
Abstract
Exosomes are a subgroup of extracellular vesicles generated by distinct cells. Tumor-derived extracellular vesicles convey immunological checkpoint molecules. TEXs as critical mediators in tumor development, metastasis, and immune escape have recently become the focus of scientific research. Exosomes are involved in the regulation of the immune system. Exosomes interact with target cells in the tumor microenvironment, changing their function based on the cargo they contain. Exosomal immune checkpoints might be exploited to track tumor immune evasion, treatment response, and patient prognosis while enhancing tumor cell proliferation and spread. This review focuses on tumor-derived exosomes, their immunosuppressive effects in mice models, and their role in cancer immunotherapy. Exosomes are being studied as possible cancer vaccines, with numerous uses in tumor immunotherapy. Exosomes can carry chemotherapeutics, siRNA, and monoclonal antibodies. Exosomes produced by macrophages might be used to treat cancer. These and other clinical consequences provide new doors for cancer treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exosomes are extracellular vehicles (EVs) with a size of 40–160 nm in diameter, which are found in body fluids and are released by normal or tumor cells. Exosomes consist of a lipid bilayer membrane enclosing a cargo of biomolecule substances. For the first time, exosomes were found as cellular waste products [1, 2]. However, exosomes are now considered bio-nano particles that contribute to intercellular communication and signaling pathways [3]. Tumor-derived exosomes (TEXs) have a critical role in tumor progression. TEXs increase cancer cells’ migration [4], are involved in tumor metastasis [5], can promote angiogenesis [6], and are also involved in tumor evasion [7]. In the tumor microenvironment, tumor evasion can result from several factors, including developing regulatory immune cells or differentiation of immune cells into exhausted phenotypes, secretion of immune inhibitory agents, and cell–cell interaction between immune checkpoints on immune cells and their ligands on tumors [8]. Many studies have shown that tumor-derived exosomes induce immunosuppression. The exosomes have ligands on their surfaces that can bind to homologous receptors on immune cells and transmit immune inhibitory signals to them [9]. Immune checkpoints (ICs) have attracted more attention in cancer immunotherapy among important immune inhibitory biomolecules. In addition to cell surface expression of ICs, secretion of exosomal immune checkpoints is determined by different sources of cells. It has been shown that some tumor-derived exosomes carry immune checkpoint molecules, including CD47, CTLA-4, TIM-3, ARG1, and PD-L1, which are known as exosomal immune checkpoints [10]. Numerous studies have shown that the expression of immune checkpoint molecules on exosomes has a critical role in mechanisms that mediate tumor immune evasion [11,12,13]. Therefore, exosomal immune checkpoints could be considered novel targets for cancer immunotherapy with immune checkpoint inhibitors.
Tumor-derived exosomes (TEX)
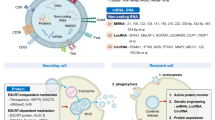
Exosomes are made from a lipid bilayer membrane that carries biomolecule cargo consisting of mRNAs, microRNAs, DNAs, proteins, lipids, and other agents, resembling the cytoplasmic content of the parent cells. After secretion of exosomes in the tumor microenvironment, they interact with target cells. Upon internalization by target cells, they release their cargo in the cytoplasm. Unlike their normal counterparts, tumor cells are intensive exosome producers, possibly because of the tumor microenvironment [14]. Following secretion of exosomes in the tumor microenvironment, they interact with target cells and depending on their containing cargos, the function of target cells will be changed. With different strategies, exosomes influence target cells. They can attach to the receptor of target cells and initiate downstream signaling cascades by ligand-receptor interaction or are taken up by target cells via fusion with the membrane of the target cell or through phagocytosis, endocytosis, or micropinocytosis and release the content of cargo into the cytoplasm of target cells [15]. In the recent decade, TEXs attracted more attention as imperative mediators in tumor progression, tumor metastasis, and tumor immune escape. Although TEXs comprise tumor-specific antigens, HSP70 and HSP90, involved in the activation of the immune response [16], they express immune inhibitory molecules leading to the inhibition of immune cells response and establishing an immunosuppressive tumor microenvironment [17]. TGF-β1 is an immunosuppressive cytokine secreted by immune cells and tumor cells. In patients with gastric cancer, expression of TGF-β1 by exosomes has been proposed to be associated with increased levels of FOXP3+ regulatory T cells in lymph nodes and increased metastatic rate [18].
Therefore, TGF-β1 levels in TEXs may be considered a predictive mediator in lymph node metastasis of gastric tumors. Being a member of the TNF family, TNF-related apoptosis-inducing ligand (TRAIL) induces cell death in a Fas-independent fashion. In colorectal carcinoma and melanoma, the TRAIL and Fas L expression by TEXs stimulates tumor-specific T cells' apoptosis. CD8+ T cells have been shown to be more susceptible to apoptosis by TEX with the membrane of FasL or PD-L1 due to the enrichment of CD95 or PD-1 on the membrane of CD8+ T cells, respectively [19, 20]. NKG2D ligand (NKG2D ligand) has been found in TEX in prostate cancer and promotes immune inhibition and tumor evasion. It has also been shown that NKG2D ligand positive TEX selectively downregulates NKG2D on NK cells and cytotoxic CD8+ T cells, leading to impaired cytotoxic activity in vitro that subsequently facilitates immune escape [21]. Investigation about TEXs and their interaction with immune cells underscore the crucial role of TEX in creating an immunosuppressive microenvironment, which can lead to the facilitation of tumor immunosurveillance, angiogenesis, tumor metastasis, and increased resistance to cancer immunotherapy.
Exosomes in immune regulation
T cells, B cells, and DCs have miRNA patterns that differ from their parent cells, according to research published in 2011 [22]. They showed that antigen-driven unidirectional transfer of miRNAs from T cells to antigen-presenting cells (APCs) on immunological synapse development is accomplished by CD63 + EVs. Moreover, mesenchymal stem cells (MSCs) significantly recruit immune cells and maintain an inflammatory milieu in the tumor microenvironment [23]. MSCs treated with EVs generated from gastric cancer (GC) cell line significantly increased the release of pro-inflammatory molecules, activated CD69 and CD25 on the T cell surface, and induced macrophage phagocytosis [24]. Following GC EV injection, the aberrant stimulation of the Nuclear Factor kappa B (NF-κB) signaling pathway in MSCs facilitated these immunomodulatory effects [25]. This was owing to the inhibited NF-κB signalling pathway, which significantly reduced the impact of MSCs triggered by GC EVs on T cells and macrophages. Toll-like Receptor 4 (TLR4) activates both innate and adaptive immune responses, and its enhanced activity in chronic infectious and inflammatory disorders contributes to cancer development [26]. When tumor cells eluded immune surveillance due to TLR4 stimulation, Domenis et al. studied the immunological modulatory features of immunosuppressive EVs released by tumor cells, hence encouraging tumor development [27]. They discovered that glioma-derived EVs decreased T-cell immunity by affecting monocyte development rather than directly interacting with T cells. In another glioma investigation, the extracellular matrix protein tenascin-C (TNC), which is correlated with EVs released by stem-like brain tumor-initiating cells (BTICs), was found to have a unique immunosuppressive effect in affecting local and distal T lymphocyte immunity [28]. They discovered that glioblastoma patients’ circulating EVs had higher TNC and T cell-suppressive activity levels than healthy people. They demonstrated that BTICs released EVs containing TNC and that TNC suppressed T cell proliferation by interacting with the integrins α5β1 and αvβ6 on T cells, thus lowering mTOR signaling using co-cultures. EVs generated from melanoma cells were shown to be enriched with coding and noncoding RNAs that did not match their source cells' abundance [29]. Melanoma-derived EVs generated dynamic alterations in the transcriptome of cytotoxic T cells after administration, resulting in altered mitochondrial respiration and upregulation of Notch signaling pathway genes. The same lab studied EVs generated from three melanoma-related cell lines [30]. Only those originating from the melanoma B16F0 cell line have both protein and mRNA for protein tyrosine phosphatase non-receptor سtype 11 (PTPN11), a T-cell proliferation inhibitor. Tung et al. reported EVs' transfer of miRNAs from Tregs to DCs for the first time [31]. Upon LPS stimulation, the high amounts of miR-150-5p and miR-142-3p in DCs caused by their association with Treg-derived EVs resulted in adopting a tolerant phenotype in these cells, with enhanced IL-10 and lowered IL-6 production.
Furthermore, Li et al. explored whether tumor-derived EVs alter the anti- and pro-tumoral balance of gamma-delta T cells in the tumor microenvironment at varied oxygen pressures [32]. They demonstrated that tumor-derived EVs might boost the growth and cytotoxicity of gamma delta T cells without needing DCs. Antigen-specific interactions between B and T cells are required for a successful immune response. MHC class II complexes on B cells must interact with the T cell receptor on antigen-specific T cells. Muntasell et al. investigated the processes governing the persistence, loss, and release of particular MHC-II complexes on activated B cells in 2007 [33]. They discovered that active B cells destroyed around 50% of MHC-II molecules daily and that EVs secreted about 12% of these molecules, which were previously expressed on the plasma membrane of B cells. MHC-II is able to avoid intracellular degradation thanks to this regulated EV release from activated B cells, and EVs can directly stimulate prepared CD4 + T cells but not naive CD4 + T cells.
Exosomal immune checkpoints
A study on the structure of exosomes has discovered the expression of several immune checkpoints by TEXs, such as Programmed cell death protein 1 (PD-1), Programmed cell death ligand-1 (PD-L1), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), and Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4). These exosomal immune checkpoints are involved in tumor immune escape [10]. Various investigations have shown that exosomal immune checkpoints induce tumor evasion and increase cancer cell proliferation and metastasis. We have reviewed each of the exosomal immune checkpoints in cancer progression in this part (Table 1). PD-L1, which is also known as (CD274), is an immune checkpoint found on immune cells’ surfaces, including Dendritic cells (DCs), macrophages, and B and T cells [34]. PD-L1 is also expressed on tumor cells and acts as a pro-tumorigenic ligand in tumor cells through ligation to PD-1 on immune cells and induction of proliferative and survival signaling pathways [35]. In addition to tumor cells’ surface, tumor cells also release PD-L1 into the extracellular tumor microenvironment and circulation. Extracellular PD-L1 includes the exosomal forms of PD-L1 and a soluble type [36, 37]. Although exosomal PD‐L1 shares a similar structure to the membranous form, exosomal PD-L1 is more stable against enzymatic degradation than soluble and membranous forms [38]. Furthermore, it seems that levels of exosomal PD‐L1 are relatively consistent with the levels of membranous PD-L1 expressed by parental cancer cells [39]. The immunoinhibitory function of exosomal PD-L1 in the progression and metastasis of tumors has attracted the attention of scholars in recent years. It has been shown that tumor-derived exosomal PD-L1 has inhibitory effects on the function of activated T cells and develops tumor growth via ligation with cell surface PD-1 [40]. Gang Chen et al. demonstrated that exosomal PD-L1 inhibited cytotoxic T cell proliferation and granzyme B production and facilitated melanoma progression in vitro and in a mouse model [41]. In Yang et al.’s research, the injection of exosomal PD-L1 into the breast cancer mouse model developed tumor growth compared to PD-L1 knockdown groups. The result indicated the systemic immunosuppressive impact of exosomal PD-L1. Consistently, PD-L1 expression was also increased in aggressive breast tumors [42]. Moreover, the increased level of exosomal PD-L1 in gastric cancer patients is associated with elevated IL-10 and TGF-β levels and poor prognosis. Exosomal PD-L1 was also negatively associated with granzyme B production [37]. Additional support on the significant role of exosomal PD-L1 in the inhibition of the anti-tumor response of activated T cells was provided by a study on Non-Small Cell Lung Cancer (NSCLC) patients. Exosomal PD-L1 isolated from lung tumor cells demonstrated high PD-L1 expression, inhibiting the anti-tumor response of Jurkat cells in production of IFN-γ. That inhibitory effect was reversed by monoclonal PD-L1 blocking antibody or PD-L1 expression knockdown and in turn, the injection of exosomal PD-L1 increased tumor development [43]. These results will provide support for the concept of exosomal PD-L1 as a biomarker of tumor immune evasion. Exosomal PD-L1 in comparison with cell surface PD-L1 appears to be a more appropriate biomarker to predict the tumor response to immunotherapy and patients’ outcome. M. Cordonnier et al. in their study detected exosomal PD-L1 in plasma samples of all melanoma patients, while only 67% of tumor biopsy samples were PD-L1 positive [44]. The exosomal PD-L1 is considered a prognostic biomarker for anti-PD1 therapy response. In patients with different cancer types, the efficacy of anti-PD1 therapy was evaluated prior and after treatment. Compared to the responders, exosomal PD-L1 of the non-responders was higher than that of the responders prior to the treatment. Simultaneously, exosomal PD-L1 and cell surface PD-L1 reduced when anti-PD-1 therapy was effective [45]. A controversial study about exosomal immune checkpoints refers to investigation of Y. Qiu et al., which indicated that activated T cells release exosomal PD-1. The exosomal PD-1 remotely interacts with either exosomal PD-L1 or cell surface PD-L1, which induces internalization of PD-L1 by clathrin-mediated endocytosis, and thus inhibits subsequent cellular PD-1: PD-L1 ligation, which in turn entails restoration of tumor surveillance via attenuating PD-L1-induced suppression of tumor-specific cytotoxic T cell activity. As a result, the anti-PD-L1 activity of exosomal PD-1 sets in motion a positive function for ameliorating cytotoxic T cell activity and a potential therapeutic strategy to modify the exosome surface with membrane-bound inhibitory immune checkpoints receptors to reduce the suppressive tumor immune microenvironment [46]. This is while the other studies have shown a supportive role of exosomal immune checkpoints in tumor growth. As such, more studies are warranted to further discover the anti-tumor function of exosomal PD-1.
Interaction of exosomal immune checkpoints on immune cells
CD8 + and CD4 + T cells
It has been shown that tumor-derived exosomes can modulate the function of immune cells [50]. Research on exosomal immune checkpoints has revealed that tumor cells release exosomal PD-L1 within the tumor microenvironment, which binds to PD-1 on T cells, leading to exhaustion of T cells. In return, the inhibition of exosomal PD-L1 can induce anti-tumor immunity with elevated levels of cells containing the activation marker Granzyme B [47]. To enable tumor immune evasion, tumor cells with the secretion of exosomal immune checkpoints can suppress cytotoxic activity and modulate the expression of immune-related genes in T cells. Estrogen receptor-binding fragment-associated antigen 9 (EBAG9) has been a tumor-promoting factor affecting the tumor-infiltrating immune cells. Miyazaki et al. have demonstrated that the EBAG9 molecule in tumor-derived exosomes suppressed cytotoxic activity and modulated immune-related gene expression, including IFNG, CXCR3, and granzyme B in T cells. Furthermore, induction of epithelial-mesenchymal transition (EMT)-related genes such as VIM, SNAI1, and SNAI2 imply that EBAG9 may promote tumor growth and metastasis via the EMT pathway [51]. Tim-3+ exosome is another exosomal immune checkpoint involved in aggressive tumor progression. Tim-3 expresses by Th1 cells, which plays a negative regulatory role in tumor immunity and correlates with T cell exhaustion. There are increased expression levels of Tim-3 on both cancer cells and tumor-infiltrating lymphocytes (TILs) are considered a predictive factor for poor prognosis [52]. TIM-3+ exosomes have inhibitory effects on the anti-tumor response of CD4 + T cells and induce macrophage M2 polarization via upregulation of CD206, CD163, and Arginase-1 to promote proliferation and metastasis of melanoma cells [48]. Wang et al. demonstrated that in tumor-derived exosomes of Spleen Deficiency-Hepatocellular carcinoma (SD-HCC) mouse model, PD-1, CTLA-4, PTEN, and AKT (also known as protein kinase B or PKB) were upregulated in comparison with HCC. Moreover, tumor progression and metastasis also increased in SD-HCC mice. Further experiments showed that CTLA-4+ exosomes increased tumor progression by modulating PTEN/CD44 signaling pathway to promote tumor growth, self-renewal, and invasion of liver cancer cells [53]. In addition, CD73+ exosomes lead to immune inhibition by eliminating T cell function. These tumor-derived exosomes dephosphorylate exogenous ATP and cAMP to form adenosine and cause T cell inhibition via the adenosine A2A receptor. Consequently, TEX modulates the anti-tumor activity of T cells with elevated levels of extracellular adenosine production [54]. Inhibition of CD4+ and CD8+ T cells derived from peripheral blood mononuclear cells by TEX was displayed at the early steps of T cells activation (activation of NFκB and NFAT) until late endpoints (secretion of IL-2, IFN-γ, and proliferation) [50, 55, 56]. Chatterjee et al., in their study, showed that TGF-β induces PD-L1 upregulation in TEXs and mediates cytotoxic CD8+ T cells’ dysfunction. They found that PD-L1 + exosomes isolated from TNBC cell lines could induce exhaustion-related genes, including PD-1 and CTLA-4, in co-cultured CD8 + T cells. In contrast, siPD-L1 exosomes (exosomes isolated from PD-L1-knocked out cancer cells) failed to promote exhaustion. Additionally, upregulation of PD-1 expression by CD8+ T cells while cultured with PD-L1+ exosomes was reduced following siPD-L1 exosomes treatment. Moreover, regulation of TCR signaling in CD8+ T cells showed enhanced dephosphorylation of SRC family molecules, PLC-γ and LAT by PD-L1 + exosomes. In contrast, TCR signaling adaptors were phosphorylated in the presence of siPD-L1 exosomes, indicating how TEX increased CD8+ T cell dysfunction [49]. According to numerous studies, exosome-associated miRNAs have been implicated in the modulation of signaling pathways in target cells in tumor microenvironments [57, 58]. Ye et al. have found that by inhibiting FGF11 and modulating the phosphorylation of the ERK and STAT proteins, exosomal miR-24-3p plays a crucial role in TEX-mediated T-cell dysfunction and indicates a new strategy of tumor immune evasion. Exosomal miR-24-3p and its target gene FGF11 may also be used as predictive indicators in nasopharyngeal carcinoma [59]. Arginase-1 (ARG-1+) tumor-derived exosomes emerge as a key regulator in cancer cell proliferation and tumor immune evasion amongst many other immune-suppressive molecules. In a mouse model of ovarian carcinomas, ARG-1+ TEXs are delivered to draining lymph nodes, where they are picked up by dendritic cells and suppress the proliferation of antigen-specific T lymphocytes. ARG1+ TEXs isolated from ovarian carcinomas patients as well as ovarian carcinoma cell lines affect the activities of T cells by inhibiting their proliferation and decreasing CD3ε and CD3ζ chain expression levels. The TCR complex’s subunits serve as important signaling molecules in T cells, so their normal expression or phosphorylation is needed for T cell activation, proliferation, and cytokine secretion [60]. In another study, Azambuja et al. have reported high levels of FasL, TRAIL, CTLA-4, CD39, and CD73 expression, with low expression of immunostimulatory molecules by glioblastoma-derived exosomes (GBex). The results indicated that in the presence of GBex, TNF-α and INF-γ secretion were inhibited, and apoptosis was triggered in CD8+ T cells.
Regulatory T cells (Tregs)
In another study, exosomes derived from a human chronic myelogenous leukemia cell line (K562 cells) altered cord blood-derived T cells properties toward differentiation regulatory phenotype with upregulation of FOXP3 gene levels and IL-10 secretion [61]. TEXs not only promote regulatory T cell differentiation but also support the function of regulatory T cells. Tumor-derived exosomes have been found to prevent CD4+ and CD8+ T cells activation upon IL-2 secretion. In the presence of tumor-derived exosomes, regulatory T cells respond to IL-2 in a TGF-β-dependent manner [50, 62]. According to this research, tumor and Treg-derived exosomes containing CTLA-4 on their surface may interfere with ipilimumab treatment. These findings imply that exosome-carried chemicals might represent a unique mechanism for patients to develop treatment resistance, as well as potential biomarkers for cancer diagnosis [63].
Natural Killer (NK) cells
Exosomes produced by NK cells have also been found to have therapeutic properties. For example, the contents of TNF-α, perforin, and FasL found in NK cell-derived exosomes suppressed melanoma growth in both in vitro and in vivo tests [64]. In addition, exosomes produced from NK cells pretreated with NB cells boosted the expression of natural killer cell receptors. Furthermore, they improved the cytotoxicity of NK cells against NB tumors in neuroblastoma (NB) tumors [65]. Furthermore, exosomes generated from Glioblastoma have been found to include a number of immunosuppressive proteins, including CTLA-4, which decrease the immunological function of CD8 + T cells, CD4 + T cells, natural killer (NK) cells, and macrophages [66].
B cells
Exosomes from patients with esophageal squamous cell carcinoma (ESCC) and ESCC cell lines decreased B cell proliferation and increased interleukin-10 + and PD1high Bregs. Meanwhile, research shows that ESCC-derived exosomes might increase PD-1 expression and IL-10 secretion in recipient B cells, which could be linked to the activation of TLR4 and MAPK signaling pathways [67]. Exosomes produced from mesenchymal stem cells have powerful regulatory effects on immunological responses involving various immune cells, including T cells and B cells [68].
Innate immune cells
Similarly, GBex inhibited the function of NK cells and CD4+ T cells. In macrophages, GBex stimulated the NF-κB pathway, enhancing their development into the M2 phenotype. Moreover, splenic CD8+ T cells, NK cells, and M1 macrophages were decreased in the mice injected with GBex, in contrast to naive and M2 macrophages developed [66] (Fig. 1). ARG1 + EVs can be phagocytosed by DCs or directly decrease T cell proliferation and antigen-specific T cell proliferation to promote tumor growth. [69].

Exosome-related immune checkpoints’ interaction with immune cells
Estrogen receptor-binding fragment-associated antigen 9 (EBAG9) as a tumor-promoting factor modulates immune-related gene expression, including IFNG, CXCR3, and granzyme B in T cells, as well as suppressing T-cells cytotoxic activity. Arginase-1+ (ARG-1+) TEXs are transported to draining lymph nodes, wherein dendritic cells collect them up and inhibit antigen-specific T lymphocyte proliferation. They also suppress T-cell proliferation and decrease CD3ε and CD3ζ chain expression levels. Tumor-derived CD73+ exosomes dephosphorylate exogenous ATP and cAMP to form adenosine and cause T-cell inhibition via the adenosine A2A receptor. Thus, TEXs modulate the anti-tumor activity of T-cells with elevated levels of extracellular adenosine production. Tumor-derived Tim-3 containing exosomes cause CD-4+ T-cells dysfunction and induce macrophage M2 polarization via upregulation of CD206, CD163, and Arginase-1 to promote proliferation and metastasis of melanoma cells. Tumor cells-derived exosomal PD-L1 binds to PD-1 and brings about CD8+ T-cells exhaustion, and the dysfunction thus facilitates tumor immune evasion. Glioblastoma-derived exosomes (GBex) with high levels of FasL, TRAIL, CTLA-4, CD39, and CD73 expression, inhibit the secretion of TNF-α and INF-γ and trigger apoptosis of CD8+ T-cells, besides decreasing NK-cells and enhancing macrophages development into M2 phenotype.
Exosomal immune checkpoints in mice models
In mouse models, tumor exosomal immune checkpoints are crucial in mediating local and systemic immunosuppression [39]. It has been shown that exosomes containing PD-L1 obtained from supernatants of murine or human head and neck squamous cell carcinomas cell lines can inhibit the infiltration of CD4 + and CD8 + T cells into the tumor environment and consequently boost tumor growth in the 4-nitroquinoline 1-oxide -induced malignant oral/esophageal injury model [70]. It has also been found that tumor-derived exosomes derived from the Lewis lung cancer model and the 4 T1 breast cancer model increase the expression of PD-L1, inhibiting the differentiation of myeloid precursors into DCs and their maturation [71]. The immunosuppressive function of exosomal PD-L1 was also seen in a syngeneic mouse model. In vitro, PDL1-containing exosomes obtained from B16-F10 tumor cells prevented the proliferation and cytotoxicity of CD8 + T cells. Intravenous delivery of exosomes obtained from the tumor cell line to mice supported tumor progression and reduced the infiltration of B16-F10 CD8 + T cells; however, anti-PD-L1 antibody therapy prevented this impact [41]. Further attempts using a mouse model of prostate cancer indicated that tumor-released exosomal PD-L1 can move to the tumor’s draining lymph nodes, where T cell activation is prevented, eventually resulting in T cell exhaustion and size reduction of the spleen [47]. Another investigation used Rab27a and nSMNase2, two important exosomal biogenesis genes, to create exosome-null B16 cells. The majority of exosomes were eliminated when both of these genes were deleted, as evidenced by CD63 marker expression and electron microscopy. Exosome deletion prevented severe lung metastases and prolonged the longevity of melanoma cell-bearing mice. In vivo, however, additional purified exosome therapy reversed the phenotype, significantly demonstrating that exosomes play a role in supporting metastasis. Furthermore, exosomes’ function is not restricted to the B16 melanoma model. In the Lewis lung carcinoma tumor model, removing exosomes with GW4869, a specific inhibitor of nSMNase2 significantly reduced metastatic growth and extended mouse survival. So, small molecule inhibitors of important enzymes like Rab27a and nSMNase2, as well as molecules found on the exosome surface like PDL1 have the potential to operate alone or in combination with current cancer treatment options. It is also revealed that exosomes secreted by melanoma cells perform an immunosuppressive activity through endogenous PD‐L1 to stimulate tumor‐specific CD8 + T cell exhaustion, leading to the elevation of metastatic growth [72]. Moreover, PD-L1 was detected in exosomes obtained from the culture media of PD-L1-secreting human breast cancer cells and 4T1 mouse mammary tumor cells. In the mouse 4T1 breast tumor model, it was found that by using Rab27a knockdown and consequently preventing the secretion of exosome in tumor cells, the effectiveness of anti-PD-1 treatment was noticeably enhanced, and the tumor development was blocked [42]. The effect of Orexin A on cellular and exosomal PD-L1 expression was studied in vitro using a mouse CT-26 model and human HCT-116 colon cancer cells. The growth rate of orthotopically transplanted colon cancer was slower in the Orexin A-treated group, which also had lower PD-L1 expression and higher immune infiltration. Orexin A has the ability to inhibit both cellular and exosomal PD-L1 expression. This study revealed that by inhibiting the JAK2/STAT3 signaling pathway, Orexin A was able to inhibit the expression of exosomal PD-L1 in colon cancer cells and promote T cell activity [73]. It is also reported that exosomes carry PD-L1 from the Bone marrow-derived cells (BMDCs) of tumor-bearing mice (BMDC-EXOsTu), not exosomes from BMDCs from healthy mice. Without the dependency on PD-L1, BMDC-EXOsTu can prevent CD8 + T cell proliferation in vitro and can support tumor development by inhibiting anti-tumor CD8 + T cell reactions. Without dependency on PD-L1, endogenous BMDC-EXOsTu can move into tumor tissues and suppress CD8 + T cell proliferation. Also, BMDC-EXOsTu is able to augment tumor metastasis through reducing the anti-tumor CD8 + T cell responses at metastatic locations via PD-L1 [74]. To confirm the anti-tumor activity of exosomal PD-1 in vivo, mouse exosomal PD-1 obtained from mouse T cells were administrated to PY8119 tumor-bearing mice. Mouse exosomal PD-1 notably reduced tumor prgression and improved survival duration in tumor-bearing mice [46]. In summary, such experimental studies support the notion that by reprinting cell-surface immune checkpoints, exosomal immune checkpoints can inhibit T cell immunity and promote tumor growth in various tumor types (Table 2).
Exosome-based cancer immunotherapy
Using exosomes as carriers to trigger anti-cancer immune responses and as carriers for anti-cancer drugs could be a promising approach to cancer treatment. Various immunotherapy ways have been used against tumors; one of the promising ways against tumors is a cancer vaccine that triggers the immune activities against tumor cells [75] (Table 3). DC-derived exosomes are used in vaccines manufactured via exosome platforms and may respond better against tumors by activating the CD8 + . When DCs catch antigens, they might pack immunostimulatory factors into exosomes along with the MHC antigen peptide complex. After that, these exosomes activate cytotoxic T-cell lymphocytes in lymph nodes which take action against tumors [76]. Some studies have been conducted on the platform of DC-derived exosomes.
A phase I/II clinical trial in patients with advanced stage of squamous cell carcinoma of esophagus showed that DC-derived exosome vaccines for esophageal squamous cell carcinoma are one of the possible treatment options [81]. Another viable option in vaccination against tumors is the macrophage-derived exosome. A study showed that M1-polarized macrophages exosomes persuade antigen-specific CTLs response using the improved activity of lipid calcium phosphate nanoparticles in melanoma cancer cells [82]. Tumor-released exosomes can also take part in vaccination platforms, but their effect may not be strong enough compared to other platforms [83]. Exosomes have gained increasing attention from researchers in recent years, along with potential cancer vaccines with broad potential in tumor immunotherapy.
Nevertheless, more studies in more extensive clinical trials are warranted to learn about their safety and effectiveness against tumors. Exosomes as nanoparticles have a better character for drug delivery in cancer immunotherapy. The nanoscale size and cargo delivery ability of exosomes make them highly suitable for penetrating tissue barriers [84]. Different types of cells can be potentially utilized. Still, recent studies showed that compared to other types of cells, mesenchymal stem cells can secrete more exosomes and can have a better effect on stimulating the immune system [85]. Accordingly, the outcome of a study using Mesenchymal stem cells-exosomes was remission of colon cancer progression via doxorubicin [86]. The primary technique in drug delivery is to perform structural engineering on the surface of exosomes [87]. In addition, engineering exosomes can be used as a carrier of combination therapy like chemo drugs, siRNA, and monoclonal antibodies [88]. Like-wise the possible option in drug delivery against tumors is macrophage-derived exosome. A study revealed that reversing the M2 macrophages tumor immunosuppression mechanism also induces anti-tumor immunity, cytotoxic T lymphocyte recruitment, and Treg downregulation, in addition to achieving significant therapeutic effects in mice with pancreatic cancer treatment [89]. Similar to the previous study’s engineered exosomes, combining GM‑CSF and IL‑12 alongside tumor-derived exosomes in renal cell carcinoma better affects CD8 + and their cytotoxic effect in the tumor microenvironment [90]. These clinical implications, alongside others, open new doors for treating cancers such as lung, prostate, renal cell, gastric, breast, gallbladder, pancreas, and rectal cancers. These engineered exosomes improve immune responses by enhancing the DC’s antigen-presenting function and stimulating the cytokine releases in tumors [91]. IFN- may cause an increase in the expression of PD-1 ligands on DEXs, a well-known immunological checkpoint that suppresses T-cell function, according to scientists. DEXs were found to successfully stimulate NK cells in an MHC-independent way, even though these vaccines were meant to activate specific MHC-restricted T-cell responses. DEX-based vaccines, however, have focused on direct CTL activation as a separate mechanism in other immune cells. Näslund et al., on the other hand, found that CD4 + T cells and B cells are required for DEX activation of the CTL anti-tumor response [92]. We summarise the modified exosomes as an immunotherapy usage in the table below.
In-silico analysis to identify involved miRNAs in regulating immune checkpoints
miRNAs have complicated roles in regulating the immune system. Hence, we used miRWalk v2 [93] to find miRNAs that could target immune checkpoints with a high affinity. Besides, these targetings were approved by miRTarBase, which finds experimentally verified miRNAs. We showed that six co-inhibitory immune checkpoints (BTLA, CD276, CYBB, HAVCR2, PDCD1, VSIR) could be targeted by 70 potential miRNAs (Fig. 2). The results demonstrated that the most interactive miRNAs are miR-6756-5p, miR-4447, miR-28-5p, miR-4472, miR-4651, and miR-6887-5p.

Interaction network of immune checkpoints and miRNAs. These predicted miRNAs could target six immune checkpoints
Validated roles of predicted miRNAs in exosomal form
MSCs treated with a miR-28-5p mimic showed increased proliferation, migration, and immunomodulation. This mimic stimulated the paracrine production of VEGF, HGF, LL-37, and Ang-1 by the body. Furthermore, the PI3K/Akt signaling pathway was shown to be greatly enhanced by the miR-28-5p mimic, demonstrating that it is activated after acute lung damage [94]. Hou et al. demonstrated that the amount of let-7a in exosomes obtained from BM-MSCs that had been transduced with Let-7a rose after being treated with exosomes. The expression of HMGA2 was downregulated in B16f10 cells, and the cell survival rate of BM-MSCs was lowered. However, neither the cell survival rate of B16f10 cells nor the amount of IGF-1 released by B16f10 cells differed significantly across the four groups. Finally, Let-7a contained in exosomes has been shown to limit the migration of Melanoma cells as well as the proliferation of BM-MSCs [95]. It was discovered that the target genes of exosomal miR-107 were predominantly associated with the PI3K-Akt signaling network, as well as the Hippo and AMPK signaling pathways. Notably, miR-107 decreased the expression of the elevated 14–3-3η (YWHAH) gene in DLBCL, suggesting that miR-107 may act as a tumor suppressor by targeting the 14–3-3η gene [96]. In addition, atrial fibrillation-derived exosomes have been shown to transfer miR-107 to human umbilical vein endothelial cells, and exosomal miR-107 has been shown to influence cell survival, migration, and apoptosis; in addition, to cell cycle progression, through the miR-107/USP14 pathway [97]. Earlier research by Ni et al. found that decreased levels of exosomal miR-30b were related to recurrence in patients with breast cancer [98]. It has also been reported that miR-204-5p was highly expressed in the PF-Exo model of pulmonary fibrosis and that PF-Exo injection accelerated the progression of PF and increased the proliferation ability of lung fibroblasts both in vivo and in vitro. Exosomal miR-204-5p derived from bronchoalveolar lavage fluid inhibits autophagy, thereby accelerating the progression of PF rats [99]. This is accomplished by targeting AP1S2. By precisely inhibiting the target genes of miR-204-5p in human cancer cells, Yao et al. demonstrated that exosomal miR-204-5p could successfully reduce cancer cell growth, induce apoptosis, and enhance chemosensitivity [100].
The prospect of exosome immunotherapy
In this fast cancer immunotherapy development period, there is much excitement in using cell-released microscopic vesicles to boost the immune system. As cell-derived nanovesicles with immunogenicity and molecular transfer functions, exosomes hold considerable potential in cancer immunotherapy. Exosome cargoes have just lately been found thanks to technical advancements, and they play a role in immune response modulation. Exosomes generated from tumor cells and immune cells, in particular, have distinct composition profiles that play a direct role in anti-cancer immunotherapy. In fact, many of the exosomes secreted by tumor cells appear to have a dual role. These exosomes can not only stimulate the immune system, but they can also inhibit the function of immune cells, promote immune suppression, and establish an immune microenvironment that is favorable to the growth of the tumor. In addition, exosomes may convey their payloads to specific cells, influencing their phenotypic and immune-regulatory roles. According to research accumulated over the previous decade, exosomes can participate in many cellular processes that contribute to cancer formation and therapeutic actions, demonstrating their dual properties of promoting and inhibiting cancer. Exosomes have much potential in the realm of cancer immunotherapy, and they might be the most effective cancer vaccines and tailored antigen/drug carriers. Many researchers have developed cancer immunotherapy approaches based on exosomes, including cancer vaccines and drug delivery vehicles. These strategies use the characteristics of exosomes that allow for flexible and rich information transmission. Exosomes derived from dendritic cells (DCs), macrophages, tumor cells, and even T cells have been investigated for their potential use in cancer immunotherapy, and the results of these studies have been very encouraging. Exosomes have implications for diagnostics and the creation of innovative treatment techniques. Thus, it is crucial to understand how they might be used in immune therapy to regulate cancer growth.
Conclusion
To sum up, the evidence from in vitro and in vivo animal studies supports the impacts of TEXs and their biochemical cargoes, such as immune checkpoint molecules on various immune cells, as well as their role in establishing an immunosuppressive microenvironment for tumor growth, invasion, metastasis, angiogenesis, apoptosis, and inducing immunotherapy resistance. Better awareness and knowledge of TEXs’ molecular composition and the complex interference among TEXs and immune cells in the tumor microenvironment could result in more effective individualized immunotherapy and better therapeutic outcomes. Further research is needed to determine if exosomes contain other immunological checkpoints besides those listed in this article. We believe that by continuing the scientific study process in this field, significant progress in cancer management could be made soon.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther. 2020;5(1):145. https://doi.org/10.1038/s41392-020-00261-0.
Johnstone RM, Adam M, Hammond J, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem. 1987;262(19):9412–20.
Bahrami A, Moradi Binabaj MA, Ferns G. Exosomes: emerging modulators of signal transduction in colorectal cancer from molecular understanding to clinical application. Biomed Pharmacother. 2021;141:111882. https://doi.org/10.1016/j.biopha.2021.111882.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977.
Sun W, Ren Y, Lu Z, Zhao X. The potential roles of exosomes in pancreatic cancer initiation and metastasis. Mol Cancer. 2020;19(1):1–18.
Ahmadi M, Rezaie J. Tumor cells derived-exosomes as angiogenenic agents: possible therapeutic implications. J Transl Med. 2020;18(1):1–17.
Gluszko A, Mirza SM, Piszczatowska K, Kantor I, Struga M, Szczepanski MJ. The role of tumor-derived exosomes in tumor angiogenesis and tumor progression. Curr Issues Pharm Med Sci. 2019;32(4):193–202.
Pan X, Zheng L. Epigenetics in modulating immune functions of stromal and immune cells in the tumor microenvironment. Cell Mol Immunol. 2020;17(9):940–53.
Olejarz W, Dominiak A, Żołnierzak A, Kubiak-Tomaszewska G, Lorenc T. Tumor-derived exosomes in immunosuppression and immunotherapy. J Immunol Res. 2020;2020:6272498. https://doi.org/10.1155/2020/6272498.
Xing C, Li H, Li R-J, Yin L, Zhang H-F, Huang Z-N, et al. The roles of exosomal immune checkpoint proteins in tumors. Mil Med Res. 2021;8(1):1–10.
Lawler SE, Nowicki MO, Ricklefs FL, Chiocca EA. Immune escape mediated by exosomal PD-L1 in cancer. Adv Biosyst. 2020;4(12):2000017.
Shimizu A, Sawada K, Kobayashi M, Yamamoto M, Yagi T, Kinose Y, et al. Exosomal CD47 plays an essential role in immune evasion in ovarian cancer. Mol Cancer Res. 2021;19(9):1583–95.
You L, Wu W, Wang X, Fang L, Adam V, Nepovimova E, et al. The role of hypoxia-inducible factor 1 in tumor immune evasion. Med Res Rev. 2021;41(3):1622–43.
Spugnini EP, Logozzi M, Di Raimo R, Mizzoni D, Fais S. A role of tumor-released exosomes in paracrine dissemination and metastasis. Int J Mol Sci. 2018;19(12):3968.
Fu S, Wang Y, Xia X, Zheng JC. Exosome engineering: current progress in cargo loading and targeted delivery. NanoImpact. 2020;20:100261. https://doi.org/10.1016/j.impact.2020.100261.
Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Can Res. 2005;65(12):5238–47.
Taghikhani A, Farzaneh F, Sharifzad F, Mardpour S, Ebrahimi M, Hassan ZM. Engineered tumor-derived extracellular vesicles: potentials in cancer immunotherapy. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.00221.
Yen E-Y, Miaw S-C, Yu J-S, Lai IR. Exosomal TGF-β1 is correlated with lymphatic metastasis of gastric cancers. Am J Cancer Res. 2017;7(11):2199–208.
Iero M, Valenti R, Huber V, Filipazzi P, Parmiani G, Fais S, et al. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15(1):80–8.
Hao Q, Wu Y, Wu Y, Wang P, Vadgama JV. Tumor-derived exosomes in tumor-induced immune suppression. Int J Mol Sci. 2022;23(3):1461.
Lundholm M, Schröder M, Nagaeva O, Baranov V, Widmark A, Mincheva-Nilsson L, et al. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: mechanism of immune evasion. PLoS ONE. 2014;9(9):e108925.
Mittelbrunn M, Gutiérrez-Vázquez C, Villarroya-Beltri C, González S, Sánchez-Cabo F, González MÁ, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2(1):1–10.
Shojaei S, Hashemi SM, Ghanbarian H, Salehi M, Mohammadi-Yeganeh S. Effect of mesenchymal stem cells-derived exosomes on tumor microenvironment: tumor progression versus tumor suppression. J Cell Physiol. 2019;234(4):3394–409.
Shen Y, Xue C, Li X, Ba L, Gu J, Sun Z, et al. Effects of gastric cancer cell-derived exosomes on the immune regulation of mesenchymal stem cells by the NF-kB signaling pathway. Stem Cells Dev. 2019;28(7):464–76.
Mulero MC, Huxford T, Ghosh G. NF-κB, IκB, and IKK: integral components of immune system signaling. Struct Immunol. 2019. https://doi.org/10.1007/978-981-13-9367-9_10.
Mishra V, Pathak C. Human Toll-Like Receptor 4 (hTLR4): Structural and functional dynamics in cancer. Int J Biol Macromol. 2019;122:425–51.
Domenis R, Cifù A, Marinò D, Fabris M, Niazi KR, Soon-Shiong P, et al. Toll-Like receptor-4 activation boosts the immunosuppressive properties of tumor cells-derived exosomes. Sci Rep. 2019;9(1):1–14.
Mirzaei R, Sarkar S, Dzikowski L, Rawji KS, Khan L, Faissner A, et al. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oxfordshire: Taylor & Francis; 2018. p. e1478647.
Bland CL, Byrne-Hoffman CN, Fernandez A, Rellick SL, Deng W, Klinke DJ. Exosomes derived from B16F0 melanoma cells alter the transcriptome of cytotoxic T cells that impacts mitochondrial respiration. FEBS J. 2018;285(6):1033–50.
Wu Y, Deng W, McGinley EC, Klinke DJ. Melanoma exosomes deliver a complex biological payload that upregulates PTPN 11 to suppress T lymphocyte function. Pigment Cell Melanoma Res. 2017;30(2):203–18.
Tung SL, Boardman DA, Sen M, Letizia M, Peng Q, Cianci N, et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci Rep. 2018;8(1):1–12.
Li L, Cao B, Liang X, Lu S, Luo H, Wang Z, et al. Microenvironmental oxygen pressure orchestrates an anti-and pro-tumoral γδ T cell equilibrium via tumor-derived exosomes. Oncogene. 2019;38(15):2830–43.
Muntasell A, Berger AC, Roche PA. T cell-induced secretion of MHC class II–peptide complexes on B cell exosomes. EMBO J. 2007;26(19):4263–72.
Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8(3):239–45.
Lotfinejad P, Kazemi T, Mokhtarzadeh A, Shanehbandi D, Niaragh FJ, Safaei S, et al. PD-1/PD-L1 axis importance and tumor microenvironment immune cells. Life Sci. 2020;259:118297.
Tang Y, Zhang P, Wang Y, Wang J, Su M, Wang Y, et al. The biogenesis, biology, and clinical significance of exosomal PD-L1 in cancer. Front Immunol. 2020;11:604.
Fan Y, Che X, Qu J, Hou K, Wen T, Li Z, et al. Exosomal PD-L1 retains immunosuppressive activity and is associated with gastric cancer prognosis. Ann Surg Oncol. 2019;26(11):3745–55.
Liang B, Hu X, Ding Y, Liu M. Tumor-derived exosomes in the PD-1/PD-L1 axis: Significant regulators as well as promising clinical targets. J Cell Physiol. 2021;236(6):4138–51.
Zhou K, Guo S, Li F, Sun Q, Liang G. Exosomal PD-L1: new insights into tumor immune escape mechanisms and therapeutic strategies. Front Cell Dev Biol. 2020. https://doi.org/10.3389/fcell.2020.569219.
Ye L, Zhu Z, Chen X, Zhang H, Huang J, Gu S, et al. The Importance of exosomal PD-L1 in cancer progression and its potential as a therapeutic target. Cells. 2021;10(11):3247.
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560(7718):382–6.
Yang Y, Li C-W, Chan L-C, Wei Y, Hsu J-M, Xia W, et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018;28(8):862–4.
Kim DH, Kim H, Choi YJ, Kim SY, Lee J-E, Sung KJ, et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp Mol Med. 2019;51(8):1–13.
Cordonnier M, Nardin C, Chanteloup G, Derangere V, Algros M-P, Arnould L, et al. Tracking the evolution of circulating exosomal-PD-L1 to monitor melanoma patients. J Extracell Vesicles. 2020;9(1):1710899.
Zhang C, Fan Y, Che X, Zhang M, Li Z, Li C, et al. Anti-PD-1 therapy response predicted by the combination of exosomal PD-L1 and CD28. Front Oncol. 2020;10:760.
Qiu Y, Yang Y, Yang R, Liu C, Hsu J-M, Jiang Z, et al. Activated T cell-derived exosomal PD-1 attenuates PD-L1-induced immune dysfunction in triple-negative breast cancer. Oncogene. 2021;40(31):4992–5001.
Poggio M, Hu T, Pai C-C, Chu B, Belair CD, Chang A, et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell. 2019;177(2):414-27. e13.
Li X, Liu Y, Yang L, Jiang Y, Qian Q. TIM-3 shuttled by MV3 cells-secreted exosomes inhibits CD4+ T cell immune function and induces macrophage M2 polarization to promote the growth and metastasis of melanoma cells. Transl Oncol. 2022;18:101334. https://doi.org/10.1016/j.tranon.2021.101334.
Chatterjee S, Chatterjee A, Jana S, Dey S, Roy H, Das MK, et al. Transforming growth factor beta orchestrates PD-L1 enrichment in tumor-derived exosomes and mediates CD8 T-cell dysfunction regulating early phosphorylation of TCR signalome in breast cancer. Carcinogenesis. 2021;42(1):38–47.
Shenoy GN, Bhatta M, Bankert RB. Tumor-associated exosomes: a potential therapeutic target for restoring anti-tumor T Cell responses in human tumor microenvironments. Cells. 2021;10(11):3155. https://doi.org/10.3390/cells10113155.
Miyazaki T, Ikeda K, Sato W, Horie-Inoue K, Inoue S. Extracellular vesicle-mediated EBAG9 transfer from cancer cells to tumor microenvironment promotes immune escape and tumor progression. Oncogenesis. 2018;7(1):7. https://doi.org/10.1038/s41389-017-0022-6.
Gao J, Qiu X, Li X, Fan H, Zhang F, Lv T, et al. Expression profiles and clinical value of plasma exosomal Tim-3 and Galectin-9 in non-small cell lung cancer. Biochem Biophys Res Commun. 2018;498(3):409–15. https://doi.org/10.1016/j.bbrc.2018.02.114.
Wang Y, Li P, Mao S, Mo Z, Cao Z, Luo J, et al. Exosome CTLA-4 regulates PTEN/CD44 signal pathway in spleen deficiency internal environment to promote invasion and metastasis of hepatocellular carcinoma. Front Pharmacol. 2021;12:757194-. https://doi.org/10.3389/fphar.2021.757194.
Li X, Liu Y, Zheng S, Zhang T, Wu J, Sun Y, et al. Role of exosomes in the immune microenvironment of ovarian cancer. Oncol Lett. 2021;21(5):1–17.
Kelleher RJ Jr, Balu-Iyer S, Loyall J, Sacca AJ, Shenoy GN, Peng P, et al. Extracellular vesicles present in human ovarian tumor microenvironments induce a phosphatidylserine-dependent arrest in the T-cell signaling cascade. Cancer Immunol Res. 2015;3(11):1269–78. https://doi.org/10.1158/2326-6066.cir-15-0086.
Shenoy GN, Loyall J, Maguire O, Iyer V, Kelleher RJ Jr, Minderman H, et al. Exosomes associated with human ovarian tumors harbor a reversible checkpoint of T-cell responses. Cancer Immunol Res. 2018;6(2):236–47. https://doi.org/10.1158/2326-6066.cir-17-0113.
Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14(3):195–208.
Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood, J Am Soc Hematol. 2014;124(25):3748–57.
Ye SB, Zhang H, Cai TT, Liu YN, Ni JJ, He J, et al. Exosomal miR-24-3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J Pathol. 2016;240(3):329–40.
Czystowska-Kuzmicz M, Sosnowska A, Nowis D, Ramji K, Szajnik M, Chlebowska-Tuz J, et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat Commun. 2019;10(1):3000. https://doi.org/10.1038/s41467-019-10979-3.
Jafarzadeh N, Gholampour MA, Alivand M-R, Kavousi S, Arzi L, Rad F, et al. CML derived exosomes promote tumor favorable functional performance in T cells. BMC Cancer. 2021;21(1):1002. https://doi.org/10.1186/s12885-021-08734-3.
Clayton A, Mitchell JP, Court J, Mason MD, Tabi Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007;67(15):7458–66. https://doi.org/10.1158/0008-5472.can-06-3456.
Theodoraki M-N, Yerneni S, Gooding WE, Ohr J, Clump DA, Bauman JE, et al. Circulating exosomes measure responses to therapy in head and neck cancer patients treated with cetuximab, ipilimumab, and IMRT. Oncoimmunology. 2019;8(7):e1593805.
Zhu L, Kalimuthu S, Gangadaran P, Oh JM, Lee HW, Baek SH, et al. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics. 2017;7(10):2732.
Shoae-Hassani A, Hamidieh AA, Behfar M, Mohseni R, Mortazavi-Tabatabaei SA, Asgharzadeh S. NK cell–derived exosomes from NK Cells previously exposed to neuroblastoma cells augment the antitumor activity of cytokine-activated NK cells. J Immunother (Hagerstown, Md: 1997). 2017;40(7):265.
Azambuja JH, Ludwig N, Yerneni S, Rao A, Braganhol E, Whiteside TL. Molecular profiles and immunomodulatory activities of glioblastoma-derived exosomes. Neuro Oncol Adv. 2020;2(1):vdaa056.
Mao Y, Wang Y, Dong L, Zhang Q, Wang C, Zhang Y, et al. Circulating exosomes from esophageal squamous cell carcinoma mediate the generation of B10 and PD-1high Breg cells. Cancer Sci. 2019;110(9):2700–10.
Seo Y, Kim H-S, Hong I-S. Stem cell-derived extracellular vesicles as immunomodulatory therapeutics. Stem cells int. 2019. https://doi.org/10.1155/2019/5126156.
Czystowska-Kuzmicz M, Sosnowska A, Nowis D, Ramji K, Szajnik M, Chlebowska-Tuz J, et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat Commun. 2019;10(1):1–16.
Razzo BM, Ludwig N, Hong C-S, Sharma P, Fabian KP, Fecek RJ, et al. Tumor-derived exosomes promote carcinogenesis of murine oral squamous cell carcinoma. Carcinogenesis. 2020;41(5):625–33.
Ning Y, Shen K, Wu Q, Sun X, Bai Y, Xie Y, et al. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol Lett. 2018;199:36–43.
Chen J, Song Y, Miao F, Chen G, Zhu Y, Wu N, et al. PDL1-positive exosomes suppress antitumor immunity by inducing tumor-specific CD8+ T cell exhaustion during metastasis. Cancer Sci. 2021;112(9):3437.
Wen J, Chang X, Bai B, Gao Q, Zhao Y. Orexin A suppresses the expression of exosomal PD-L1 in colon cancer and promotes T cell activity by inhibiting JAK2/STAT3 signaling pathway. Dig Dis Sci. 2022;67(6):2173–2181. https://doi.org/10.1007/s10620-021-07077-0.
Sun Y, Guo J, Yu L, Guo T, Wang J, Wang X, et al. PD-L1+ exosomes from bone marrow-derived cells of tumor-bearing mice inhibit antitumor immunity. Cell Mol Immunol. 2021;18(10):2402–9.
Zeng L, Liao Z, Li W, Yuan Q, Wu P, Gu Z, et al. Non-covalent glycosylated gold nanoparticles/peptides nanovaccine as potential cancer vaccines. Chin Chem Lett. 2020;31(5):1162–4.
Gu X, Erb U, Büchler MW, Zöller M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int J Cancer. 2015;136(4):E74–84.
Shin K, Kim J, Park J, Kim OR, Kang N, Kim I-H. Prognostic significance of exosomal programmed death-ligand 1 in advanced gastric cancer patients treated with first-line chemotherapy. American Society of Clinical Oncology; 2022.
Shimada Y, Matsubayashi J, Kudo Y, Maehara S, Takeuchi S, Hagiwara M, et al. Serum-derived exosomal PD-L1 expression to predict anti-PD-1 response and in patients with non-small cell lung cancer. Sci Rep. 2021;11(1):1–10.
Cheng Z, Wang L, Wu C, Huang L, Ruan Y, Xue W. Tumor-derived exosomes induced M2 macrophage polarization and promoted the metastasis of osteosarcoma cells through Tim-3. Arch Med Res. 2021;52(2):200–10.
Ren W, Zhang X, Li W, Feng Q, Feng H, Tong Y, et al. Exosomal miRNA-107 induces myeloid-derived suppressor cell expansion in gastric cancer. Cancer Manag Res. 2019;11:4023.
Narita M, Kanda T, Abe T, Uchiyama T, Iwafuchi M, Zheng Z, et al. Immune responses in patients with esophageal cancer treated with SART1 peptide-pulsed dendritic cell vaccine. Int J Oncol. 2015;46(4):1699–709.
Cheng L, Wang Y, Huang L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol Ther. 2017;25(7):1665–75.
Zhang L, Yu D. Exosomes in cancer development, metastasis, and immunity. Biochim Biophys Acta. 2019;1871(2):455–68.
Andaloussi SE, Lakhal S, Mäger I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev. 2013;65(3):391–7.
Zhang F, Guo J, Zhang Z, Qian Y, Wang G, Duan M, et al. Mesenchymal stem cell-derived exosome: a tumor regulator and carrier for targeted tumor therapy. Cancer Lett. 2022;526:29–40.
Bagheri E, Abnous K, Farzad SA, Taghdisi SM, Ramezani M, Alibolandi M. Targeted doxorubicin-loaded mesenchymal stem cells-derived exosomes as a versatile platform for fighting against colorectal cancer. Life Sci. 2020;261:118369.
Liu J, Ye Z, Xiang M, Chang B, Cui J, Ji T, et al. Functional extracellular vesicles engineered with lipid-grafted hyaluronic acid effectively reverse cancer drug resistance. Biomaterials. 2019;223:119475.
Zhao Y, Liu L, Sun R, Cui G, Guo S, Han S, et al. Exosomes in cancer immunoediting and immunotherapy. Asian J Pharm Sci. 2022. https://doi.org/10.1016/j.ajps.2021.12.001.
Zhou W, Zhou Y, Chen X, Ning T, Chen H, Guo Q, et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials. 2021;268:120546. https://doi.org/10.1016/j.biomaterials.2020.120546.
Xu HY, Li N, Yao N, Xu XF, Wang HX, Liu XY, et al. CD8+ T cells stimulated by exosomes derived from RenCa cells mediate specific immune responses through the FasL/Fas signaling pathway and combined with GM-CSF and IL-12, enhance the anti-renal cortical adenocarcinoma effect. Oncol Rep. 2019;42(2):866–79.
Lu Z, Zuo B, Jing R, Gao X, Rao Q, Liu Z, et al. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J Hepatol. 2017;67(4):739–48.
Näslund TI, Gehrmann U, Qazi KR, Karlsson MC, Gabrielsson S. Dendritic cell–derived exosomes need to activate both T and B cells to induce antitumor immunity. J Immunol. 2013;190(6):2712–9.
Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: an online resource for prediction of microRNA binding sites. PLoS ONE. 2018;13(10):e0206239.
Xu N, He D, Shao Y, Qu Y, Ye K, Memet O, et al. Lung-derived exosomes in phosgene-induced acute lung injury regulate the functions of mesenchymal stem cells partially via miR-28-5p. Biomed Pharmacother. 2020;121:109603.
Hou X, Wan W, Wang J, Li M, Wang Y, Yao Y, et al. Let-7a inhibits migration of melanoma cells via down-regulation of HMGA2 expression. Am J Transl Res. 2016;8(9):3656.
Liu J, Han Y, Hu S, Cai Y, Yang J, Ren S, et al. Circulating exosomal MiR-107 restrains tumorigenesis in diffuse large B-Cell lymphoma by targeting 14-3-3η. Front Cell Dev Biol. 2021;9:845.
Wang S, Li L, Hu X, Liu T, Jiang W, Wu R, et al. Effects of atrial fibrillation-derived exosome delivery of miR-107 to human umbilical vein endothelial cells. DNA Cell Biol. 2021;40(4):568–79.
Ni Q, Stevic I, Pan C, Müller V, Oliveira-Ferrer L, Pantel K, et al. Different signatures of miR-16, miR-30b and miR-93 in exosomes from breast cancer and DCIS patients. Sci Rep. 2018;8(1):1–10.
Zhu L, Chen Y, Chen M, Wang W. Mechanism of miR-204–5p in exosomes derived from bronchoalveolar lavage fluid on the progression of pulmonary fibrosis via AP1S2. AnnTransl Med. 2021. https://doi.org/10.21037/atm-20-8033.
Yao S, Yin Y, Jin G, Li D, Li M, Hu Y, et al. Exosome-mediated delivery of miR-204-5p inhibits tumor growth and chemoresistance. Cancer Med. 2020;9(16):5989–98.
Acknowledgements
The authors of the present review article appreciate the sincere efforts of the esteemed researchers of the Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran. Furthermore, the writers of this review investigation know and respect the use of sample images given by https://smart.servier.com/ in the creation of Fig. 1.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Data collection, figure designing, and analysis were performed by Seyed Ziaeddin Rasihashemi, Hadi Sahrai, and Parisa Lotfinejad. The first draft of the manuscript was written by Parisa Lotfinejad, Erfan Rezazadeh-Gavgani, Yalda Yazdani, and Amirreza Khalaji. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
None.
Consent to participate
None.
Consent to publish
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rasihashemi, S.Z., Sahrai, H., Rezazadeh-Gavgani, E. et al. Exosomes carrying immune checkpoints, a promising therapeutic approach in cancer treatment. Med Oncol 39, 183 (2022). https://doi.org/10.1007/s12032-022-01781-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-022-01781-1