samples obtained from pMCAO-induced animals exhibited a different response to the E/P treatments. As we observed in the parietal cortex, the total levels of Akt, GSK3 and β-catenin did not show any significant variation between the experimental groups (Fig. 6a, d and f, respectively). A significant reduction of Akt activity after pMCAO was inferred by the reduction of pAktSer473 (37 ± 4.3 % reduction vs SV levels; p ≤ 0.01), and pAktThr308 (66 ± 10 % reduction vs SV levels; p ≤ 0.05) phospho-epitopes. In this case, the E/P treatments caused a significant increase in the levels of pAktSer473 and pAktThr308; suggesting that E/P administration was able to revert the inactivation pMCAO-induced on Akt in the hippocampus. Similar findings were found when the GSK3 activity in this area was evaluated. IV rats showed an important decrease in the pGSK3Ser21/9 levels (38.5 ± 6.5 % reduction vs SV levels; p ≤ 0.01); but E/P administration partially recovered GSK3 phosphorylation, reaching a value of 81.88 ± 5.5 % of SV levels (p ≤ 0.05; Fig. 6e).

The activity levels of Akt and GSK3 were recovered by estradiol/progesterone treatment post-pMCAO in the hippocampus. Akt and GSK3 activity levels were measured in the ipsilateral hippocampus by Western blotting using phospho-specific antibodies against active/inactive forms of both kinases. The bar graph represent the levels of total Akt (a), pAktSer473 (b), pAktThr308 (c), total GSK3 (d), pGSK3Ser21/9 (e) and β-catenin (f) normalized to β-actin/β-tubulin. A representative blot is shown above each graph . The data represent the mean of 3–4 independent experiments. The levels of pAktSer473 (b), pAktThr308 (c) and pGSK3Ser21/9 (e) showed a significant decrease compared with the SV group after ischemia induction. The treatment using estradiol/progesterone (IEP group) significantly recovered the levels of pAktSer473 (b), pAktThr308 (c) and pGSK3Ser21/9 (e). Total levels of Akt (a), GSK3 (d) and β-catenin (f) did not change in any experimental group. The data were expressed as the mean ± SEM; *p ≤ 0.05 **p ≤ 0.01; n = 5 rats per group. SV, Sham-vehicle; SEP, Sham-estradiol/progesterone; IV: pMCAO-vehicle; IEP, pMCAO-estradiol/progesterone; r.u., relative units
Progesterone Addition After Oxygen-glucose Deprivation Induction (OGD) Avoids the Estradiol Effects on Neuron Viability and PI3K/Akt/GSK3/β-catenin Activity
To confirm that some of in vivo results are due to a neuronal response to the hormones, we subjected primary neuronal cultures to periods of OGD and we determined cell viability at 24 h by MTT assay. Preliminary experiments using cells deprived of oxygen and glucose for various intervals (1 to 17 h) were carried out. The cells were analysed 24 h after incubation with the regeneration medium and the results indicated that a period of 17-h OGD caused a substantial, 50 % reduction in neuronal viability (data not shown). We analysed the effect of the regeneration medium supplemented with estradiol and estradiol/progesterone treatments after OGD (Fig. 7a). The results showed that the presence of estradiol at 200 nM induced a significant increase on neuronal viability, but the addition of progesterone at 20 μM impeded this estradiol-induced effect.

Progesterone prevents the beneficial effect of estradiol in the cortical neurons after OGD. After overnight OGD, estradiol, estradiol/progesterone or vehicle were applied for 24 h. a MTT assay to assess cell viability after 17-h OGD and 24-h regeneration in the presence of estradiol, estradiol/progesterone or vehicle (NB). The addition of progesterone attenuated the estradiol-induced increase in cell viability. Data are expressed as the percentage values for non-OGD condition (n = 6). b pAktSer473, pAktThr308 and pGSK3Ser21/9 levels after 17-h OGD following regeneration for 24-h. Estradiol treatment significantly increased the OGD-induced reduction in pAktSer473, pAktThr308 and pGSK3Ser21/9 levels. The presence of progesterone abolished estradiol-induced effect. The bar graphs represent the quantification of phospho-proteins levels normalized to β-actin levels and are expressed as a percentage of the values of the controls (non-OGD).Bars represent the means ± SEM of three samples per group. A representative blot is shown above the bar graph . (*p < 0.05, **p < 0.01). NB: regeneration in presence of vehicle; E: regeneration in presence of estradiol; E/P: regeneration in the presence of estradiol/progesterone; r.u., relative units
Secondly, we examined the effect of OGD and ovarian hormone treatments on the PI3K/Akt/GSK3/β-catenin pathway testing Akt and GSK3 activity using a Western blot (Fig. 7b). An analysis of the cell extracts from cortical neurons exposed to OGD revealed a downregulation of this pathway, reflected by a significant decrease in pAktSer473, pAktThr308 and pGSK3Ser21/9 levels (78.7 ± 11, 56.3 ± 15 and 22.2 ± 4.3 % compared with the non-OGD controls, respectively).
The addition of estradiol to the regeneration medium for 24 h abolished the Akt inactivation caused by the OGD but did not completely abolish the decrease in pGSK3Ser21/9 levels. Moreover, the combination of E/P resulted in a non-significant variation in the pAktSer473, pAktThr308 and pGSK3Ser21/9 levels compared with the OGD condition.
Discussion
At the current time, there are contradictory data in the literature regarding the neuroprotective effects of combined hormone administration compared with the treatment of estradiol or progesterone alone. It has been described that progesterone reduces the oestrogen-mediated neuroprotection against excitotoxicity [35] and inhibits some beneficial effects of estradiol in animal models [22]. However, epidemiological data obtained from humans and evidence from animal models of experimental stroke, suggest a neuroprotective effect of endogenous ovarian hormones against the cerebral ischemic damage [1, 6]. With regard to stroke models, there are only a few articles that use the combined treatment and point to the beneficial effects preventing cell damage, improving behavioural recovery and reducing the oxidative stress [25, 24, 36]. The majority of these studies have been performed using transient MCAO (tMCAO), where focal blood flow deprivation for a short time, from 30 min to 2 h, is followed by reperfusion. Some researchers believe that permanent MCAO (pMCAO), where reperfusion is not permitted, has a greater clinical relevance than tMCAO [3]. In addition, most studies have established the potential of sexual hormones as preventive agents, treating animals before ischemic insult [37, 25, 36]. However, exploring the effects of these hormones administered after ischemic injury is more relevant and poorly investigated.
Our present data showed that the addition of progesterone combined with estradiol avoided some of the protective effect of estradiol both in vivo (pMCAO) and in vitro (OGD). In fact, we previously described that the administration of three injections of estradiol (0.04 mg/kg) after pMCAO, attenuated the ischemic injury in neuronal and glial cells, especially in the parietal cortex [13]. We evaluated whether progesterone combined with estradiol treatments modified the neuroprotective effects observed using estradiol alone. Six hours after pMCAO induction, a significant decrease in the IDI score was found. Administration of E/P showed a trend towards accelerating the recovery of maximum IDI. The maximum neurological score in the IEP group was recovered earlier than in the IV group, suggesting an improvement in sensory-motor functions after the E/P treatment. These results are consistent with those obtained by others 24 h after ischemia onset in a tMCAO model [25, 38].
It has been well-established that astrocytes and microglia activation after ischemia is an adaptive response to regulate the neuronal death because they play a key role in repairs in the CNS [34]. There is evidence that astrocytes express all types of receptors for both steroid hormones [39, 40]. However, the expression of these receptors in microglia has not been well characterized. Several studies have indicated the presence of estradiol receptors in resting and activated microglia [41] but until now, the presence of classical progesterone receptors have not been detected in any physiological state [34]. In this study, we found that after 54-h pMCAO induction, the amount of astrocytes and microglia increased significantly compared with the SV group. The administration of three doses of E/P starting at 6 h post-ischemia decreased reactive gliosis, measured by the expression of GFAP and Iba-1. These data concur with our recently published work, using estradiol alone [13]. An identical reduction in Iba-1 levels was found with E/P treatment, indicating that it could mainly be mediated by the direct action of estradiol, because classical progesterone receptors have not been identified in microglia. However, the reduction in GFAP levels after E/P therapy was slightly lower than with estradiol alone [13]. This seems to indicate that progesterone could be partially blocking some of the effects of estradiol in astrocytes.
After blood flow deprivation, the neurons localized in the area affected suffered detrimental processes which triggered cell death. In the MCAO model, the striatum and the parietal cortex are considered to be the ischemic core and the hippocampus a region that suffers secondary damage [5]. In order to evaluate the degree of neuronal damage after pMCAO induction and the effect of E/P administration, we stained coronal sections with a neuron-specific antibody, NeuN. Our results revealed that the percentage of NeuN-positive labelled area in the parietal cortex was significantly decreased after 54-h pMCAO induction, but no significant variations were found in the hippocampus (CA1 and CA2-CA3) compared with the SV group. E/P treatments after an ischemic insult had no impact on neuronal numbers in the parietal cortex. The effects observed are somewhat similar to those obtained using an in vitro OGD model with primary cortical neurons. Neuronal cultures subjected to OGD conditions for 17 h decreased the cell viability by 50 %. The treatment using E/P did not cause any significant variation over the OGD group. Whilst the presence of estradiol in the regeneration media improved the cell viability to control levels (non-OGD). This suggests that, in our paradigm, progesterone could be blocking some of the neuroprotective effects of estradiol. These findings were consistent with previous results obtained by Jayarama and Pike using cortical primary neurons [23]. They demonstrated the inhibitory effects of progesterone on estradiol-mediated neuroprotective actions and suggested that this effect could be mediated through downregulation of ERα and ERβ expression. However, it has been described that E/P administration after reperfusion induces a reduction of approximately 70 % of the infarction volume (measured by hematoxylin-eosin staining or by NeuN immunohistochemistry) at 2 weeks after insult, using a tMCAO model [38]. This apparent discrepancy with our findings could be due to some factors that may strongly affect the degeneration/regeneration of injured brain areas: duration of artery occlusion; reperfusion vs non-reperfusion; hormonal doses and ratio used (E/P = 0.025/10 vs 0.04/4 mg/kg); the time at which the first dose is administered (after reperfusion vs 6 h after the onset of ischemic insult); and the time when the tissue was analyzed (2 weeks vs 54 h after MCAO induction). It is well known that reperfusion, depending of damage period, help tissue regeneration. In our model, reperfusion is not allowed and therefore, the parietal cortex is more affected that in a tMCAO model. Also, the timing of pharmacological intervention affect neuronal rescue, an early intervention help to improve neuronal recovery. All these factors did not permit a straight comparison between our model system and this reported by Ulbrich et al. [38].
Nonetheless, treatments using E/P after pMCAO induced a significant increase in the percentage of NeuN-positive labelled area in CA2-CA3, with respect to the control, mostly in the stratum lucidum and radiatum. We also found that the NeuN-positive neurons in these layers decreased after pMCAO, whereas the quantity of neurons in the CA2-CA3 pyramidal layer (IV group) was similar to the SV group. The presence of some interneurons in the stratum lucidum implicated in the local circuits modulation has been described [42]. These neurons could be particularly vulnerable to ischemia and their degeneration affects the CA3 pyramidal neurons. Consequently, it could cause a delayed degeneration of the CA1 pyramidal cells [42]. We suggest that the increment in NeuN-positive neurons on stratum lucidum induced by E/P treatment could be due to an increase in NeuN expression in pre-existing interneurons. Additional experiments will be required to further confirm this hypothesis and finally decipher the E/P effect in the hippocampi.
It has been reported that the PI3K/Akt/GSK3/β-catenin survival pathway is downregulated after an ischemic insult [33, 13]. In this and in our previous work, we have shown that ischemia induced a diminution of the Akt activity (determined by levels of phosphorylated residues: Ser-473 and Thr-308) which produces a reduction in pGSK3Ser21/9 levels, associated with an activation of GSK3 activity. We previously reported that estradiol, administered after pMCAO, recovered the Akt activity predominantly in the cerebral cortex and to a lesser extent in the hippocampus, which correlates with an increment of GSK3 inhibition [13].
These results are in agreement with those obtained using our in vitro OGD model. The addition of E after OGD induced a recovery in Akt activity with the subsequent increase in pGSK3Ser21/9 levels, which may mediate the recovery of neuronal viability described above. Our in vivo and in vitro results are fully in agreement and enable us to postulate an important modulatory role of the estradiol-dependent PI3K/Akt/GSK3 pathway after ischemia. Administration of three doses of E/P after pMCAO slightly recovered the pAktThr308 epitope but not pAktSer473 in the cerebral cortex. This correlates with the effect observed in the pGSK3Ser21/9 levels and therefore maintained the GSK3 activity as seen in the IV group. Moreover, in vitro experiments using cortical neurons indicated that E/P treatment did recover either Akt activity or the degree of viability in the cortical neurons. Thus, both our present and previous work support the hypothesis that the post-pMCAO administration of estradiol reduces some of the negative effects of ischemic injury in the parietal cortex, both in vivo and in vitro, acting as a recovery agent. However, the presence of progesterone abolished some of the beneficial effects observed.
In the hippocampus, E/P treatments induced a recovery in the Akt activity levels, as demonstrated by the significant increase of pAktSer473 and pAktThr308 with respect to IV. This correlates with a significant increase in the pGSK3Ser21/9 levels and therefore an inhibition of GSK3 activity which can improve cell survival. In a previous report, we described that post-pMCAO-estradiol treatments were unable to reduce the GSK3 activity in the hippocampus [13]. The phosphorylation of both Akt-residues is necessary for the maximal Akt activity [43]. Different kinases are implicated in the phosphorylation of each residue: PDK1 (Thr-408) and mTORC2 (Ser-473) although the regulatory mechanisms, and physiological importance of each phosphorylation site, are still not fully understood [43]. According to our data, we hypothesize that post-pMCAO E/P treatment modulated PDK1 in the cortex and hippocampus, but mTORC2 only in the hippocampus through a mechanism that has not been unidentified. The results obtained in NeuN-positive cells suggests that E/P induced upregulation of pAktThr308 which occurred in the cortex, probably mediated by PDK1, but was not able to reach the Akt/GSK3 activity levels necessary for the recovery of the neurons in the ischemic core. However the pAktThr308 and pAktSer473 upregulation in the hippocampus (reflecting full activation) was able to achieve the suitable activity levels of Akt/GSK3 preventing the pMCAO-induced neuronal loss.
Therefore, we suggest that the combined administration of E/P reduces some of the beneficial effects triggered by estradiol, mediated in part by the survival pathway PI3K/Akt/GSK3/β-catenin, in the cerebral cortex. By contrast, this survival pathway is upregulated by E/P in the hippocampus, as shown by the increased levels of pAktThr308, pAktSer473 and pGSK3Ser21/9. The apparent discrepancy in the E/P effects observed in the cortex and hippocampus may be explained by variations in the ratio between progesterone/estradiol receptors between both cerebral territories. In a non-pathological condition, cortical and hippocampal neurons exhibits differences in expression of estradiol and progesterone receptors [44, 45]. These differences could be exacerbated by an ischemic insult because both territories are affected differently by pMCAO and the underlying cause of damage is not the same. In fact, after MCAO, the parietal cortex suffers a dramatic reduction in the glucose and oxygen supply whereas hippocampus underwent damage mainly by deafferentation. Nevertheless, further studies are required to reveal the detailed mechanism of E/P interactions.
In summary, we have used two experimental approaches and examined the effects of combined treatment using E/P after pMCAO induction. We have found that E/P administration after pMCAO induction (1) accelerated the recovery of neurological score in male rats; (2) reduced the degree of reactive gliosis; (3) did not avoid the neuronal loss in cerebral cortex, but induced an evident response of the neurons in the stratum lucidum/radiatum of the hippocampus CA2-CA3 region; (4) caused a significant recovery in the PI3K/Akt/GSK3/β-catenin pathway in the hippocampus but not in the cerebral cortex. However, this hormonal treatment had no remarkable impact on synapse-associated proteins at this time.
A complementary approach was performed in vitro exposing primary neurons to OGD. We found that estradiol treatments recovered neuronal viability and recuperated the PI3K/Akt/GSK3 pathway downregulated by OGD. Moreover, the combination of progesterone and estradiol avoided the protective effect of estradiol. In conclusion, we propose that the post-ischemia administration of estradiol can reduce some neurodegenerative processes associated with ischemic damage in the cortex, both in vivo (pMCAO) and in vitro (OGD). But the presence of progesterone abolished the beneficial effects in the cortex.
References
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, on behalf of the American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2013) Heart Disease and Stroke Statistics—2013 Update A Report From the American Heart Association. Circulation 127:e6–e245. doi:10.1161/CIR.0b013e31828124ad
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22(9):391–397. doi:10.1016/S0166-2236(99)01401-0
Hossmann K-A (2006) Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol 26(7):1055–1081. doi:10.1007/s10571-006-9008-1
Green A, Odergren T, Ashwood T (2003) Animal models of stroke: do they have value for discovering neuroprotective agents? Trends Pharmacol Sci 24(8):402–408
Liu F, McCullough LD (2011) Middle cerebral artery occlusion model in rodents: methods and potential pitfalls. J Biomed Biotechnol. doi:10.1155/2011/464701
Alkayed N, Harukuni I, Kimes A, London E, Traystman R, Hurn P (1998) Gender-linked brain injury in experimental stroke. Stroke 29:159–165
Takaba H, Fukuda K, Yao H (2004) Substrain differences, gender, and age of spontaneously hypertensive rats critically determine infarct size produced by distal middle cerebral artery occlusion. Cell Mol Neurobiol 24(5):589–598
Schumacher M, Mattern C, Ghoumari A, Oudinet JP, Liere P, Labombarda F, Sitruk-Ware R, De Nicola AF, Guennoun R (2014) Revisiting the roles of progesterone and allopregnanolone in the nervous system: resurgence of the progesterone receptors. Prog Neurobiol 113:6–39. doi:10.1016/j.pneurobio.2013.09.004
Pérez-Alvarez MJ, Wandosell F (2013) In: Palmeri Rand Grimaudo S (ed) Estradiol in CNS: role in neurodegeneration. Estradiol: synthesis, health effects and drug interaction, 1st edn. Nova, New York, pp 35–68
Schumacher M, Guennoun R, Robert F, Carelli C, Gago N, Ghoumari A, Gonzalez Deniselle MC, Gonzalez SL, Ibanez C, Labombarda F, Coirini H, Baulieu E-E, De Nicola AF (2004) Local synthesis and dual actions of progesterone in the nervous system: neuroprotection and myelination. Growth Hormon IGF Res 14:18–33. doi:10.1016/j.ghir.2004.03.007
Azcoitia I, Arevalo MA, De Nicola AF, Garcia-Segura LM (2011) Neuroprotective actions of estradiol revisited. Trends Endocrinol Metab 22(12):7. doi:10.1016/j.tem.2011.08.002
McCullough L, Hurn P (2003) Estrogen and ischemic neuroprotection: an integrated view. Trends Endocrinol Metab 14(5):228–235. doi:10.1016/S1043-2760(03)00076-6
Perez-Alvarez M, Maza M, Anton M, Ordonez L, Wandosell F (2012) Post-ischemic estradiol treatment reduced glial response and triggers distinct cortical and hippocampal signaling in a rat model of cerebral ischemia. J Neuroinflammation 9(1):157. doi:10.1186/1742-2094-9-157
Jia J, Guan D, Zhu W, Alkayed NJ, Wang MM, Hua Z, Xua Y (2009) Estrogen inhibits Fas-mediated apoptosis in experimental stroke. Exp Neurol 215:5. doi:10.1016/j.expneurol.2008.09.015
Singer CA, Rogers KL, Strickland TM, Dorsa DM (1996) Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett 212(1):13–16. doi:10.1016/0304-3940(96)12760-9
Stein DG (2008) Progesterone exerts neuroprotective effects after brain injury. Brain Res Rev 57(2):386–397
Baudry M, Bi X, Aguirre C (2013) Progesterone–estrogen interactions in synaptic plasticity and neuroprotection. Neuroscience 239:14. doi:10.1016/j.neuroscience.2012.10.051
Wieloch T, Nikolich K (2006) Mechanisms of neural plasticity following brain injury. Curr Opin Neurobiol 16(3):258–264. doi:10.1016/j.conb.2006.05.011
Cesca F, Baldelli P, Valtorta F, Benfenati F (2010) The synapsins: key actors of synapse function and plasticity. Prog Neurobiol 91(4):313–348
Ohtani-Kaneko R, Iwafuchi M, Iwakura T, Muraoka D, Yokosuka M, Shiga T, Watanabe C (2010) Effects of estrogen on synapsin I distribution in developing hypothalamic neurons. Neurosci Res 66(2):180–188. doi:10.1016/j.neures.2009.10.012
Sato K, Akaishi T, Matsuki N, Ohno Y, Nakazawa K (2007) β-Estradiol induces synaptogenesis in the hippocampus by enhancing brain-derived neurotrophic factor release from dentate gyrus granule cells. Brain Res 1150:108–120. doi:10.1016/j.brainres.2007.02.093
Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ (2007) Progesterone and estrogen regulate alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci 27(48):13357–13365. doi:10.1523/jneurosci. 2718-07.2007
Jayaraman A, Pike CJ (2014) Differential effects of synthetic progestagens on neuron survival and estrogen neuroprotection in cultured neurons. Mol Cell Endocrinol 384(1–2):12. doi:10.1016/j.mce.2014.01.003
Toung TJ, Chen T-Y, Littleton-Kearney MT, Hurn PD, Murphy SJ (2004) Effects of combined estrogen and progesterone on brain infarction in reproductively senescent female rats. J Cereb Blood Flow Metab 24(10):1160–1166
Dang J, Mitkari B, Kipp M, Beyer C (2011) Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain Behav Immun 25(4):715–726. doi:10.1016/j.bbi.2011.01.013
Cai W, Zhu Y, Furuya K, Li Z, Sokabe M, Chen L (2008) Two different molecular mechanisms underlying progesterone neuroprotection against ischemic brain damage. Neuropharmacology 55(2):127–138
Jiang N, Chopp M, Stein D, Feit H (1996) Progesterone is neuroprotective after transient middle cerebral artery occlusion in male rats. Brain Res 735(1):101–107
Yrjänheikki J, Koistinaho J, Kettunen M, Kauppinen RA, Appel K, Hüll M, Fiebich BL (2005) Long-term protective effect of atorvastatin in permanent focal cerebral ischemia. Brain Res 1052(2):174–179
Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinates, 6th edn. Academic, San Diego, California
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7):676–682. doi:10.1038/nmeth.2019
Arevalo MA, Santos-Galindo M, Acaz-Fonseca E, Azcoitia I, Garcia-Segura LM (2013) Gonadal hormones and the control of reactive gliosis. Horm Behav 63(2):216–221. doi:10.1016/j.yhbeh.2012.02.021
Costain WJ, Rasquinha I, Sandhu JK, Rippstein P, Zurakowski B, Slinn J, MacManus JP, Stanimirovic DB (2007) Cerebral ischemia causes dysregulation of synaptic adhesion in mouse synaptosomes. J Cereb Blood Flow Metab 28(1):99–110
Ishrat T, Sayeed I, Atif F, Hua F, Stein DG (2012) Progesterone is neuroprotective against ischemic brain injury through its effects on the phosphoinositide 3-kinase/protein kinase B signaling pathway. Neuroscience 210:442–450. doi:10.1016/j.neuroscience.2012.03.008
Johann S, Beyer C (2013) Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J Steroid Biochem Mol Biol 137:71–81. doi:10.1016/j.jsbmb.2012.11.006
Aguirre C, Jayaraman A, Pike C, Baudry M (2010) Progesterone inhibits estrogen-mediated neuroprotection against excitotoxicity by down-regulating estrogen receptor-β. J Neurochem 115(5):1277–1287. doi:10.1111/j.1471-4159.2010.07038.x
Ozacmak VH, Sayan H (2009) The effects of 17β estradiol, 17α estradiol and progesterone on oxidative stress biomarkers in ovariectomized female rat brain subjected to global cerebral ischemia. Physiol Res 58(6):909–912
Toung TJK, Traystman RJ, Hurn PD (1998) Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke 29:1666–1670
Ulbrich C, Zendedel A, Habib P, Kipp M, Beyer C, Dang J (2012) Long-term cerebral cortex protection and behavioral stabilization by gonadal steroid hormones after transient focal hypoxia. J Steroid Biochem Mol Biol 131(1–2):10–16. doi:10.1016/j.jsbmb.2012.01.007
Jung-Testas I, Do Thi A, Koenig H, Désarnaud F, Shazand K, Schumacher M, Baulieu EE (1999) Progesterone as a neurosteroid: synthesis and actions in rat glial cells. J Steroid Biochem Mol Biol 69(1–6):97–107. doi:10.1016/S0960-0760(98)00149-6
Pawlak J, Karolczak M, Krust A, Chambon P, Beyer C (2005) Estrogen receptor-α is associated with the plasma membrane of astrocytes and coupled to the MAP/Src-kinase pathway. Glia 50(3):270–275. doi:10.1002/glia.20162
Habib P, Dreymueller D, Ludwig A, Beyer C, Dang J (2013) Sex steroid hormone-mediated functional regulation of microglia-like BV-2 cells during hypoxia. J Steroid Biochem Mol Biol 138:195–205. doi:10.1016/j.jsbmb.2013.06.003
Spruston N, Lübke J, Frotscher M (1997) Interneurons in the stratum lucidum of the rat hippocampus: an anatomical and electrophysiological characterization. J Comp Neurol 385(3):427–440
Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B (2006) SIN1/MIP1 Maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127(1):125–137
Shughrue PJ, Lane MV, Merchenthaler I (1997) Comparative distribution of estrogen receptor-α and -β mRNA in the rat central nervous system. J Comp Neurol 388(4):507–525. doi:10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6
Kato J, Hirata S, Nozawa A, Yamada-Mouri N (1994) Gene expression of progesterone receptor isoforms in the rat brain. Horm Behav 28(4):454–463. doi:10.1006/hbeh.1994.1043
Acknowledgments
We are grateful to all members of Lab 206 at the Centro de Biología Molecular “Severo Ochoa” (CBM-SO) for thoughtful discussions during the preparation of this manuscript. This work was supported in part by grants from the CIBERNED (an initiative of ISCIII), the Plan Nacional DGCYT [SAF2012-39148-C03-01], and EU-FP7-2009-CT222887 and the Autonomous Government of Madrid (S20/BMD-2331). In addition CBM-SO was supported by an Institutional grant from the ‘Fundación Areces”.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Perez-Alvarez, M.J., Mateos, L., Alonso, A. et al. Estradiol and Progesterone Administration After pMCAO Stimulates the Neurological Recovery and Reduces the Detrimental Effect of Ischemia Mainly in Hippocampus. Mol Neurobiol 52, 1690–1703 (2015). https://doi.org/10.1007/s12035-014-8963-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8963-7