
Abstract
miR-146b-5p has been studied to be highly expressed in bronchopulmonary dysplasia (BPD), but whether it is involved in regulating the process of BPD in premature infants remains unclear. This study was to explore miR-146b-5p in premature BPD and reveal its molecular mechanism. BPD mouse model and high-oxygen MLE-12 cell model were established. HE staining, TUNEL staining, and IF staining were conducted to evaluate the pathological injury and protein expression in mouse lung tissue. LDH assay, MMT assay, and flow cytometry were achieved to evaluate cytotoxicity, cell viability, and apoptosis. ELISA and immunoblotting were performed to evaluate inflammatory cytokines and Wnt pathway proteins in lung tissues and cells. Dual-luciferase reporter assay and RIP assay were needed to examine the targeting relationship between miR-146b-5p and KDM6B. miR-146b-5p was abundantly expressed in BPD and KDM6B was lowly expressed. miR-146b-5p knockdown improved hyperoxia-induced lung epithelial cell inflammation and apoptosis in both models. miR-146b-6p upregulation or KDM6B downregulation aggravated hyperoxia-induced inflammation and apoptosis of lung epithelial cells. This effect of overexpressing miR-146b-5p was rescued by forcing KDM6B. MiR-146b-5p activated Wnt signaling by regulating KDM6B. miR-146b-5p activates the Wnt pathway through targeted regulation of KDM6B, thereby aggravating hyperoxia-induced inflammation and apoptosis of lung epithelial cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As a chronic respiratory disease, bronchopulmonary dysplasia (BPD) is common in preterm infants [1]. For preterm infants younger than 29 weeks, about 45% suffers from BPD [2]. It is worth noting that hyperoxia-induced acute lung injury is a pathogenic factor for BPD [3]. Reduced alveolation in premature infants results in less efficient gas exchange in the lungs. Although there are several treatment strategies for BPD, the morbidity and survival rate of BPD still face great challenges [4]. Therefore, a more comprehensive and in-depth understanding of BPD pathogenesis is of great significance to develop therapeutic strategies and targets.
miRNAs are endogenous non-coding RNAs that can participate in the post-transcriptional expression of eukaryotic genes [5]. Due to their high expression and stability in tissues and cells, miRNAs have become the focus of research on the pathogenesis of various diseases [6]. Studies have noticed the involvement of miRNA in BPD. For example, miR-574-3p protects preterm infants with BPD by regulating adrenomedullin [7]. In addition, miR34a is a novel small-molecule regulator involved in BPD [8]. Recently, a miRNA microarray analysis shows highly expressed miR-146b in BPD [9]. However, it is an uncertainty considering the involvement of miR-146b-5p in BPD.
This study hypothesized that miR-146b-5p participates in BPD development, based on which the biological function of miR-146b-5p in BPD was studied from mouse and cell models, thereby revealing its downstream molecular mechanism.
Materials and Methods
BPD Mouse Model
The animal experiment program was approved by the Animal Care and Use Committee of The First Affiliated Hospital of Shaoyang University. Thirty-two neonatal C57BL/6J mice were provided by Animal Laboratory Center of The First Affiliated Hospital of Shaoyang University. To establish the BPD model, 24 mice were kept in a hyperoxic environment with 85% oxygen one day after birth. Mice for control were exposed to 21% oxygen. To limit oxygen toxicity, mice for BPD modeling were fed alternately in hyperoxic and normal-oxic environments every 24 h. To knock down miR-146b-5p, mmu-miR-146b-5p antagomir (80 mg/kg) was injected through the caudal vein 1 week before BPD modeling, and antagomir NC was considered a negative control. After 14 days, euthanasia was performed on mice by intraperitoneal injection of pentobarbital sodium (50 mg/kg) and lung tissue was obtained (some were fixed with 4% paraformaldehyde and the rest were frozen at −80 ℃ for RNA or protein extraction).
HE Staining
The fixed lung tissue was embedded in paraffin and cut into continuous sections. After removing the wax, the sections were hydrated and stained with HE to evaluate the pathological changes under a light microscope (Nikon, Japan).
TUNEL Staining
Apoptotic cells in the lung tissue were measured using One‐Step TUNEL Apoptosis Assay kit (Roche). After dewaxing and hydration, the fixed paraffin sections were digested with protease K for 20 min and combined with dUTP solution and TdT enzymes at 37 °C for 1 h. Meanwhile, DNase I and dUTP were taken as positive control and negative control, respectively. Then, DBA-developed sections were stained with DAPI (Sigma-Aldrich; Merck KGaA), dehydrated with a gradient ethanol, aluminized with xylene, loaded with neutral balsam, and viewed with a microscope (Carl Zeiss, LSM700).
Immunofluorescence
The fixed lung tissue was sealed with 10% blocking buffer for 1 h after permeation with 0.25% Triton X-100 for 10 min and subsequently incubated overnight at 4 °C with primary antibodies: cleaved caspase-3 (ab2302, Abcam) and α-SMA (55135-1-AP, Proteintech). After PBS washing, the lung tissue was incubated at 37 °C for 2 h in a secondary antibody coupled with Alexa Fluor 594, and the nuclei were stained with DAPI. Finally, the lung tissue was observed under a laser confocal scanning microscope.
ELISA
IL-1β, IL-6, and TNF-α in lung tissue and cell culture medium supernatant were measured using ELISA kits (Gibco), as per the manufacturerʹs method.
Cell Culture
MLE-12 cells (ATCC, USA) were treated with 15% FBS (Gibco; Thermo Fisher Scientific) and 100-U/ml penicillin and streptomycin (Thermo Fisher Scientific) in Hites medium (Cytiva). The culture environment was conditioned to 37 °C and 5% CO2. MLE-1 cells were exposed to hyper oxygen (85% oxygen) for 6 h to establish a BPD cell model. MLE-1 cells exposed to normal oxygen were used as controls.
Construction of Overexpression Vector
The CDS region of KDM6B gene was cloned into pcDNA 3.1 plasmid (RiboBio) to upregulate KDM6B in cells. Target sequences with cho1 and BamH1 restriction sites were amplified by PCR (Takara, Japan). The target sequence was visualized on agarose gel and then purified with DNA gel extraction kit (Sinaclon, Iran). The carrier and insert were cut by restriction enzyme, respectively. The construct was transformed into a suitable bacterial host DH5αby heat shock conversion. Simply put, after a short incubation on ice, a mixture of chemically active bacteria and DNA was placed at 42 °C for 45 s and then placed back on the ice. SOC medium was added, and the transformed cells were incubated at 37 ℃ for 30 min. The constructs were extracted using the mini-prep plasmid extraction kit (Bioneer, Korea).
Cell Transfection
MiR-146b-5p mimic/inhibitor, si-KDM6B, and pcDNA 3.1-KDM6B (RiboBio) were transfected into MLE-1 cells with Lipofectamine 3000 (Invitrogen) as requested. The transfection efficiency was verified after 48 h.
LDH Detection
LDH release in cell culture medium was assessed using the Pierce LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific) to assess cytotoxicity.
MTT Experiment
MLE-12 cells inoculated into 96-well plates (1 × 104 cells/well) were incubated for 24 h, exposed to MTT (10 µl, 5 mg/ml) for 4 h, and incubated with 100 µl of DMSO in each well. After 15 min, a plate reader (BioTek China) was employed for reading optical density at 570 nm.
Flow Cytometry
Apoptosis in MLE-12 cells was assessed using Annexin V/FITC Apoptosis assay kit (Southern Biotech, USA). In short, MLE-12 cells were cleaned with PBS (Invitrogen) and re-suspended in a binding buffer. Later on, 5-μl Annexin V-FITC/PI (Bender Med System) was added for 15 min, and the staining was performed on the FACSan flow cytometer (BD Bioscience).
RT-qPCR
Total RNA from mouse lung tissue and MLE-12 cells was isolated using a Trizol kit (Thermo Fisher Scientific). To prepare cDNA, miRNA First-Strand cDNA Synthesis (Tailing Reaction) and MightyScript First-Strand cDNA Synthesis Master Mix (B639251, Sangon) were utilized, and RT-qPCR was performed using SYBR Green PCR kit (Takara). The primer sequence is shown in Table 1. RNA expression quantification was performed using 2−ΔΔCt method.
Immunoblotting
Total proteins were extracted from mouse lung tissue and MLE-12 cells using RIPA buffer (Beyotime Shanghai, China). After measurements of protein concentration based on BCA kit, 30-µg protein underwent 10% SDS-PAGE and was loaded onto PVFD membrane. After 5% skim milk powder treatment for 1 h, the membrane was incubated with primary antibody overnight and re-detected with secondary antibody labeled by HRP for 1 h. The immune complexes on membranes were developed using ECL fluorescence detection kit (BB-3501, Amersham, UK) and quantified in the Bio-Rad image analysis system with Quantity One v4.6.2 software. Primary antibodies: cleaved caspase-3 (ab2302, Abcam), KDM6B (9212, CST), GAPDH (2118, CST), α-SMA (55135-1-AP, Proteintech), p-p65 (3031, CST), AXIN2 (ab32197, Abcam), and Wnt5a (AF645, R&D Systems).
Luciferase Activity Analysis
Wild-type and mutant sequences of KDM6B (WT/MUT-KDM6B) were constructed using KDM6B 3ʹUTR containing the putative binding site of miR-146b-5p. These sequences (Sangon) were cloned into the pmirGLO Dual-Luciferase miRNA vector (Promega), which was co-transfected with miR-146b-5p mimic or mimic NC into MLE-12 cells using Lipofectamine 3000 (Thermo Fisher Scientific). After 48 h, the cell lysates were assessed in the Dual-Luciferase® Reporter Assay System (Promega).
RIP Experiment
MLE-12 cell lysate was detected with RIP buffers, in which magnetic beads labeled with human anti-AgO2 or mouse IgG were supplementary. The complex was harvested with protease K and the immunoprecipitated RNA was then isolated. RNA concentration measurements and RNA quality assessment were performed by NanoDrop spectrophotometers (Thermo Fisher Scientific). Purified RNA was collected and tested by RT-qPCR.
Data Analysis
All experiments were biologically replicated at least three times. GraphPad Prism 9.0 software was selected for data analysis. Data were shown as mean ± standard deviation. The student t test detected two group comparisons and one-way ANOVA detected multiple group comparisons. *P < 0.05 was considered statistically significant.
Results
Knocking Down miR-146b-5p Improves Lung Inflammation and Apoptosis in BPD Mice
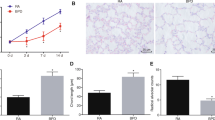
miR-146b-5p expression was enhanced in BPD mice, while miR-146b-5p antagomir injection decreased its expression (Fig. 1A). HE staining observed that the lung tissue of BPD mice had inflammation and abnormal lung morphology, which was effectively improved by silencing miR-146b-5p (Fig. 1B). The number of TUNEL-positive cells was increased in the lung tissue of BPD mice, and knockdown of miR-146b-5p lowered this number (Fig. 1C). Immunofluorescence staining noticed that BPD mice expressed higher cleaved caspase-3 and α-SMA than control mice, but downregulating miR-146b-5p decreased their expression levels (Fig. 1D). ELISA results presented that TNF-α, IL-1β, and IL-6 were increased in the lung tissue of BPD mice, while downregulating miR-146b-5p restored their levels (Fig. 1E). Immunoblotting measured that cleaved caspase-3, α-SMA, and p-p65 were higher in the lung tissue of BPD mice, but suppressing miR-146b-5p inhibited expression of these three proteins (Fig. 1F).

miR-146b-5p knockdown improves lung inflammation and apoptosis in BPD mice. An in vivo BPD mouse model was established, and miR-146b-5p antagomir was injected into BPD mice. A RT-qPCR measured miR-146b-5p in mouse lung tissue; B lung tissue of mice represented by HE staining; C lung tissue of mice represented by TUNEL staining; D cleaved caspase-3 and α-SMA expression detected by IF staining; E inflammatory cytokines in mouse lung tissue determined by ELISA; F protein expression of cleaved caspase-3, α-SMA, and p-p65 detected by immunoblotting. Data were expressed as mean ± SD (n = 8). *P < 0.05
Knocking Down miR-146b-5p Improves Hyperoxia-Induced Lung Epithelial Cell Inflammation and Apoptosis
A hyperoxic MLE-12 cell model was configured to simulate BPD in vitro and transfected with miR-146b-5p inhibitor. Hyperoxia increased miR-146b-5p levels, while miR-146b-5p inhibitor suppressed miR-146b-5p expression (Fig. 2A). LDH detection showed that hyperoxia promoted the release of LDH, but knocking down miR-146b-5p reduced LDH release (Fig. 2B). In addition, hyperoxia inhibited cell viability, but suppressing miR-146b-5p restored cell viability (Fig. 2C). Flow cytometry showed that hyperoxia resulted in increased apoptosis, while miR-146b-5p inhibition effectively reduced apoptosis rate (Fig. 2D). ELISA manifested that hyperoxia elevated the levels of inflammatory cytokines, but silencing miR-146b-5p reduced the levels of inflammatory cytokines (Fig. 2E). Immunoblotting presented that hyperoxia augmented the expression of cleaved caspase-3, α-SMA, and p-p65, but inhibiting miR-146b-5p significantly inhibited these three proteins (Fig. 2F).

miR-146b-5p knockdown reduces hyperoxia-induced inflammation and apoptosis of lung epithelial cells. miR-146b-5p inhibitor was transfected into hyperoxic MLE-12 cells. A miR-146b-5p levels detected by RT-qPCR; B LDH levels detected by commercial kits; C cell viability detected by MMT method; D cell apoptosis detected by flow cytometry; E inflammatory cytokines in cells determined by ELISA; F protein expression of cleaved caspase-3, α-SMA, and p-p65 detected by immunoblotting. Data were expressed as mean ± SD (N = 3). *P < 0.05
miR-146b-5p Targets KDM6B
Bioinformatics website starbase predicted the potential binding sites of miR-146b-5p and KDM6B (Fig. 3A). Subsequently, the expression pattern of KDM6B in BPD was detected by immunoblotting. KDM6B was underexpressed in both BPD mice and hyper-oxygenated lung epithelial cells, but knockdown of miR-146b-5p could increase KDM6B protein expression (Fig. 3B, C). Dual-luciferase assay showed that co-transfection of miR-146b-5p mimic and WT-KDM6B could reduce luciferase activity (Fig. 3D). RIP experiments confirmed that miR-146b-5p and KDM6B were highly enriched in Ago2 magnetic beads (Fig. 3E).

Targeted regulation of KDM6B by miR-146b-5p. A Bioinformatics sites predicted the potential binding sites of miR-146b-5p and KDM6B; B, C KDM6B expression in BPD mice and hyperoxic MLE-12 cells detected by immunoblotting; D, E the targeting relationship between miR-146b-5p and KDM6B assayed by dual-luciferase reporting experiment and RIP experiment. Data were expressed as mean ± SD (B, n = 8; C–E, N = 3). * P < 0.05
KDM6B Downregulation Aggravates Hyperoxia-Induced Lung Epithelial Cell Injury
si-KDM6B was transfected into hyperoxia-treated MLE-12 cells to investigate the biological function of KDM6B in BPD. si-KDM6B successfully reduced KDM6B levels (Fig. 4A). Furthermore, reducing KDM6B promoted the release of LDH from lung epithelial cells (Fig. 4B). MTT assay observed that reducing KDM6B inhibited cell viability (Fig. 4C), while flow cytometry indicated that reducing KDM6B promoted apoptosis (Fig. 4D). In addition, KDM6B knockdown increased levels of inflammatory cytokines and increased cleaved caspase-3, α-SMA, and p-p65 protein expression (Fig. 4E, F).

KDM6B knockdown aggravates hyperoxia-induced lung epithelial cell injury. si-KDM6B was transfected into hyperoxic MLE-12 cells. A KDM6B expression detected by immunoblotting; B LDH levels detected by commercial kits; C cell viability detected by MMT method; D cell apoptosis detected by flow cytometry; E inflammatory cytokines in cells determined by ELISA; F protein expression of cleaved caspase-3, α-SMA, and p-p65 detected by immunoblotting. Data were expressed as mean ± SD (N = 3). * P < 0.05
MiR-146b-5p Affects Hyperoxia-Induced Lung Epithelial Cell Injury Via KDM6B
miR-146b-5p mimic and pcDNA 3.1-KDM6B were co-transfected into high-oxygen-treated MLE-12 cells. miR-146b-5p mimic inhibited KDM6B protein expression (Fig. 5A), but this effect was reversed by pcDNA 3.1-KDM6B. Functional rescues tests demonstrated that miR-146b-5p mimic increased LDH levels, inhibited cell viability, promoted apoptosis, increased levels of cellular inflammatory cytokines, and promoted cleaved caspase-3, α-SMA, and p-p65, but these effects were rescued by pcDNA 3.1-KDM6B (Fig. 5B–F).

MiR-146b-5p affects hyperoxia-induced lung epithelial cell injury via KDM6B. miR-146b-5p mimic and pcDNA 3.1-KDM6B were co-transfected into hyperoxic MLE-12 cells. A KDM6B expression detected by immunoblotting; B LDH levels detected by commercial kits; C cell viability detected by MMT method; D cell apoptosis detected by flow cytometry; E inflammatory cytokines in cells determined by ELISA; F protein expression of cleaved caspase-3, α-SMA, and p-p65 detected by immunoblotting. Data were expressed as mean ± SD (N = 3). *P < 0.05
MiR-146b-5p Regulates Wnt Pathway by Targeting KDM6B
Wnt pathway is a key pathway for normal lung development and changes in its signaling are related to BPD progression [10]. Wnt pathway-related proteins Wnt5a and AXIN2 were examined. Hyperoxia elevated Wnt5a and AXIN2 protein expression, while inhibiting miR-146b-5p effectively reduced Wnt5a and AXIN2 protein expression, but silencing of KDM6B mitigated the effect of miR-146b-5p inhibitor (Fig. 6).

MiR-146b-5p regulates Wnt pathway by targeting KDM6B. A, B Wnt5a and AXIN2 in MLE-12 cells detected by immunoblotting. Data were expressed as mean ± SD (N = 3). *P < 0.05
Discussion
BPD is still a major threat to premature infants worldwide [11]. There has been convincing evidence that miRNAs are correlated with BPD development. In BPD models, the study confirmed that miR-146b-5p activated the Wnt pathway by targeting KDM6B, thereby promoting hyperoxia-induced inflammation and apoptosis of lung epithelial cells.
miRNAs have been observed to be involved in regulating BPD by targeting post-transcriptional expression of downstream mRNAs. These miRNAs include miR-29a-3p [12], miR-421 [13], and miR-214 [14]. The discovery of these genes has important implications for understanding BPD pathogenesis. A former report has discussed miR-146b high expression in BPD [9]. In this study, consistent results were obtained, indicating that miR-146b-5p was highly expressed in both lung tissue of BPD mice and hyperoxia-treated MLE-12 cells, and depleting miR-146b-5p effectively inhibited hyperoxia-induced lung epithelial cell inflammation and apoptosis, and decreased the expression of α-SMA, a marker of myoblast. miR-146b-5p expression is high in hepatic fibrosis tissues, which can upregulate TGF-β to promote fibrosis progression [15]. This study speculates that high levels of miR-146b-5p may exacerbate pulmonary fibrosis in BPD, which may lead to difficulties in gas exchange. Recently, a study identified that miR-146b-5p is up-regulated after LPS treatment and can increase the number of neutrophils, enhance myeloperoxidase activity, and promote the production of inflammatory cytokines [16]. Similar results were obtained in this research, suggesting that miR-146b-5p exacerbates hyperoxiagen-induced inflammatory damage in lung epithelial cells.
This study confirmed KDM6B as the downstream target gene of miR-146b-5p. It was further found that silencing KDM6B aggravated hyperoxia-induced apoptosis and inflammatory damage in lung epithelial cells under oxygen enrichment conditions. Notably, KDM6B can promote macrophage M2 polarization [17], indicating that KDM6B has positive anti-inflammatory significance. In this study, KDM6B reduced the release of inflammatory cytokines. This way may be involved in promoting macrophage M2 polarization, which will be beneficial to ameliorating hyperoxia-induced lung tissue inflammation.
At present, the clinical application of miR-146b-5p/KDM6B axis still faces great challenges. The results only studied miR-146b-5p/KDM6B axis in both BPD models. Whether KDM6B affects lung tissue damage in BPD mice remains to be determined. In future studies, it is necessary to further explore whether the clinical regulation of miR-146b-5p/KDM6B is significant.
Conclusion
This study confirms that the miR-146b-5p/KDM6B axis can regulate BPD through the Wnt pathway. These data provide new molecular targets for developing clinical drugs for BPD, as well as a new insight into the mechanism of BPD.
Data availability
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
References
Gilfillan, M., Bhandari, A., & Bhandari, V. (2021). Diagnosis and management of bronchopulmonary dysplasia. BMJ, 375, n1974.
Stoll, B. J., et al. (2015). Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993–2012. JAMA, 314(10), 1039–1051.
Hwang, J. S., & Rehan, V. K. (2018). Recent advances in bronchopulmonary dysplasia: Pathophysiology, prevention, and treatment. Lung, 196(2), 129–138.
Principi, N., Di Pietro, G. M., & Esposito, S. (2018). Bronchopulmonary dysplasia: Clinical aspects and preventive and therapeutic strategies. Journal of Translational Medicine, 16(1), 36.
Correia de Sousa, M., et al. (2019). Deciphering miRNAsʹ action through miRNA editing. International Journal of Molecular Sciences, 20(24), 6249
Fabian, M. R., & Sonenberg, N. (2012). The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nature Structural & Molecular Biology, 19(6), 586–593.
Gong, X., et al. (2020). Adrenomedullin regulated by miRNA-574-3p protects premature infants with bronchopulmonary dysplasia. Bioscience Reports, 40(5), BSR20191879
Das, P., Shah, D., & Bhandari, V. (2021). miR34a: A novel small molecule regulator with a big role in bronchopulmonary dysplasia. American Journal of Physiology. Lung Cellular and Molecular Physiology, 321(1), L228–L235.
Yang, Y., et al. (2012). Expression profile of microRNAs in fetal lung development of Sprague–Dawley rats. International Journal of Molecular Medicine, 29(3), 393–402.
Lecarpentier, Y., et al. (2019). Bronchopulmonary dysplasia: Crosstalk between PPARgamma, Wnt/beta-catenin and TGF-beta pathways; the potential therapeutic role of PPARgamma agonists. Frontiers in Pediatrics, 7, 176.
Tracy, M. K., & Berkelhamer, S. K. (2019). Bronchopulmonary dysplasia and pulmonary outcomes of prematurity. Pediatric Annals, 48(4), e148–e153.
Zhong, Q., et al. (2020). Long non-coding RNA TUG1 modulates expression of elastin to relieve bronchopulmonary dysplasia via sponging miR-29a-3p. Frontiers in Pediatrics, 8, 573099.
Tao, X., Fang, Y., & Huo, C. (2021). Long non-coding RNA Rian protects against experimental bronchopulmonary dysplasia by sponging miR-421. Experimental and Therapeutic Medicine, 22(1), 781.
Zhang, Z. Q., et al. (2021). MicroRNA-214 promotes alveolarization in neonatal rat models of bronchopulmonary dysplasia via the PlGF-dependent STAT3 pathway. Molecular Medicine, 27(1), 109.
Xie, J., et al. (2022). miR-146b-5p activation of hepatic stellate cells contributes to the progression of fibrosis by directly targeting HIPK1. Experimental and Therapeutic Medicine, 24(2), 537.
Gao, W., & Zhang, Y. (2021). Depression of lncRNA MINCR antagonizes LPS-evoked acute injury and inflammatory response via miR-146b-5p and the TRAF6-NFkB signaling. Molecular Medicine, 27(1), 124.
Salminen, A., et al. (2014). Histone demethylase Jumonji D3 (JMJD3/KDM6B) at the nexus of epigenetic regulation of inflammation and the aging process. Journal of Molecular Medicine (Berlin, Germany), 92(10), 1035–1043.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Long, Y., Luo, Y., Hu, L. et al. Targeting miR-146b-5p to Regulate KDM6B Expression Aggravates Bronchopulmonary Dysplasia. Mol Biotechnol 66, 2078–2086 (2024). https://doi.org/10.1007/s12033-023-00849-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-023-00849-1