
1. Stroke is the neurological evidence of a critical reduction of cerebral blood flow in a circumscribed part of the brain, resulting from the sudden or gradually progressing obstruction of a large brain artery. Treatment of stroke requires the solid understanding of stroke pathophysiology and involves a broad range of hemodynamic and molecular interventions. This review summarizes research that has been carried out in many laboratories over a long period of time, but the main focus will be on own experimental research.
2. The first chapter deals with the hemodynamics of focal ischemia with particular emphasis on the collateral circulation of the brain, the regulation of blood flow and the microcirculation. In the second chapter the penumbra concept of ischemia is discussed, providing a detailed list of the physiological, biochemical and structural viability thresholds of ischemia and examples of how these thresholds can be applied for imaging the penumbra. The third chapter summarizes the pathophysiology of infarct progression, focusing on the role of peri-infarct depolarisation, the multitude of putative molecular injury pathways, brain edema and inflammation. Finally, the fourth chapter provides an overview of currently discussed therapeutic approaches, notably the effect of mechanical or thrombolytic reperfusion, arteriogenesis, pharmacological neuroprotection, ischemic preconditioning and regeneration.
3. The main emphasis of the review is placed on the balanced differentiation between hemodynamic and molecular factors contributing to the manifestation of ischemic injury in order to provide a rational basis for future therapeutic interventions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
HEMODYNAMICS OF FOCAL ISCHEMIA
Collateral Circulation
The six main supplying arteries of the brain, i.e. the right and left anterior, middle and posterior cerebral arteries, are interconnected by two major collateral systems: the circle of Willis at the base of the brain which provides low-resistance connections between the origins of these arteries, and Heubner's leptomeningeal anastomoses which interconnect the distal cortical branches (for review see Zülch, 1985; Liebeskind, 2003).
The circle of Willis is responsible for the redistribution of blood supply under conditions of extracranial vascular occlusion, notably the constriction or occlusion of the carotid and vertebral arteries. With increasing flow resistance in these vessels, blood perfusion pressure decreases throughout the circle of Willis, and when the autoregulatory range of blood perfusion is exhausted, blood flow declines first in the most peripheral branches of the brain arteries. This mechanism is referred to as the “last meadow phenomenon” and explains the induction of border zone or watershed lesions between the territories of the main supplying arteries of the brain (Zülch, 1985).
The pial network of Heubner's anastomoses critically determines the volume and severity of focal ischemia induced by the constriction or occlusion of a brain artery distal to its origin from the circle of Willis (Fig. 1). The larger the number and diameter of these anastomoses is, the more efficient is the collateral supply from the adjacent unobstructed vascular territories. In contrast to the circle of Willis, these anastomoses built up the highest perfusion pressure in the peripheral branches of the brain arteries, resulting in the preferential protection of the borderzone areas. The individual variability of these anastomoses is responsible for the fact that under clinical conditions vascular occlusion may result in a wide range of injury volumes from small lesions located in the central region (minimal infarct) to large infarcts involving the total vascular supplying territory (maximal infarct; for review see Zülch, 1985).

Collateral circulation of the brain. Heubner's leptomeningeal anastomoses connect the peripheral branches of the brain arteries and provide collateral blood flow to the peripheral parts of the adjacent vascular territories (modified from Zülch, 1985).
The functional capacity of the anastomoses depends not only on the site, but also on the speed of vascular occlusion. After abrupt occlusion induced experimentally by clot embolism or mechanical obstruction, the maximal possible collateral blood supply is not immediately established because the vascular resistance of the small anastomotic vessels is much higher than that of the main supplying artery. However, if vascular occlusion develops slowly, collaterals may undergo arteriogenesis, i.e. an active outward remodelling of the vascular wall, leading to the increase in vascular diameter and conductance (Busch et al., 2003). Arteriogenesis of collateral vessels is initiated by a change in intravascular shear stress, followed by adherence and transmigration of monocytes across the vessel wall (Buschmann et al., 2003). According to Hagen–Poiseuille's law, vascular conductance increases with the 4th power of vessel diameter. Arteriogenesis is, therefore, a powerful mechanism to adjust collateral blood supply to the flow requirements of the hypoperfused brain tissue.
Regulation of Blood Flow and Hemodynamic Reserve Capacity
When collateral blood supply does not suffice to maintain a normal blood perfusion pressure within the territory of the obstructed artery, the beginning reduction of blood supply can be compensated for some time by the physiological mechanism of flow regulation. The decline of blood perfusion pressure causes first an autoregulatory dilation of the resistance vessels. Later the beginning stimulation of anaerobic metabolism causes lactacidosis and, in consequence, a further pH-mediated enhancement of vasodilation (for review see Dirnagl and Pulsinelli, 1990). Once the resistance vessels are fully dilated, both autoregulation and CO2 reactivity are abolished, and blood flow follows passively the fluctuations of the systemic blood pressure. The abolishment of CO2 reactivity also causes an uncoupling from metabolic activity which explains the dissociation between blood flow and metabolism during peri-infarct depolarisations (see later).
The measurement of the response of cerebral blood flow to an increase in arterial pCO2—induced either by inhalation of CO2 or by the application of the carboanhydrase inhibitor acetazolamide—is a convenient way to estimate the hemodynamic reserve capacity of the autoregulatory response (Marshall et al., 2003). Once the brain vessels are fully dilated, CO2 reactivity disappears and any further reduction of blood supply cannot be longer compensated by an autoregulatory adjustment of the vascular resistance. The loss of CO2 reactivity is therefore a serious predictor of impending brain ischemia that requires immediate therapeutic interventions (Markus and Cullinane, 2001).
Microcirculation
In the intact brain the capillary network is evenly perfused with blood (for review see Hudetz, 1997). Although the suggestion had been made that capillaries may intermittently open and close, sensitive measurements revealed that under physiological conditions all capillaries are perfused with blood at all times (Göbel et al., 1990). In focal ischemia, however, microcirculation is progressively disturbed (Vogel et al., 1999). This disturbance is caused by at least three different pathophysiological mechanism: by the adhesion of white blood cells to the vessel wall; by an increase in the blood viscosity due to the aggregation of blood corpuscles; and by the compression of the capillaries by swollen astroglia. Although it is difficult to establish which of these factors is the most important, the combined effect results in heterogeneous microcirculation with greatly varying topical oxygen pressures within the ischemic tissue. This has been demonstrated by oxygen micropressure recordings which revealed a shift from the normal Gaussian distribution to both subnormal and supranormal values (Leniger-Follert and Lübbers, 1977). Some neurons may therefore be exposed to lethal tissue hypoxia at mean blood flow values which are still in the penumbral range, resulting in the phenomenon of disseminated neuronal loss in the periphery of an ischemic brain lesion (Mies et al., 1983).
THE PENUMBRA CONCEPT OF ISCHEMIA
Definitions
The intact mammalian brain covers its energy needs almost exclusively by oxidation of glucose. Opitz and Schneider (1950) were the first to draw attention to the fact that an impairment of energy production induced by a reduction of oxygen supply affects the energy-consuming processes in a sequential way: First, the functional activity of the brain is impaired followed, at a more severe degree of hypoxia, by the suppression of the metabolic activity required to maintain its structural integrity. The concept of two different thresholds of hypoxia for the preservation of functional and structural integrity was later refined by Symon et al. (1977) who used a model of focal ischemia to establish the respective rates of blood flow. These studies revealed that EEG and evoked potentials are disturbed at substantially higher flow rates than the ion gradients across the plasma membranes (Fig. 2). Since the preservation of these gradients is a sign of cell viability, Symon and his colleagues concluded that neurons located in the flow range between “electrical” and “membrane” failure are functionally silent, but structurally intact. In focal ischemia, this flow range corresponds to a crescent-shaped region intercalated between the necrotic tissue and the normal brain; it has been termed “penumbra” in analogy to the partly illuminated area around the complete shadow of the moon in full eclipse (Astrup et al., 1981).

Thresholds of metabolic (left) and electrophysiological (right) disturbances during graded reduction of cortical blood flow. The infarct core is the region in which blood flow decreases below the threshold of energy failure, and the penumbra is the region of constrained blood supply in which energy state is preserved. SEP: somatically evoked potentials; EEG: electroencephalogram (modified from Symon et al., 1977; Hossmann, 1994b).
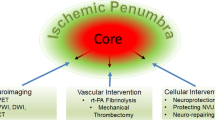
The penumbra concept of focal ischemia is of considerable interest for the understanding of stroke pathophysiology because it is the conceptual basis not only for the progressive evolution of ischemic injury, but also for the therapeutic reversal of the acute neurological symptomatology arising from stroke (for reviews see Hossmann, 1994b; Heiss, 2000; Ginsberg, 2003; Fisher, 2004; Guadagno et al., 2004). Hakim (1987) defined the penumbra as “fundamentally reversible,” but he stressed that this reversibility is time-limited. Memezawa et al. (1992) described the penumbra as the difference between the ischemic infarct developing after 1 and 24 h vascular occlusion. Other characterisations include the mismatch between perfusion and diffusion imaging (Schlaug et al., 1999), the preservation of oxygen extraction or receptor binding (Heiss, 2000), intermediate staining with neutral red as an indicator of beginning acidosis (Selman et al., 1987), or the loss of calmodulin staining (Degraba et al., 1993) as an indicator of increased intracellular calcium uptake. The common denominator of these and other definitions of the penumbra is (a) the reduction in blood flow and (b) the fundamental viability of the ischemic tissue. Since viability of brain tissue requires maintenance of energy-dependent metabolic processes, we proposed to define the penumbra as a region of constrained blood supply in which energy metabolism is preserved (Hossmann, 1994b).
Viability Thresholds of Ischemia
During the initial few hours of vascular occlusion, different brain functions break down at widely varying flow levels (for references see Heiss, 1992; Hossmann, 1994b). Progressing from the periphery to the core of the infarct, the most sensitive parameter is protein synthesis which is inhibited by 50% at about 0.55 mL g−1 min−1, and is completely suppressed below 0.35 mL g−1 min−1 (Fig. 3). These values are clearly above the disturbances of glucose utilisation and energy metabolism which begin to evolve at distinctly lower flow values. Glucose utilisation transiently increases at a flow rate below 0.35 mL g−1 min−1 before it sharply declines below 0.25 mL g−1 min−1. This range corresponds to the beginning acidosis and the beginning accumulation of lactate. At flow rates below 0.26 mL g−1 min−1 tissue acidosis becomes very pronounced and both PCr and ATP begin to decline.

Effect of focal ischemia on brain metabolism (above), neuronal release of neurotransmitters into the extracellular compartment (middle) and the water and ion homoiostasis of cerebral cortex (below). Note marked dissociation between the suppression of protein synthesis and energy failure, and between the narrowing of extracellular space and disturbances of water and electrolyte homoiostasis. ATP: tissue ATP content; Na/K ratio: quotient of the sodium and potassium content (data from Schuier and Hossmann, 1980; Paschen et al., 1992; Shimada et al., 1993).
Anoxic depolarisation occurs at even lower flow values. The sodium/potassium ratio of brain tissue increases at flow values below 0.10–0.15 mL g−1 min−1 and extracellular ion changes occur between 0.06 and 0.15 mL g−1 min−1. At the same threshold extracellular calcium declines due to the opening of calcium channels. The metabolic and ionic disturbances in the periphery of focal ischemia thus proceed in the following order: Initially protein synthesis is inhibited (at a threshold of about 0.55 mL g−1 min−1), followed by a stimulation of anaerobic glycolysis (below 0.35 mL g−1 min−1), a breakdown of energy state (at about 0.20 mL g−1 min−1) and anoxic depolarisation of the cell membranes (below 0.15 mL g−1 min−1).
As far as the functional disturbances are concerned, amplitudes of EEG and evoked potentials begin to decline at 0.23–0.25 mL g−1 min−1 and are suppressed below 0.15 mL g−1. Spontaneous unit activity disappears at a mean value of 0.18 mL g−1 min−1. Neurological studies suggest that reversible hemiparalysis appears at about 0.23 mL g−1 min−1, followed by irreversible paralysis below 0.17–0.18 mL g−1 min−1. All these values are distinctly below the threshold of the suppression of protein synthesis and even below that of the beginning activation of anaerobic glycolysis, but they fall into the range of the beginning energy crisis.
This is also true for the release of neurotransmitters into the extracellular compartment, as measured by interstitial dialysis techniques. According to these investigations, both inhibitory and excitatory neurotransmitters are released at about 0.2 mL g−1 min−1 with a possibly slightly higher threshold for glycine, adenosine and GABA than for glutamate. The release of neurotransmitters is probably unspecific because other intracellular metabolites are co-released.
A direct consequence of the metabolic disturbances associated with focal ischemia is the rise of cell osmolality which causes a shift of water from the extracellular into the intracellular compartment. The resulting decline in the fluid volume of the extracellular space can be detected by measurement of electrical impedance or by diffusion-weighted NMR imaging, both of which are sensitive to cell volume changes. Two hours after vascular occlusion the threshold of the beginning rise of electrical impedance is about 0.30 mL g−1 min−1 and that of the rise of signal intensity in diffusion-weighted imaging is 0.41 mL g−1 min−1. These thresholds are distinctly higher than the threshold of brain edema—defined as the volumetric increase of water content—which is close to 0.1 mL g−1 min−1 and which corresponds to that of anoxic depolarisation. This difference is the reason for the fact that T2-weighted NMR imaging which detects only alterations of total tissue water content, is less sensitive to mild ischemic changes than diffusion-weighted imaging.
In contrast to the biochemical and functional changes which appear shortly after vascular occlusion, histological lesions require some time before they become visible. The threshold of histological changes, therefore, depends on both the density and the duration of the flow reduction. Under conditions of permanent ischemia, the threshold of pan-necrosis is between 0.17 and 0.24 mL g−1 min−1. When ischemia lasts only a few hours, the tissue is able to survive a reduction of flow to 0.12 mL g−1 min−1. At flow values below 0.80 mL g−1 min−1, i.e. far above the threshold of pan-necrosis, selective neuronal loss may occur. Interestingly, this loss is not threshold-dependent. The flow rate correlates linearly with the number of surviving neurons which suggests a coupled decrease in parallel to the reduced metabolic requirements of the tissue. This interpretation is in line with the hypothesis that the peri-infarct brain tissue suffers pathological changes which go beyond those induced by the reduction of blood flow (see later).
Most of these thresholds have been determined at only one time point, i.e. after a few hours of vascular occlusion. However, studies dealing with the dynamics of infarct development clearly indicate that the thresholds may change with time. The threshold of ATP depletion increases from 0.13 mL g−1 min−1 after 30 min to 0.19 mL g−1 min−1 after 2 h vascular occlusion and further to 0.23 and 0.32 mL g−1 min−1 after 6 and 12 h, respectively. Similarly, the threshold of glutamate release rises from 0.2 mL g−1 min−1 after 1 h to 0.3 mL g−1 min−1 after 6–15 h ischemia. The threshold of the irreversible suppression of spontaneous neuronal unit activity rises from 0.05 to 0.12 mL g−1 min−1 during the initial 2 h of vascular occlusion, and that of the signal intensity in diffusion weighted imaging—which reflects alterations in the intra/extracellular water compartmentation—from 0.41 to 0.47 mL g−1 min−1 between 30 min and 2 h vascular occlusion. In contrast to these gradually increasing threshold values, the threshold for the suppression of protein synthesis remains remarkably stable at about 0.55 mL g−1 min−1 during the initial 12 h of ischemia and, therefore, is a robust predictor of the final size of ischemic infarction.
Imaging of the Penumbra
Based on the threshold concept of brain ischemia, the penumbra can be imaged in various ways. Under experimental conditions a precise delineation of the penumbra is obtained by imaging the mismatch between tissue ATP depletion (for detecting the infarct core) and any biochemical disturbance that evolves at flow values in the penumbral range, such as lactacidosis or the inhibition of protein synthesis. The penumbra is the difference between the respective lesion areas (Fig. 4). The exactness of this approach is documented by co-imaging gene transcripts that are selectively expressed in the penumbra, such as the stress protein hsp72 (Hata et al., 1998) or by demonstrating the gradual disappearance of the penumbra with increasing ischemia times (see later).

Imaging of infarct core and penumbra after middle cerebral artery occlusion in rat. The areas of disturbed energy metabolism (ATP) and protein synthesis are outlined and projected onto the images of blood flow (above) and the in situ hybridisation autoradiogram of hsp72 (below). Note correspondance of the biochemically characterised penumbra with hypoperfusion and the area of hsp72 upregulation (data from Mies et al., 1991; Hata et al., 1998).
Imaging of the penumbra can also be achieved by non-invasive methods, although with less regional resolution. Widely used PET parameters are the increase in oxygen extraction or the mismatch between reduced blood flow and the preservation of viability markers, such as the binding of flumazenil to central benzodiazepine receptors (Heiss, 2000). A widely used NMR method for the imaging of the penumbra is the mismatch between the signal intensities in perfusion and diffusion weighted images (Schlaug et al., 1999), but the validity of this approach is questioned by the known decline of the apparent diffusion coefficient (ADC) at flow values which are in the penumbra range (see earlier). A more precise demarcation is obtained by tissue segmentation using quantitative ADC thresholds which exhibit robust correlation with the biochemically characterised penumbra at values between 90 and 77% of control (Hoehn-Berlage et al., 1995).
PROGRESSION OF ISCHEMIC INJURY
With the advent of non-invasive imaging techniques unequivocal evidence has been provided that brain infarcts grow (Fig. 5). This growth is not due to the progression of ischemia because the activation of collateral blood supply and spontaneous thrombolysis tend to improve blood flow over time. Infarct progression can be differentiated into three phases. During the acute phase, tissue injury is the direct consequence of the ischemia-induced energy failure and the resulting terminal depolarisation of cell membranes. This injury is established within a few minutes after the onset of ischemia. During the subsequent subacute phase, the infarct core expands into the peri-infarct penumbra until, after 4–6 h, it becomes congruent with it. The main mechanisms of this subacute infarct expansion are peri-infarct spreading depressions and a multitude of cell biological disturbances, collectively referred to as molecular cell injury. Finally, a delayed phase of injury evolves which may last for several days or even weeks. During this phase, secondary phenomena such as vasogenic edema, inflammation and possibly programmed cell death may contribute to a further progression of injury.

Monitoring of infarct growth after middle cerebral artery occlusion of rat. Non-invasive imaging of the apparent diffusion coefficient of water (ADC) at three coronal slices to demonstrate the time-dependent expansion of the region in which ADC falls below the threshold of energy failure (marked by the blue colour) (modified from Hoehn-Berlage et al., 1995).
The largest increment of infarct volume occurs during the subacute phase in which the infarct core expands into the penumbra. Using multiparametric imaging techniques for the differentiation between core and penumbra, evidence could be provided that shortly after occlusion of the middle cerebral artery the penumbra is approximately of the same size as the infarct core (Hata et al., 2000a). After 3 h more than 50% and between 6 and 8 h almost all of the penumbra has disappeared and is now part of the irreversibly damaged infarct core. In the following, the most important mediators of infarct progression will be discussed.
Peri-Infarct Spreading Depression
A functional disturbance contributing to the growth of the infarct core into the penumbra zone is the generation of peri-infarct spreading depression like depolarisations. As first described by Nedergaard and Astrup (1986), such depolarisations are initiated by the infarct core from where they spread into the peripheral zone (for review see Hossmann, 1996). During spreading depression the metabolic rate of the tissue markedly increases in response to the greatly enhanced energy demands of the activated ion exchange pumps. In the healthy brain the associated increase of glucose and oxygen demands are coupled to a parallel increase of blood flow which may rise to more than twice the base level. This flow response is suppressed in the peri-infarct penumbra because the reduced hemodynamic capacity of the collateral system prevents the adequate coupling of blood supply to the metabolic needs of the tissue (Back et al., 1994). As a result, a misrelationship arises between the increased metabolic workload and the low-oxygen supply, leading to transient episodes of hypoxia and the stepwise increase in lactate during the passage of each depolarisation (Gyngell et al., 1994; Norris et al., 1998).
The pathogenic importance of peri-infarct depolarisations for the progression of ischemic injury is supported by the linear relationship between the number of depolarisations and infarct volume (Mies et al., 1993). Correlation analysis of this relationship suggests that during the initial 3 h of vascular occlusion each depolarisation increases the infarct volume by more than 20%. This is probably one of the reasons that glutamate antagonists reduce the volume of brain infarcts because these drugs are potent inhibitors of spreading depression (Iijima et al., 1992).
Molecular Mechanisms of Injury Progression
In the border zone of permanent focal ischemia or in the central part transient vascular occlusion, cellular disturbances may evolve that cannot be explained by a lasting impairment of blood flow or energy metabolism. These disturbances are referred to as molecular injury, where the term “molecular” does not anticipate any particular injury pathway. The molecular injury cascades are interconnected in complex ways, which makes it difficult to predict their relative pathogenic importance in different ischemia models (Fig. 6). In particular, molecular injury induced by transient focal ischemia is not equivalent to the alterations that occur in the penumbra of permanent ischemia. The relative contribution of the following injury mechanisms differ therefore in different types of ischemia (for review see Siesjö and Siesjö, 1996; Lipton, 1999; Nicotera, 2003).

Schematic representation of molecular injury pathways leading to ischemic cell death. Injury pathways can be blocked at numerous sites, providing multiple approaches for the amelioration of both necrotic and apoptotic tissue injury.
Excitotoxicity
Shortly after the onset of ischemia, excitatory and inhibitory neurotransmitters are released, resulting in the activation of their specific receptors. Among these, particular attention has been attributed to glutamate, which at high concentrations is known to produce excitotoxicity. The activation of ionotropic glutamate receptors results in the inflow of calcium from the extracellular into the intracellular compartment, leading to mitochondrial calcium overload and the activation of calcium-dependent catabolic enzymes. The activation of metabotropic glutamate receptors induces the IP3-dependent signal transduction pathway, leading to the stress response of endoplasmic reticulum (Paschen, 1996), and—via the induction of immediate-early-genes (IEG)—to adaptive genomic expressions (Kiessling and Gass, 1994). At high concentration, glutamate results in primary neuronal necrosis. However, following pharmacological inhibition of ionotropic glutamate receptors, an apoptotic injury mechanism evolves that may prevail under certain pathophysiological conditions (Choi, 1996). The importance of excitotoxicity for ischemic cell injury has been debated (Hossmann, 1994a), but this does not invalidate the beneficial effect of glutamate antagonists for the treatment of focal ischemia (Prass and Dirnagl, 1998). An explanation for this incongruity is the above-described pathogenic role of peri-infarct depolarisations in infarct expansion. As glutamate antagonists inhibit the spread of these depolarisations, the resulting injury is also reduced.
Calcium Toxicity
In the intact cell, highly efficient calcium transport systems assure the maintenance of a steep calcium concentration gradient between the extra- and the intracellular compartment on the one hand, and between the cytosol and the endoplasmic reticulum (ER) on the other. Following anoxic depolarisation, these gradients break down, resulting in the sharp rise of calcium ion activity in the cytoplasm, and its decline in the ER. Both events are pathogenic: The rise of calcium in the cytoplasm leads to the activation of catabolic enzymes and mitochondrial disturbances (Siesjö et al., 1999), and the fall of calcium in the ER evokes an ER stress response, which mediates a great number of ER-dependent functional disturbances (Paschen, 1996). Calcium-dependent pathological events are therefore more complex than widely assumed and should also consider ER-mediated effects.
Free Radicals
In brain regions with low or intermittent blood perfusion, reactive oxygen species (ROS) are formed which produce peroxidative injury of plasma membranes and intracellular organelles (Chan, 1996). The reaction with nitric oxide leads to the formation of peroxynitrate, which also causes violent biochemical reactions (Eliasson et al., 1999). Secondary consequences of free radical reactions are the release of biologically active free fatty acids such as arachidonic acid (Hillered and Chan, 1988), the induction of endoplasmic reticulum stress (Paschen, 2003), the induction of mitochondrial disturbances (Siesjö et al., 1999) and fragmentation of DNA (Lipton and Nicotera, 1998). The latter may induce apoptosis and thus enhance molecular injury pathways related to mitochondrial dysfunction (see later).
Nitric Oxide Toxicity
Nitric oxide (NO) is a product of NO synthase (NOS) acting on argenin. There are at least three isoforms of NOS: eNOS is constitutively expressed in endothelial cells, nNOS in neurons and the inducible isoform iNOS mainly in macrophages (Samdani et al., 1997). Pathophysiologically, NO has two opposing effects (Dalkara and Moskowitz, 1994). In endothelial cells the generation of NO leads to vascular dilation, an improvement of blood flow and the alleviation of hypoxic injury, whereas in neurons it contributes to glutamate excitotoxicity and—by formation of peroxynitrate—to free radical-induced injury. The net effect of NO thus depends on the individual pathophysiological situation and is difficult to predict.
Zinc Toxicity
Zinc is an essential catalytic and structural element of numerous proteins and a secondary messenger which is released from excitatory synapses during neuronal activation (Choi and Koh, 1998). Recent evidence points to the contribution of zinc to the manifestation of ischemic cell injury, one of the possible mechanisms being the formation of free radicals (Kim et al., 1999). However, the precise injury pathways are not fully understood and have to await further clarification.
Dysfunction of Endoplasmic Reticulum
Similar to many other forms of cellular stress, cerebral ischemia causes the release of calcium from the endoplasmic reticulum (ER). The resulting dysfunction of the ER is associated with various cell biological abnormalities such as misfoldings of proteins, expression of stress proteins and disturbances of global protein synthesis (Paschen, 2003). The latter is due to the activation of protein kinase R (PKR) which causes the phosphorylation and inactivation of the eukaryotic initiation factor eIF2α (DeGracia et al., 1999). This again leads to the selective inhibition of polypeptide chain initiation, disaggregation of ribosomes and inhibition of protein synthesis at the translational level.
Autoradiographic studies of regional protein synthesis revealed that all hitherto described forms of delayed ischemic cell death are preceded by irreversible inhibition of protein synthesis (Hossmann, 1993). It is therefore likely that factors responsible for the inhibition of protein synthesis play a crucial role in the initiation of delayed ischemic cell death.
Mitochondrial Disturbances
The concurrence of an increased cytosolic calcium activity with the generation of reactive oxygen species leads to the increase in permeability of the inner mitochondrial membrane (mitochondrial permeability transition, MPT), which has been associated with the formation of MPT pores (Siesjö et al., 1999). The increase in permeability of the inner mitochondrial membrane has two pathophysiologically important consequences. The breakdown of the electrochemical gradient interferes with mitochondrial respiration and, in consequence, the oxidative phosphorylation of adenine nucleotides. Furthermore, the equilibration of mitochondrial ion gradients causes swelling of the mitochondrial matrix, which eventually will cause disruption of the outer mitochondrial membrane and the release of pro-apoptotic mitochondrial proteins (Green and Kroemer, 2004). Among these, cytochrome C and caspase 9 are of particular importance because they activate the cysteine protease caspase 3, which is directly involved in the execution of apoptotic cell death (Krajewski et al., 1999). Ischemia induced mitochondrial disturbances thus contribute to delayed cell death both by impairment of the energy state and the activation of apoptotic injury pathways.
Brain Edema
An important modulator of focal ischemic injury is brain edema which can be differentiated into two phases: An early cytotoxic type of edema, followed after some time by a late vasogenic type of edema (for review see Hossmann, 1989; Rosenberg, 1999; Rosand and Schwamm, 2001). The cytotoxic type of edema is threshold dependent. It is initiated at flow values similar to 30% of control when stimulation of anaerobic metabolism causes an increase of brain tissue osmolality and, hence, an osmotically obliged cell swelling. At flow values below 20% of control, anoxic depolarisation and equilibration of ion gradients across the cell membranes further enhance intracellular osmolality and the associated cell swelling. The intracellular uptake of sodium is also associated with a coupled movement of water that is independent of an osmotic gradient and which is referred to as “anomalous osmosis” (Tomita, 2005).
In the absence of blood flow, cell swelling occurs at the expense of the extracellular fluid volume, leading to the shrinkage of the extracellular compartment, but not to a change in the net water content. The shift of fluid is reflected by a decrease of the apparent diffusion coefficient of water which underlies the increase of signal intensity in diffusion-weighted MR imaging (Hossmann and Hoehn-Berlage, 1995). However, if some residual blood flow persists, water is taken up from the blood, and the net tissue water content increases. After MCA occlusion this increase starts within a few minutes after the onset of ischemia and within 4 h may raise the water content of cerebral cortex from 4.2 to 4.9 mL g−1 d.w. and that of white matter from 2.1 to 2.6 mL g−1 d.w.
With the manifestation of tissue necrosis after 4–6 h of ischemia, the blood-brain barrier breaks down and serum proteins begin to leak from the blood into the brain. This disturbance initiates a vasogenic type of edema which further enhances the water content of the tissue. Vasogenic edema reaches its peak at 1–2 days after the onset of ischemia and may cause an increase of tissue water content to as much as 8 mL g−1 d.w. If brain infarcts are large, the volume increase of the edematous brain tissue may be so pronounced that transtentorial herniation results in compression of the midbrain. Under clinical conditions, this “malignant” form of brain infarction is the by far most dangerous complication of stroke and an indication for decompressive craniectomy (Walz et al., 2002).
Vasogenic edema, in contrast to the early cytotoxic type of edema, is isoosmotic and accumulates mainly in the extracellular compartment. This reverses the narrowing of the extracellular space and explains the “pseudonormalisation” of the signal intensity observed in diffusion-weighted MR imaging (Lansberg et al., 2001). However, as the total tissue water content is increased at this time, the high signal intensity in T2-weighted images clearly distinguishes this situation from a “real” recovery to normal (Warach et al., 1995).
Recently, much attention has been directed to the fact that the formation of cytotoxic—and to a lesser degree also vasogenic—edema requires the passage of water through aquaporin channels located in the plasma membrane (Badaut et al., 2002). Inhibition of aquaporin water conductance may, therefore, reduce the severity of ischemic brain edema (Griesdale and Honey, 2004). Similarly, the inhibition of sodium transport across sodium channels has been suggested to reduce edema formation (Betz et al., 1994). However, as the driving force for the generation of edema is the gradient of osmotic and ionic concentration differences built up during ischemia, aquaporin channels may modulate the speed of edema generation, but not the final extent of tissue water accumulation. Their pathophysiological importance is, therefore, limited.
Inflammation
Brain infarcts evoke a strong inflammatory response which is thought to contribute to the progression of ischemic brain injury (Del Zoppo and Hallenbeck, 2000). Gene expressions related to this response have, therefore, been extensively investigated to search for possible interventional targets (for review see Touzani et al., 1999; Rothwell and Luheshi, 2000). The pro-inflammatory cytokines interleukin (IL)-1 beta, IL-6, and IL-10 are massively upregulated both during permanent and after transient focal ischemia. The importance of IL-1 beta for the progression of focal ischemic injury is strongly supported by evidence that application or overexpression of the interleukin 1-beta receptor antagonist IL-1ra reduces infarct size (Stroemer and Rothwell, 1997; Yang et al., 1999). Similarly, blockade of interleukin-1 beta converting enzyme (ICE, caspase-1), which reduces the formation of IL-1, also reduces brain injury (Hara et al., 1997; Rabuffetti et al., 2000). IL-1 beta expression is closely associated with an upregulation of ICAM and ELAM which reach a peak between 6 and 12 h after the onset of ischemia (Wang and Feuerstein, 1995). ICAM-1 deficient mice suffer smaller infarcts after transient MCA occlusion, suggesting that part of the IL-1 beta dependent injury is mediated by activation of ICAM-1 (Soriano et al., 1996).
The inflammatory response of the ischemic tissue has been associated, among others, with the generation of free radicals in reperfused or critically hypoperfused brain tissue (see earlier). NF-kappa B, a transcription factor that is responsive to oxidative stress is upregulated in the penumbra, but declines in the centre of the ischemic lesion (Aronowski et al., 2000; Seegers et al., 2000). The prostaglandin synthesizing enzyme cyclo-oxygenase-2 (COX-2) is also strongly upregulated in the peri-infarct penumbra and can be detected in neutrophils, vascular cells and neurons (Iadecola et al., 1999; Bidmon et al., 2000). The pathogenic importance of this protein is supported by the beneficial effect of a selective COX-2 inhibitor (Nagayama et al., 1999). Infarct reduction was also observed after genetic or pharmacological inhibition of matrix metalloproteinase (MMP)-9 (Asahi et al., 2000). Inflammatory reactions and the associated free radical-mediated injury are, therefore, important modulators of ischemic injury that should be treated to improve postischemic outcome.
THERAPY OF ISCHEMIC INJURY
In principle, brain injury can be reversed after normothermic focal ischemia of at least 1 h, but recovery depends crucially on how fast nutritive blood flow is restored (Hossmann, 1997). As post-ischemic recirculation dynamics differ greatly after transient mechanical vascular occlusion, on one hand, and spontaneous or thrombolytic reperfusion following thromboembolic stroke, on the other, the two pathophysiological situations will be discussed separately.
Reversal of Mechanical Occlusion
Recirculation after transient clip or filament occlusion of the middle cerebral artery restores blood flow almost instantaneously, provided the occlusion does not cause structural damage of the vessel. As a result, oxidative glucose metabolism and brain energy state recover rapidly throughout the MCA territory, even after occlusion of as long as 1 h (Fig. 7). If vascular occlusion is further prolonged, recovery of energy metabolism depends on the residual flow rate during ischemia and fails first in the central parts of the vascular territory where blood flow is lowest (Lust et al., 2002). After more than 3–6 h occlusion, recovery also fails in the peripheral parts of the vascular territory and the size of injury approaches that of permanent ischemia (Buchan et al., 1992).

Reversal of focal ischemia after mechanical occlusion of the middle cerebral artery for 1 h. Simultaneous imaging of the tissue content of ATP and protein synthesis. Note rapid restoration of energy metabolism, but not of protein synthesis. A few hours after restoration of blood flow, energy metabolism secondarily fails in the areas in which protein synthesis did not recover (data from Hata et al., 2000b).
In contrast to energy metabolism, recovery of protein synthesis is much slower. It also depends on both the duration of ischemia and the residual flow rate, but the longest ischemia time after which protein synthesis recovers throughout the vascular territory is only about 30 min. After longer ischemia times recovery fails in a gradually expanding core region until, after about 2 h of vascular occlusion, recovery is absent (Mies et al., 2001).
The late reversal of protein synthesis is one of the reasons that after long ischemia times recovery of energy metabolism is only transient. Depending again on both the duration and the residual flow rate during ischemia, secondary failure occurs first in the central parts and then spreads to the more peripheral regions until it merges with the area in which protein synthesis has not returned (Mies et al., 2001) (Fig. 7). The delay between the beginning of recirculation and secondary energy failure correlates inversely with the duration of ischemia and after 3 h vascular occlusion may be as long as 3 weeks (Du et al., 1996).
There is good evidence that secondary energy failure and the resulting delayed tissue necrosis is initiated by the molecular injury cascade discussed above. In fact, most of the pharmacological interventions interfering successfully with this cascade, were tested in models of transient mechanical occlusion and, therefore, are directed against this particular pathophysiology. It should be noted, however, that reversal of mechanical occlusion does not replicate naturally occurring reperfusion patterns and that the observations made using this experimental paradigm are not readily applicable to clinical stroke.
Reversal of Thromboembolic Occlusion
In most instances of stroke, collateral blood supply and spontaneous thrombolysis result in some degree of reperfusion. The speed and rate of reperfusion is greatly accelerated by the application of thrombolytic agents but even under optimal conditions, reperfusion is not instantaneous (Kilic et al., 2000). In the core of the ischemic territory, the slowly increasing, heterogeneously distributed supply of blood promotes edema formation and reoxygenation injury before oxidative metabolism is restored, preventing the recovery of tissue damage (Fig. 8). In the penumbra, in contrast, even minor improvement of blood flow increases the hemodynamic reserve capacity of the still undamaged tissue and enhances the tolerance to additional hemodynamic or functional loads, such as spurious fluctuations of blood pressure or peri-infarct spreading depressions. As a result, infarct expansion into the penumbra slows down, leading to the relative reduction of the final infarct volume as compared to untreated ischemia. If thrombolysis—and hence recirculation—is delayed, the therapeutic outcome depends on how far the infarct core has expanded into the penumbra prior to the initiation of recirculation. This explains that with increasing delay the therapeutic effect gradually declines until, after 3–6 h, improvement cannot be longer expected (Brinker et al., 1999).

Thrombolytic reversal of focal ischemia at 1 h after clot embolism of middle cerebral artery. MR angiography of the circle of Willis (left), perfusion-weighted imaging (middle) and imaging of the apparent diffusion coefficient of water (ADC). Thrombolysis was started 1 h after embolism. Recanalisation of the occluded vessel and restoration of blood flow prevents progression, but does not reverse the ADC-detectable brain lesion (data from Hilger et al., 2002).
Arteriogenesis of Collateral Vessels
In the vertebrate brain three modes of vascular growth have been identified: vasculogenesis (the formation of vessels by angioblasts during early ontogenesis), angiogenesis (the production of capillary networks by sprouting or de novo growth) and arteriogenesis (the outward remodelling of pre-existing arteries and arterioles). Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine that is known to induce arteriogenic growth of collateral vessels after occlusion of major cardiac or peripheral arteries. Recently, evidence has been provided that arteriogenesis also occurs in the brain under conditions of reduced arterial blood supply (Busch et al., 2003). Hemispheric hypoperfusion induced by the combination of unilateral common carotid and bilateral vertebral artery occlusions (three-vessel occlusion, 3-VO) led to the slowly progressing growth of the anterior and posterior segments of the circle of Willis which is the main collateral pathway between the origins of the anterior, middle and posterior cerebral arteries. GM-CSF applied subcutaneously at daily doses of 40 μg kg−1 resulted in the marked acceleration of this process (Buschmann et al., 2003) (Fig. 9). Within 1 week after the onset of treatment, the diameter of the posterior segment of the circle of Willis enlarged to 170% of control, leading to the normalisation of blood flow in the hypoperfused hemisphere. Arteriogenesis even fully restored the hemodynamic reserve capacity of the brain which immediately after 3-VO was completely abolished. As a result, the severity of hemodynamic stroke induced after 3-VO by systemic hypotension, was significantly reduced, indicating that the improved hemodynamic reserve capacity was functionally relevant (Schneeloch et al., 2004). GM-CSF induced stimulation of arteriogenesis in the hypoperfused brain thus provides powerful protection against ischemic stroke

Arteriogenesis of the posterior cerebral artery for the prevention of hemodynamic stroke. Rats were submitted to three-vessel (bilateral vertebral and unilateral carotid artery) occlusion, followed by treatment with granulocyte-monocyte (GM) colony stimulating factor for the induction of arteriogenic growth of the posterior part of the circle of Willis. Treatment restored CO2 reactivity and greatly reduced hemodynamic infarction induced by systemic hypotension (data from Busch et al., 2003; Buschmann et al., 2003; Schneeloch et al., 2004).
Neuroprotection
In the wider sense, neuroprotection is the preservation of the structural and functional integrity of the brain by any kind of intervention that interferes with the deleterious effects of ischemia. The general use of this term, however, refers to pharmacological treatments that alleviate the molecular injury cascades leading to neuronal death (Fig. 6). For obvious reasons, neuroprotection is useless in a brain region in which blood flow has declined below the threshold of energy failure, but it may contribute to the temporary preservation of the penumbra, bridging the interval between the onset of ischemia and the restitution of blood flow, and/or preventing secondary neuronal death during reperfusion (Ginsberg, 1997; Wahlgren and Ahmed, 2004). However, as the penumbra can be effectively protected by improvement of blood flow alone (see earlier), neuroprotective interventions are mediated not only by interference with molecular injury cascades, but also by reducing the mismatch between blood flow and metabolism. Examples of this are the suppression of peri-infarct depolarisations by glutamate antagonists (Gill et al., 1992; Iijima et al., 1992), the alleviation of mitochondrial oxidative insufficiency by inhibiting the mitochondrial permeability transition (Kuroda and Siesjö, 1997; Green and Kroemer, 2004), or the prevention of NAD depletion by PARP inhibitors (Endres et al., 1997; Takahashi et al., 1999). A hemodynamically mediated molecular intervention is also the application of statins which improve blood flow by upregulating eNOS (Amin-Hanjani et al., 2001). Neuroprotection thus contributes to the temporary preservation of the penumbra and may bridge the interval between the onset of ischemia and the restitution of blood flow.
During reperfusion after transient vascular occlusion, a great number of neuroprotective interventions are able to ameliorate outcome, apparently by preventing delayed injury. These interventions include anti-excitotoxic (Prass and Dirnagl, 1998), anti-apoptotic (Mattson et al., 2000), anti-inflammatory (Beech et al., 2001) and anti-oxidant and free-radical inhibitory approaches (Hall, 1997; Clark et al., 2001). Protective effects have also been observed by interference with calcium homeostasis (Kristian et al., 1998) and the erythropoietin receptor (Siren and Ehrenreich, 2001). However, as these interventions are only effective if energy metabolism recovers, i.e. under conditions of unimpaired reperfusion, they may be of limited relevance for clinical stroke.
Ischemic Preconditioning
The molecular signalling cascades initiated by brain ischemia are not solely destructive, but may also exert a neuroprotective effect (for review see Kirino, 2002). In fact, most of the above-described injury pathways including ischemia itself, induce a transient state of increased ischemic tolerance, provided the initial injury remains subliminal for tissue destruction (Stagliano et al., 1999). This effect is called “ischemic preconditioning” and can be differentiated into three phases: during the induction phase molecular sensors which respond to the preconditioning stimulus are activated by transcription factors; the transduction phase results in the amplification of the signal; and during the effector phase proteins with a protective impact are switched on (Dirnagl et al., 2003).
An important preconditioning pathway is the upregulation of the hypoxia-inducible factor 1 (HIF-1) in astrocytes. HIF-1 is a transcription factor that among others induces the expression of erythropoietin (EPO) which binds to the neuronal EPO receptor and which exhibits potent neuroprotective effects (Prass et al., 2003). Another putative mechanism is the endoplasmic reticulum stress response (Paschen, 2001). Depletion of ER calcium stores causes accumulation of unfolded proteins in the ER lumen and induces the activation of two highly conserved stress responses, the ER overload response (EOR) and the unfolded protein response (UPR). EOR triggers activation of the transcription factor NF-kappa B, and UPR causes a suppression of the initiation of protein synthesis. As the latter contributes to delayed ischemic injury, its reduction may have a neuroprotective effect (Burda et al., 2003; Paschen, 2003).
Ischemia tolerance increases 2–3 days after the preconditioning stimulus and slowly disappears after 1 week (Kirino, 2002). This rather long effect provides the unique opportunity to protect patients with reduced hemodynamic reserve capacity against injury until blood supply can be restored.
Regeneration

Cell replacement therapy of ischemic stroke: migration and differentiation of transplanted murine ES cells in the ischemia damaged rat brain. Grafted cells were labelled with iron oxide nanoparticles and green fluorescent protein (GFP), and are detected by magnetic resonance imaging and histochemistry. In the border zone of the ischemic lesion a GFP-positive cell (red) colocalizes with the neuronal marker NeuN (red) (data from Hoehn et al., 2002).
With the discovery of functionally active stem cells in the hippocampus and the subventricular zone of the adult brain, the possibility of endogenous regeneration of brain infarcts has been evoked (Imitola et al., 2004). In fact, neurogenesis has been documented in several focal ischemia models, notably after photothrombotic necrosis of cerebral cortex (Gu et al., 2000). The number of spontaneously regenerating neurons is so low that up to now the possibility of a regeneration of brain infarcts has been excluded. However, it is conceivable that functionally relevant neurogenesis is promoted by trophic factors, such as brain derived neurotrophic factor (BDNF) or other growth factors (Dempsey et al., 2003; Suzuki et al., 2003). Regeneration therapy has also been attempted by transplantation of immortalised neuroepithelial cells (Modo et al., 2002), neural stem cells (Chu et al., 2004) and stem cells derived from fetal brain tissue (Borlongan et al., 1997), bone marrow (Grabowski et al., 1995; Chen et al., 2003) or umbilical cord blood (Willing et al., 2003). However, the reported functional improvements are probably unspecific effects which cannot be explained by the small number of surviving cells (Fig. 10). What kind of unspecific effects these are, is difficult to predict but recent evidence of spontaneous neurogenesis, angiogenesis and synaptogenesis distant from the ischemic lesion points to a remodelling of the surviving tissue which may promote post-lesional brain plasticity (Roitberg, 2004). Understanding these processes may unveil hitherto unknown mechanisms that may become targets of future therapeutic interventions.
REFERENCES
Amin-Hanjani, S., Stagliano, N. E., Yamada, M., Huang, P. L., Liao, J. K., and Moskowitz, M. A. (2001). Mevastatin, an HMG-CoA reductase inhibitor, reduces stroke damage and upregulates endothelial nitric oxide synthase in mice. Stroke 32:980–985.
Aronowski, J., Strong, R., Kang, H. S., and Grotta, J. C. (2000). Selective up-regulation of I kappa B-alpha in ischemic penumbra following focal cerebral ischemia. Neuroreport 11:1529–1533.
Asahi, M., Asahi, K., Jung, J. C., del Zoppo, G. J., Fini, M. E., and Lo, E. H. (2000). Role for matrix metalloproteinase 9 after focal cerebral ischemia, effects of gene knockout and enzyme inhibition with BB-94. J. Cereb. Blood Flow Metab. 20:1681–1689.
Astrup, J., Symon, L., and Siesjö, B. K. (1981). Thresholds in cerebral ischemia—The ischemic penumbra. Stroke 12:723–725.
Back, T., Kohno, K., and Hossmann, K. -A. (1994). Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression—Effect on blood flow, tissue oxygenation, and electroencephalogram. J. Cereb. Blood Flow Metab. 14:12–19.
Badaut, T., Lasbennes, T., Magistretti, P. J., and Regli, L. (2002). Aquaporins in brain: Distribution, physiology, and pathophysiology. J. Cereb. Blood Flow Metab. 22:367–378.
Beech, J. S., Reckless, J., Mosedale, D. E., Grainger, D. J., Williams, S. C. R., and Menon, D. K. (2001). Neuroprotection in ischemia-reperfusion injury: An antiinflammatory approach using a novel broad-spectrum chemokine inhibitor. J. Cereb. Blood Flow Metab. 21:683–689.
Betz, A. L., Keep, R. F., Beer, M. E., and Ren, X. D. (1994). Blood-brain barrier permeability and brain concentration of sodium, potassium, and chloride during focal ischemia. J. Cereb. Blood Flow Metab. 14:29–37.
Bidmon, H. J., Oermann, E., Schiene, K., Schmitt, M., Kato, K., Asayama, K., Witte, O. W., and Zilles, K. (2000). Unilateral upregulation of cyclooxygenase-2 following cerebral, cortical photothrombosis in the rat: Suppression by MK-801 and co-distribution with enzymes involved in the oxidative stress cascade [review]. J. Chem. Neuroanat. 20:163–176.
Borlongan, C. V., Koutouzis, T. K., Jorden, J. R., Martinez, R., Rodriguez, A. I., Poulos, S. G., Freeman, T. B., McKeown, P., Cahill, D. W., Nishino, H., and Sanberg, P. R. (1997). Neural transplantation as an experimental treatment modality for cerebral ischemia. Neurosci. Biobehav. Rev. 21:79–90.
Brinker, G., Franke, C., Hoehn, M., Uhlenkuken, U., and Hossmann, K. A. (1999). Thrombolysis of cerebral clot embolism in rat: Effect of treatment delay. Neuroreport 10:3269–3272.
Buchan, A. M., Xue, D., and Slivka, A. (1992). A new model of temporary focal neocortical ischemia in the rat. Stroke 23:273–279.
Burda, J., Hrehorovska, M., Bonilla, L. G., Danielisova, V., Cizkova, D., Burda, R., Nemethova, M., Fando, J. L., and Salinas, M. (2003). Role of protein synthesis in the ischemic tolerance acquisition induced by transient forebrain ischemia in the rat. Neurochem. Res. 28:1213–1219.
Busch, H. J., Buschmann, I. R., Mies, G., Bode, C., and Hossmann, K. -A. (2003). Arteriogenesis in hypoperfused rat brain. J. Cereb. Blood Flow Metab. 23:621–628.
Buschmann, I. R., Busch, H. J., Mies, G., and Hossmann, K. -A. (2003). Therapeutic induction of arteriogenesis in hypoperfused rat brain via granulocyte-macrophage colony-stimulating factor. Circulation 108:610–615.
Chan, P. H. (1996). Role of oxidants in ischemic brain damage. Stroke 27:1124–1129.
Chen, J. L., Li, Y., Katakowski, M., Chen, X. G., Wang, L., Lu, D. Y., Lu, M., Gautam, S. C., and Chopp, M. (2003). Intravenous bone marrow stromal cell therapy reduces apoptosis and promotes endogenous cell proliferation after stroke in female rat. J. Neurosci. Res. 73:778–786.
Choi, D. W. (1996). Ischemia-induced neuronal apoptosis. Curr. Opin. Neurobiol. 6:667–672.
Choi, D. W., and Koh, J. Y. (1998). Zinc and brain injury [review]. Ann. Rev. Neurosci. 21:347–375.
Chu, K., Kim, M., Park, K. I., Jeong, S. W., Park, H. K., Jung, K. H., Lee, S. T., Kang, L., Lee, K., Park, D. K., Kim, S. U., and Roh, J. K. (2004). Human neural stem cells improve sensorimotor deficits in the adult rat brain with experimental focal ischemia. Brain Res. 1016:145–153.
Clark, W. M., Rinker, L. G., Lessov, N. S., Lowery, S. L., and Cipolla, M. J. (2001). Efficacy of antioxidant therapies in transient focal ischemia in mice. Stroke 32:1000–1004.
Dalkara, T., and Moskowitz, M. A. (1994). The complex role of nitric oxide in the pathophysiology of focal cerebral ischemia. Brain Pathol. 4:49–57.
Degraba, T. J., Ostrow, P. T., and Grotta, J. C. (1993). Threshold of calcium disturbances after focal cerebral ischemia in rats—Implications of the window of therapeutic opportunity. Stroke 24:1212–1217.
DeGracia, D. J., Adamczyk, S., Folbe, A. J., Konkoly, L. L., Pittman, J. E., Neumar, R. W., Sullivan, J. M., Scheuner, D., Kaufman, R. J., White, B. C., and Krause, G. S. (1999). Eukaryotic initiation factor 2 alpha kinase and phosphatase activity during postischemic brain reperfusion. Exp. Neurol. 155:221–227.
Del Zoppo, G. J., and Hallenbeck, J. M. (2000). Advances in the vascular pathophysiology of ischemic stroke. Thromb. Res. 98:V73–V81.
Dempsey, R. J., Sailor, K. A., Bowen, K. K., Tureyen, K., and Vemuganti, R. (2003). Stroke-induced progenitor cell proliferation in adult spontaneously hypertensive rat brain: effect of exogenous IGF-1 and GDNF. J. Neurochem. 87:586–597.
Dirnagl, U., and Pulsinelli, W. (1990). Autoregulation of cerebral blood flow in experimental focal brain ischemia. J. Cereb. Blood Flow Metab. 10:327–336.
Dirnagl, U., Simon, R. P., and Hallenbeck, J. M. (2003). Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 26:248–254.
Du, C., Hu, R., Csernansky, C. A., Hsu, C. Y., and Choi, D. W. (1996). Very delayed infarction after mild focal cerebral ischemia: A role for apoptosis?. J. Cereb. Blood Flow Metab. 16:195–201.
Eliasson, M. J. L., Huang, Z. H., Ferrante, R. J., Sasamata, M., Molliver, M. E., Snyder, S. H., and Moskowitz, M. A. (1999). Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J. Neurosci.e 19:5910–5918.
Endres, M., Wang, Z. Q., Namura, S., Waeber, C., and Moskowitz, M. A. (1997). Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 17:1143–1151.
Erdo, F., Buhrle, C., Blunk, J., Hoehn, M., Xia, T., Fleischmann, B., Focking, M., Kustermann, E., Kolossov, E., Hescheler, T., Hossmann, K. A., and Trapp, T. (2003). Host-dependent tumorigenesis of embryonic stem cell transplantation in experimental stroke. J. Cereb. Blood Flow Metab. 23:780–785.
Fisher, M. (2004). The ischemic penumbra: Identification, evolution and treatment concepts. Cerebrovasc. Dis. 17:1–6.
Gill, R., Andine, P., Hillered, L., Persson, L., and Hagberg, H. (1992). The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischemia in the rat. J. Cereb. Blood Flow Metab. 12:371–379.
Ginsberg, M. D. (1997). Injury mechanisms in the ischaemic penumbra—approaches to neuroprotection in acute ischaemic stroke. Cerebrovasc. Dis. 7(2):7–12.
Ginsberg, M. D. (2003). Adventures in the pathophysiology of brain ischemia: Penumbra, gene expression, neuroprotection. The 2002 Thomas Willis Lecture. Stroke 34:214–223.
Göbel, U., Theilen, H., and Kuschinsky, W. (1990). Congruence of total and perfused capillary network in rat brains. Circ. Res. 66:271–281.
Grabowski, M., Sorensen, J. C., Mattsson, B., Zimmer, J., and Johansson, B. B. (1995). Influence of an enriched environment and cortical grafting on functional outcome in brain infarcts of adult rats. Exp. Neurol. 133:96–102.
Green, D. R., and Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science 305:626–629.
Griesdale, D. E. G., and Honey, C. R. (2004). Aquaporins and brain edema. Surg. Neurol. 61:418–421.
Gu, W. G., Brannstrom, T., and Wester, P. (2000). Cortical neurogenesis in adult rats after reversible photothrombotic stroke. J. Cereb. Blood Flow Metab. 20:1166–1173.
Guadagno, J. V., Donnan, G. A., Markus, R., Gillard, J. H., and Baron, J. C. (2004). Imaging the ischaemic penumbra. Curr. Opin. Neurol. 17:61–67.
Gyngell, M. L., Back, T., Hoehn-Berlage, M., Kohno, K., and Hossmann, K. -A. (1994). Transient cell depolarization after permanent middle cerebral artery occlusion: An observation by diffusion-weighted MRI and localized 1H-MRS. Magn. Reson. Med. 31:337–341.
Hakim, A. M. (1987). The cerebral ischemic penumbra. Can. J. Neurol. Sci. 14:557–559.
Hall, E. D. (1997). Acute therapeutic interventions—free radical scavengers and antioxidants. Neurosurg. Clin. North Am. 8:195.
Hara, H., Friedlander, R. M., Gagliardini, V., Ayata, C., Fink, K., Huang, Z. H., Shimizusasamata, M., Yuan, J. Y., and Moskowitz, M. A. (1997). Inhibition of interleukin 1-beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. U. S. A. 94:2007–2012.
Hata, R., Maeda, K., Hermann, D., Mies, G., and Hossmann, K. -A. (2000a). Dynamics of regional brain metabolism and gene expression after middle cerebral artery occlusion in mice. J. Cereb. Blood Flow Metab. 20:306–315.
Hata, R., Maeda, K., Hermann, D., Mies, G., and Hossmann, K. -A. (2000b). Evolution of brain infarction after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 20:937–946.
Hata, R., Mies, G., Wiessner, C., and Hossmann, K. -A. (1998). Differential expression of c-fos and hsp72 mRNA in focal cerebral ischemia of mice. Neuroreport 9:27–32.
Heiss, W. D. (1992). Experimental evidence of ischemic thresholds and functional recovery. Stroke 23:1668–1672.
Heiss, W. D. (2000). Ischemic penumbra: Evidence from functional imaging in man [review]. J. Cereb. Blood Flow Metab. 20:1276–1293.
Hilger, T., Niessen, F., Diedenhofen, M., Hossmann, K. –A., and Hoehn, M. (2002). Magnetic resonance angiography of thromboembolic stroke in rats: Indicator of recanalization probability and tissue survival after recombinant tissue plasminogen activator treatment. J. Cereb. Blood Flow Metab. 22:652–662.
Hillered, L., and Chan, P. H. (1988). Role of arachidonic acid and other free fatty acids in mitochondrial dysfunction in brain ischemia. J. Neurosci. Res. 20:451–456.
Hoehn, M., Kustermann, E., Blunk, J., Wiedermann, D., Trapp, T., Wecker, S., Focking, M., Arnold, H., Hescheler, J., Fleischmann, B. K., Schwindt, W., and Buhrle, C. (2002). Monitoring of implanted stem cell migration in vivo: A highly resolved in vivo magnetic resonance imaging investigation of experimental stroke in rat. Proc. Natl. Acad. Sci. U. S. A. 99:16267–16272.
Hoehn-Berlage, M., Norris, D. G., Kohno, K., Mies, G., Leibfritz, D., and Hossmann, K. -A. (1995). Evolution of regional changes in apparent diffusion coefficient during focal ischemia of rat brain: The relationship of quantitative diffusion NMR imaging to reduction in cerebral blood flow and metabolic disturbances. J. Cereb. Blood Flow Metab. 15:1002–1011.
Hossmann, K. -A. (1989). The pathophysiology of experimental brain edema. Neurosurg. Rev. 12:263–280.
Hossmann, K. -A. (1993). Disturbances of cerebral protein synthesis and ischemic cell death. Prog. Brain Res. 96:161–177.
Hossmann, K. -A. (1994a). Glutamate-mediated injury in focal cerebral ischemia: The excitotoxin hypothesis revised. Brain Pathol. 4:23–36.
Hossmann, K. -A. (1994b). Viability thresholds and the penumbra of focal ischemia. Ann. Neurol. 36:557–565.
Hossmann, K. -A. (1996). Periinfarct depolarizations. Cerebrovasc. Brain Metab. Rev. 8:195–208.
Hossmann, K. -A. (1997). Reperfusion of the brain after global ischemia—hemodynamic disturbances. Shock 8:95–101.
Hossmann, K. -A., and Hoehn-Berlage, M. (1995). Diffusion and perfusion MR imaging of cerebral ischemia. Cerebrovasc. Brain Metab. Rev. 7:187–217.
Hudetz, A. G. (1997). Blood flow in the cerebral capillary network—a review emphasizing observations with intravital microscopy [review]. Microcirculation-London 4:233–252.
Iadecola, C., Forster, C., Nogawa, S., Clark, H. B., and Ross, M. E. (1999). Cyclooxygenase-2 immunoreactivity in the human brain following cerebral ischemia. Acta Neuropathol. 98:9–14.
Iijima, T., Mies, G., and Hossmann, K. -A. (1992). Repeated negative DC deflections in rat cortex following middle cerebral artery occlusion are abolished by MK-801. Effect on volume of ischemic injury. J. Cereb. Blood Flow Metab. 12:727–733.
Imitola, J., Park, K. I., Teng, Y. D., Nisim, S., Lachyankar, M., Ourednik, J., Mueller, F. J., Yiou, R., Atala, A., Sidman, R. L., Tuszynski, M., Khoury, S. J., and Snyder, E. Y. (2004). Stem cells: Cross-talk and developmental programs. Philos. Trans.R. Soc. Lond.B Biol. Sci. 359:823–837.
Kiessling, M., and Gass, P. (1994). Stimulus-transcription coupling in focal cerebral ischemia. Brain Pathol. 4:77–83.
Kilic, E., Hermann, D. M., and Hossmann, K .-A. (2000). Recombinant tissue-plasminogen activator-induced thrombolysis after cerebral thromboembolism in mice. Acta Neuropathol. 99:219–222.
Kim, Y. H., Kim, E. Y., Gwag, B. J., Sohn, S., and Koh, J. Y. (1999). Zinc-induced cortical neuronal death with features of apoptosis and necrosis, mediation by free radicals. Neuroscience 89:175–182.
Kirino, T. (2002). Ischemic tolerance. J. Cereb. Blood Flow Metab. 22:1283–1296.
Krajewski, S., Krajewska, M., Ellerby, L. M., Welsh, K., Xie, Z. H., Deveraux, Q. L., Salvesen, G. S., Bredesen, D. E., Rosenthal, R. E., Fiskum, G., and Reed, J. C. (1999). Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. U. S. A. 96:5752–5757.
Kristian, T., Gido, G., Kuroda, S., Schutz, A., and Siesjö, B. K. (1998). Calcium metabolism of focal and penumbral tissues in rats subjected to transient middle cerebral artery occlusion. Exp. Brain Res. 120:503–509.
Kuroda, S., and Siesjö, B. K. (1997). Reperfusion damage following focal ischemia—pathophysiology and therapeutic windows [review]. Clin. Neurosci. 4:199–212.
Lansberg, M. G., Thijs, V. N., O’Brien, M. W., Ali, J. O., de Crespigny, A. J., Tong, D. C., Moseley, M. E., and Albers, G. W. (2001). Evolution of apparent diffusion coefficient, diffusion-weighted, and T2-weighted signal intensity of acute stroke. Am. J. Neuroradiol. 22:637–644.
Leniger-Follert, E., and Lübbers, D. W. (1977). Regulation of microflow and behaviour of local tissue PO2 during activation and anoxia of the brain cortex. Bibl. Anat. 15:345–349.
Liebeskind, D. S. (2003). Collateral circulation. Stroke 34:2279–2284.
Lipton, P. (1999). Ischemic cell death in brain neurons [review]. Physiol. Rev. 79:1431–1568.
Lipton, S. A., and Nicotera, P. (1998). Calcium, free radicals and excitotoxins in neuronal apoptosis [review]. Cell Calcium 23:165–171.
Lust, W. D., Taylor, C., Pundik, S., Selman, W. R., and Ratcheson, R. A. (2002). Ischemic cell death: Dynamics of delayed secondary energy failure during reperfusion following focal ischemia. Metab. Brain Dis. 17:113–121.
Markus, H., and Cullinane, M. (2001). Severely impaired cerebrovascular reactivity predicts stroke and TIA risk in patients with carotid artery stenosis and occlusion. Brain 124:457–467.
Marshall, R. S., Rundek, T., Sproule, D. M., Fitzsimmons, B. F. M., Schwartz, S., and Lazar, R. M. (2003). Monitoring of cerebral vasodilatory capacity with transcranial Doppler carbon dioxide inhalation in patients with severe carotid artery disease. Stroke 34:945–949.
Mattson, M. P., Culmsee, C., and Yu, Z. F. (2000). Apoptotic and antiapoptotic mechanisms in stroke [review]. Cell Tissue Res. 301:173–187.
Memezawa, H., Smith, M. -L., and Siesjö, B. K. (1992). Penumbral tissues salvaged by reperfusion following middle cerebral artery occlusion in rats. Stroke 23:552–559.
Mies, G., Auer, L. M., Ebhardt, G., Traupe, H., and Heiss, W. -D. (1983). Flow and neuronal density in tissue surrounding chronic infarction. Stroke 14:22–27.
Mies, G., Iijima, T., and Hossmann, K. -A. (1993). Correlation between periinfarct DC shifts and ischemic neuronal damage in rat. NeuroReport 4:709–711.
Mies, G., Ishimaru, S., Xie, Y., Seo, K., and Hossmann, K. -A. (1991). Ischemic thresholds of cerebral protein synthesis and energy state following middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 11:753–761.
Mies, G., Trapp, T., Kilic, E., Oláh, L., Hata, R., Hermann, D. M., and Hossmann, K. -A. (2001). Relationship between DNA fragmentation, energy state, and protein synthesis after transient focal cerebral ischemia in mice. In: Maturation Phenomenon in Cerebral Ischemia IV, Springer-Verlag, Berlin, Heidelberg, pp. 85–92.
Modo, M., Rezaie, P., Heuschling, P., Patel, S., Male, D. K., and Hodges, H. (2002). Transplantation of neural stem cells in a rat model of stroke: Assessment of short-term graft survival and acute host immunological response. Brain Res. 958:70–82.
Nagayama, M., Niwa, K., Nagayama, T., Ross, M. E., and Iadecola, C. (1999). The cyclooxygenase-2 inhibitor NS-398 ameliorates ischemic brain injury in wild-type mice but not in mice with deletion of the inducible nitric oxide synthase gene. J. Cereb. Blood Flow Metab. 19:1213–1219.
Nedergaard, M., and Astrup, J. (1986). Infarct rim: effect of hyperglycemia on direct current potential and (14C)- deoxyglucose phosphorylation. J. Cereb. Blood Flow Metab. 6:607–615.
Nicotera, P. (2003). Molecular switches deciding the death of injured neurons. Toxicol. Sci. 74:4–9.
Norris, D. G., Hoehn-Berlage, M., Dreher, W., Kohno, K., Busch, E., and Schmitz, B. (1998). Characterization of middle cerebral artery occlusion infarct development in the rat using fast nuclear magnetic resonance proton spectroscopic imaging and diffusion-weighted imaging. J. Cereb. Blood Flow Metab. 18:749–757.
Opitz, E., and Schneider, M. (1950). Über die Sauerstoffversorgung des Gehirns und den Mechanismus der Mangelwirkungen. Ergebn. Physiol. 46:126–260.
Paschen, W. (1996). Disturbances of calcium homeostasis within the endoplasmic reticulum may contribute to the development of ischemic- cell damage. Med. Hypotheses 47:283–288.
Paschen, W. (2001). Dependence of vital cell function on endoplasmic reticulum calcium levels: implications for the mechanisms underlying neuronal cell injury in different pathological states [review]. Cell Calcium 29:1–11.
Paschen, W. (2003). Shutdown of translation: Lethal or protective? Unfolded protein response versus apoptosis. J. Cereb. Blood Flow Metab. 23:773–779.
Paschen, W., Mies, G., and Hossmann, K. -A. (1992). Threshold relationship between cerebral blood flow, glucose utilization, and energy metabolites during development of stroke in gerbils. Exp. Neurol. 117:325–333.
Prass, K., and Dirnagl, U. (1998). Glutamate antagonists in therapy of stroke [review]. Restorative Neurol. Neurosci. 13:3–10.
Prass, K., Scharff, A., Ruscher, K., Lowl, D., Muselmann, C., Victorov, I., Kapinya, K., Dirnagl, U., and Meisel, A. (2003). Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 34:1981–1986.
Rabuffetti, M., Sciorati, C., Tarozzo, G., Clementi, E., Manfredi, A. A., and Beltramo, M. (2000). Inhibition of caspase-1-like activity by Ac-Tyr-Val-Ala-Asp-chloromethyl ketone induces long-lasting neuroprotection in cerebral ischemia through apoptosis reduction and decrease of proinflammatory cytokines. J. Neurosci. 20:4398–4404.
Roitberg, B. (2004). Transplantation for stroke. Neurol. Res. 26:256–264.
Rosand, J., and Schwamm, L. H. (2001). Management of brain edema complicating stroke [review]. J. Intensive Care Med. 16:128–141.
Rosenberg, G. A. (1999). Ischemic brain edema [review]. Prog. Cardiovasc. Dis. 42:209–216.
Rothwell, N. J., and Luheshi, G. N. (2000). Interleukin I in the brain: biology, pathology and therapeutic target [review]. Trends Neurosci. 23:618–625.
Samdani, A. F., Dawson, T. M., and Dawson, V. L. (1997). Nitric oxide synthase in models of focal ischemia [review]. Stroke 28:1283–1288.
Schlaug, G., Benfield, A., Baird, A. E., Siewert, B., Lovblad, K. O., Parker, R. A., Edelman, R. R., and Warach, S. (1999). The ischemic penumbra—operationally defined by diffusion and perfusion MRI. Neurology 53:1528–1537.
Schneeloch, E., Mies, G., Busch, H. J., Buschmann, L. R., and Hossmann, K. A. (2004). Granulocyte-macrophage colony-stimulating factor-induced arteriogenesis reduces energy failure in hemodynamic stroke. Proc. Natl. Acad. Sci. U. S. A. 101:12730–12735.
Schuier, F. J., and Hossmann, K. -A. (1980). Experimental brain infarcts in cats. II. Ischemic brain edema. Stroke 11:593–601.
Seegers, H., Grillon, E., Trioullier, Y., Vath, A., Verna, J. M., and Blum, D. (2000). Nuclear factor-kappa B activation in permanent intraluminal focal cerebral ischemia in the rat. Neurosci. Lett. 288:241–245.
Selman, W. R., VanderVeer, C., Whittingham, T. S., LaManna, J. C., and Lust, W. D. (1987). Visually defined zones of focal ischemia in the rat brain. Neurosurgery 21:825–830.
Shimada, N., Graf, R., Rosner, G., and Heiss, W.-D. (1993). Ischemia-induced accumulation of extracellular amino acids in cerebral cortex, white matter, and cerebrospinal fluid. J. Neurochem. 60:66–71.
Siesjö, B. K., Elmer, E., Janelidze, S., Keep, M., Kristian, T., Ouyang, Y. B., and Uchino, H. (1999). Role and mechanisms of secondary mitochondrial failure. Curr. Prog. Understanding Sec. Brain Damage Trauma Ischemia 73:7–13.
Siesjö, B. K., and Siesjö, P. (1996). Mechanisms of secondary brain injury [review]. Eur. J. Anaesthesiol. 13:247–268.
Siren, A. L., and Ehrenreich, H. (2001). Erythropoietin—a novel concept for neuroprotection. Eur. Arch. Psych. Clin. Neurosci. 251:179–184.
Soriano, S. G., Lipton, S. A., Wang, Y. M. F., Xiao, M., Springer, T. A., Gutierrezramos, J. C., and Hickey, P. R. (1996). Intercellular adhesion molecule-1-deficient mice are less susceptible to cerebral ischemia—Reperfusion injury. Ann. Neurol. 39:618–624.
Stagliano, N. E., Perez-Pinzon, M. A., Moskowitz, M. A., and Huang, P. L. (1999). Focal ischemic preconditioning induces rapid tolerance to middle cerebral artery occlusion in mice. J. Cereb. Blood Flow Metab. 19:757–761.
Stroemer, R. P., and Rothwell, N. J. (1997). Cortical protection by localized striatal injection of IL-1ra following cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 17:597–604.
Suzuki, T., Ooto, S., Akagi, T., Amemiya, K., Igarashi, R., Mizushima, Y., and Takahashi, M. (2003). Effects of prolonged delivery of brain-derived neurotrophic factor on the fate of neural stem cells transplanted into the developing rat retina. Biochem. Biophys. Res. Commun. 309:843–847.
Symon, L., Branston, N. M., Strong, A. J., and Hope, T. D. (1977). The concepts of thresholds of ischaemia in relation to brain structure and function. J. Clin. Pathol. 30(Suppl. 11):149–154.
Takahashi, K., Pieper, A. A., Croul, S. E., Zhang, J., Snyder, S. H., and Greenberg, J. H. (1999). Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Res. 829:46–54.
Tomita, M. (2005). Pathophysiology of brain edema. In: Kalimo, H. (Ed.), Cerebrovascular Diseases, ISN Neuropath, Basel, Switzerland, pp. 33–46.
Touzani, O., Boutin, H., Chuquet, J., and Rothwell, N. (1999). Potential mechanisms of interleukin-1 involvement in cerebral ischaemia. J. Neuroimmunol. 100:203–215.
Vogel, J., Hermes, A., and Kuschinsky, W. (1999). Evolution of microcirculatory disturbances after permanent middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 19:1322–1328.
Wahlgren, N. G., and Ahmed, N. (2004). Neuroprotection in cerebral ischaemia: Facts and fancies. The need for new approaches. Cerebrovasc. Dis. 17:153–166.
Walz, B., Zimmermann, C., Bottger, S., and Haberl, R. L. (2002). Prognosis of patients after hemicraniectomy in malignant middle cerebral artery infarction. J. Neurol. 249:1183–1190.
Wang, X. K., and Feuerstein, G. Z. (1995). Induced expression of adhesion molecules following focal brain ischemia. J. Neurotrauma 12:825–832.
Warach, S., Gaa, J., Siewert, B., Wielopolski, P., and Edelman, R. R. (1995). Acute human stroke studied by whole brain echo planar diffusion-weighted magnetic resonance imaging. Ann. Neurol. 37:231–241.
Willing, A. E., Lixian, J., Milliken, M., Poulos, S., Zigova, T., Song, S., Hart, C., Sanchez-Ramos, J., and Sanberg, P. R. (2003). Intravenous versus intrastriatal cord blood administration in a rodent model of stroke. J. Neurosci. Res. 73:296–307.
Yang, G. Y., Schielke, G. P., Gong, C., Mao, Y., Ge, H. L., Liu, X. H., and Betz, A. L. (1999). Expression of tumor necrosis factor-alpha and intercellular adhesion molecule-1 after focal cerebral ischemia in interleukin-1 beta converting enzyme deficient mice. J. Cereb. Blood Flow Metab. 19:1109–1117.
Zülch, K. -J. (1985). The Cerebral Infarct. Pathology, Pathogenesis, and Computed Tomography, Springer-Verlag, Berlin, Heidelberg, New York, Tokyo.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hossmann, KA. Pathophysiology and Therapy of Experimental Stroke. Cell Mol Neurobiol 26, 1055–1081 (2006). https://doi.org/10.1007/s10571-006-9008-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-006-9008-1