
Abstract
Large amount of expression data were generated by high-throughput experimental techniques such as microarray. Single algorithm cannot be widely accepted as suitable method for mining of gene expression data. Therefore, integration of different algorithms and extraction of more useful information from the expression data are the key problems for identification of biomarkers. Here, we used three machine learning algorithms to select feature genes based on gene profiling data of gastric cancer (GC). Then, a common divisor was extracted as candidate feature genes aggregation for Tree Building and Tree Pruning analysis by Decision Tree (DT) algorithm. Real-time quantitative PCR and immunohistochemistry (IHC) staining were used to validate the relative expression levels of the candidate feature genes. Receiver operating characteristic curves were used to analyse the classification sensitivity and specificity of the feature genes. A total of 174, 202, 149 feature genes were selected by Class Information Index, Information Gain Index and Relief algorithms, with a common divisor consisting of 32 genes. Using a DT algorithm to contribute to the classification rule sets, we identified COL2A1 and ATP4B as candidate biomarkers of GC. The expression levels of these two genes were validated by real-time PCR and IHC with high sensitivity (>90 %) and specificity (>90 %) in both training and test samples. We first introduced an integral and systematic data-mining model for identification of biomarkers based on gene expression data. The two-gene signature obtained by our predictive model could be used for recognizing the biological characteristic of GC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the last decades, cancer genomics and proteomics have extensively penetrated into biomedical research and clinical application. After the gene chip and microarray technology were introduced, many researchers used these techniques to find new subclasses in disease states [1, 2], identify new biomarkers associated with diseases [3–6], classify subtypes of tumours [7] and predict the outcome of a disease [8–11]. Gene expression profiling from microarray studies have been used to understand the development mechanism of human diseases. However, most of the traditional statistical methods are not suitable for processing high-dimensionality and high-noise gene expression data. Usually, one may choose a gene with a higher fold-change (FC) value and lower P value for further research, but may overlook some more important and useful information in the profiling.
Unsupervised classification algorithms and unbiased approach to searching for subgroups in the expression data were among of the first statistical techniques to be applied to microarray and gene expression profiling data analysis [12]. While these techniques always produce a clustering of genes, the expression patterns of the genes observed in the sample data do not always characterize the pattern in the whole population [13]. With the development of supervised classification algorithms and machine learning algorithms, many researchers started to use these methods to identify feature gene sets which allow for the classification of the available samples. Compared to the unsupervised methods, the genes selected by supervised machine learning algorithms have more stable expression patterns both in training and test samples. Since no single algorithm is widely accepted as the optimal method for mining gene expression data [13], several algorithms are used in combination to mine the gene expression data from a small study of gastric cancer (GC).
GC is one of the most frequent cancers in the world. Almost two-thirds of gastric cancer cases and deaths occur in less developed regions. Despite remarkable declines in GC mortality were noticed in almost the entire population during the last decade in China, it remains a significant cancer burden currently and is one of the key issues in cancer prevention and control strategy in China [14]. We previously used a 22-K oligonucleotide microarray with optimized experimental protocols and analytical tools to identify transcriptional expression profiles of GC from a Chinese cohort, and identified a total of 1,519 differentially expressed genes by comparing 20 samples with GC samples against matched normal samples [15]. These gene expression profiling data provide significant research materials for biomarkers identification that could be associated with the biological characteristic of GC.
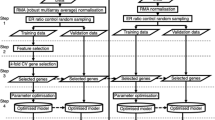
In this study, three different algorithms (CII, IGI and Relief) were used to select feature genes based on differentially expressed gene profiling of gastric cancer. Subsets of these candidate biomarkers were used in a decision tree (DT) classifiers, and the expression levels for a pair of genes sufficiently distinguished GC samples from matched normal ones. Moreover, real-time quantitative PCR was used to validate the expression levels of these two genes in 30 validation cases. Other 58 validation cases were used for immunohistochemistry staining. Finally, the sensitivity and specificity of the candidate biomarkers were calculated using ROC curves analyses.
Methods
Microarray data
The human genome oligonucleotide microarray was prepared in CapitalBio Corporation (Beijing, China). A Human Genome Oligo Set version 2.1 consisting of about 22,000 human genes was purchased from Qiagen Operon Company. A total of 20 GC and matched normal samples were obtained from Beijing Cancer Hospital for microarray experiments. Fluorescent dye–labelled DNA was produced through a RNA amplification method and subsequent enzymatic reaction. Arrays were scanned with a confocal LuxScan scanner (CapitalBio Corp.), and images were analysed with SpotData software (CapitalBio Corp.). The raw data were normalized based on a LOWESS in the R language package (http://www.R-project.org/). For details of the microarray experiments, the readers are referred to the paper by Zang et al. [15].
Clinical samples for laboratory validation
A total of 88 patients undergoing gastrectomy for potentially curable GC at the Wuhan General Hospital of Guangzhou Command from May 2010 to December 2012 were selected as subjects in this study. The patients in this research project have given informed consent, and the project has been approved by a suitably constituted Ethics Committee of Wuhan General Hospital of Guangzhou Command. The names of patients in this project were anonymized.
CII algorithms
A basic idea of feature genes selection is to select genes with high accuracy in classifying different types of samples. Divisibility can be computed via examining the similarity of sample properties in the same category (within-class distance), as well as the difference in the properties for samples in different categories (between-class distance). “Signal-to-noise ratio”, a statistical t test, which was proposed by Golub [16], as well as the genetic Classification Information Index (CII) algorithm [17], can better reflect the above-mentioned ideas and can serve as measures to estimate how much classification information each gene contains. Here, we used CII algorithm for feature genes selection based on gene expression profiling of GC.
IGI algorithm
In order to assess the importance of genes contributing to sample classification and provide a criterion of measuring the value of genetic classification, we adopt another algorithm, the Information Gain Index (IGI), to evaluate the ability of classification by a given gene [18].
Relief algorithm
The Relief algorithm evaluates the importance of attribute classification based on within-class and between-class distance [19]. This algorithm starts from a random sample rather than from the statistical characteristics of the whole class to estimate sample class separability. For any learning sample S in the training set, the algorithm searches out K (K > 1) same-class samples closest to S (nearest Hit) and K heterogeneous samples (nearest Miss). For the attribute \(A_{i}\), if the difference between S and heterogeneous sample is larger and the difference between S and same-class sample is smaller, then the separability of sample S on attribute \(A_{i}\) is greater and the classification weight of \(A_{i}\) is also greater.
DT algorithm
DT is a tree structure applied to classification in which each internal node performs a test on a certain attribute and each edge represents a test result. Each terminal leaf node represents a class or the class distribution, and the top node is the root node [19]. DT construction involves two steps: Tree Building and Tree Pruning. Tree Pruning is to reduce fluctuation due to the existence of noise in training set after Tree Building. Decision trees are sensitive to small samples and can result in additional overhead when dealing with continuous value discretization. So, intelligent threshold search and intelligent pruning strategy are used to reduce the computational complexity and increase classification accuracy. The algorithm is described below:
Pathway and gene ontology (GO) analyses
To investigate the signalling pathway involved by the feature genes, we used an integrated signalling pathway database at http://www.biorag.org. This signalling pathway database contains metabolic, cellular and regulatory pathways for human and mouse gene products from three different open source pathway resources: KEGG (http://www.genome.ad.jp), BioCarta (http://www.biocarta.com) and GenMAPP (http://www.genmapp.org). An integral gene ontology database MAS (Molecular Annotation System, http://www.capitalbio.com) was also used to annotate the function of the productions encoded by these genes.
RNA extraction and real-time quantitative PCR
The relative expression levels of the candidate biomarkers were tested using real-time PCR based on 30 GC specimens and matched normal tissues. Total RNA was extracted from the tissue samples according to a standard Trizol protocol (Invitrogen, Carlsbad, CA, USA). 5 µg of total RNA was reverse-transcribed to cDNA with 200U M-MLV reverse transcriptase (Promega, Madison). RT reaction was set as following conditions: 37 centigrade for 60 min, 72 centigrade for 10 min. Quantitative real-time PCRs were performed in a total 20-µl reaction volume containing 2 µl of cDNA, 0.6 µl 20× Eva Green (CapitalBio Corp., Beijing, China), 0.5 µl of each 10uM forward and reverse primers, 0.5 µl of 2.5 mM dNTP, 1.5U Cap Taq polymerase (CapitalBio Corp., Beijing, China), 10 µl 2 × PCR Buffer for Eva Green and 6.1 µl of H2O. PCRs were carried out with the following programmed parameters, heating at 95 centigrade for 5 min followed by 40 cycles of a three-stage temperature profile of 95 centigrade for 30 s, 57 centigrade for 30 s and 72 centigrade for 30 s. All reactions were performed in triplicates, and the final Ct values were determined by the average Ct value of the three reaction. The melting curves for each PCR were carefully analysed to avoid nonspecific amplifications in PCR products. The expression of each gene was transformed using the 2−ΔΔCt formula and normalized with β-actin expression [20].
Tissue microarray and immunohistochemistry (IHC) staining
A total of 58 human gastric specimens were obtained from the Wuhan General Hospital of Guangzhou Command. The patients were fully informed and gave consents for collection of clinical samples. Tissue microarray blocks were constructed in Beijing Cancer Hospital. For each case, we sampled five tissue cores at 1.0 mm in diameter, including two tumours and one matched-adjacent normal mucosa tissues to construct the tissue microarray. IHC staining was performed using EnVision+ Kit (Dako, Denmark). The section was incubated with anti-COL2A1 and anti-ATP4B at 4° overnight. More than 5 % stained cells in the tissue was defined as positive reaction in this experiment.
Receiver operating characteristic (ROC) curve and statistical analyses
ROC curve analysis was conducted using the MedCalc software packages (version 8.2.1.0; Mariakerke, Belgium). The area under the curves (AUC) value provided a measure of the overall performance of a diagnostic test. The ratio of gene signal intensities and Ct value of each gene were used for ROC calculation in training and test samples, respectively. For all statistical analyses, a difference with P < 0.05 was considered statistically significant based on t test approach.
Results
Feature gene selection using three machine learning algorithms
The Classification Information Index (CII) algorithm was first used for feature gene selection. Each gene was marked as i (i = {i 1, i 2… i 1519}), as shown in Fig. 1a, and was distributed in 5 intervals. One hundred and seventy-four candidate genes with high CII value (i > 0.5) were selected (Table 1). Information Gain Index (IGI) was also used to select feature genes. The IGI of each gene was marked as g (g = {g 1, g 2… g 1519}), as shown in Fig. 1b, and was distributed in 5 intervals. Two hundred and two candidate genes with the small IGI values (g < 0.6) were selected (Table 1). Finally, the Relief Algorithm employed the Mahalanobis squared distance as classifying index. The classification weight of each gene was marked as w (w = {w 1, w 2,… w 1519}), as shown in Fig. 1c. All 1,519 genes were distributed in 6 intervals, and the 149 candidate genes with high classification weight (w > 20) were selected (Table 1).

Feature gene selection based on three machine learning algorithms. a Feature gene selection using CII algorithm; b feature gene selection using IGI algorithm; c feature gene selection using Relief algorithm; d extraction of the common divisor from the results of the three algorithms above. Thirty-two genes were composed of the common divisors
Combining the results of the three algorithms described above, 32 genes were identified as candidate biomarkers to further validation (Fig. 1d), including 11 genes down-regulated and 21 genes up-regulated in GC samples compared with normal ones. Fold-change values of all the candidate biomarkers are shown in Table 2.
Biological significance of the candidate biomarker genes
The signalling pathways and annotated functions of the proteins encoded by these genes were investigated. The down-regulated gene group contained two gene families, ATP4 and KCN, and participated in ion transport and ATPase activity. The up-regulated gene group contained six members (COL2A1, COL3A1, COL4A1, COL6A3, COL8A1 and COL11A1) of the collagen family and participated in ion transport activity, cell–cell adhesion and inflammatory response pathways (Table 3). Different functions were found in the different gene groups. ATP4 and KCN members were classified in potassium ion transport (GO: 0006813) and metabolism (GO: 0008152), and collagen family members were classified in cell adhesion (GO: 0007155), phosphate transport (GO: 0006817) and negative regulation of cell proliferation (GO: 0008285) (Table 4).
Extraction of the classification rules using DT
Subsets of the 32 candidate genes were then used in DT classifiers. The samples were separated into 4 sets containing 5 GC samples and 5 matched normal samples. Three sets were used as the training set, and the remaining set became the test set. The verification was repeated 4 times so that each set became the testing set. The results showed that DT composed of two genes, COL2A1 and ATP4B, obtained the best classification accurate rate (100 %). If the expression levels of COL2A1 and ATP4B are represented by E c and E a, respectively, a sample is classified as cancerous only if E c > 1.302 and E a < 3.102 (Fig. 2).

Extraction of the rule sets using Decision Tree algorithm. a The most accurate rule set contains two genes: COL2A1 and ATP4B. The expression levels of COL2A1 and ATP4B are represented by E c and E a. If E c < 1.302, we recognize the samples as normal; if E c > 1.302, E a > 3.102, we recognize the samples as normal; only if E c > 1.302 and E a < 3.102, we recognize the samples as GC. b All of the samples were classified correctly by using our two-gene classifier
Validation of the feature genes using real-time quantitative PCR and IHC staining
In order to validate the candidate feature genes (COL2A1 and ATP4B) from our prediction model, quantitative real-time PCR was used to measure the level of expression using additional 30 validation cases containing 30 GC samples and matched normals. The results showed that COL2A1 was expressed high in GC samples and expressed low in matched normal ones; ATP4B was expressed low in GC samples and expressed high in matched normal ones. Among these 30 matched GC samples, COL2A1 was up-regulated in 25 GC samples (83.3 %) and ATP4B was down-regulated in 26 GC samples (86.7 %) (Fig. 3a, b).

COL2A1 and ATP4B were differential expressed in GC samples and matched normal ones using real-time PCR. a COL2A1 was up-regulated in 25 of 30 GC samples; b ATP4B was down-regulated in 26 of 30 GC samples. “X-axis” represents the samples; “Y-axis” represents the FC value
In addition, other 58 validation cases were used for IHC staining. The results showed that COL2A1 was expressed high in 44 GC samples with the positive rate of 75.9 % (44/58); ATP4B was expressed low in 47 GC samples with the negative rate of 81.0 % (47/58) (Fig. 4, Table 5). The IHC results were matched with PCRs that COL2A1 was high-expressed and ATP4B was low-expressed in GC samples.

IHC staining results. COL2A1 was negatively expressed in normal cases (a) and positively expressed in GC cases (b). ATP4B was positively expressed in normal case (c) and negatively expressed in GC case (d)
Sensitivity and specificity analyses of the candidate biomarkers
A total of 40 training (including 20 GC samples and 20 matched normal samples) and 176 test samples (including 88 GC samples and 88 matched normal samples) were used for ROC analysis according to the microarray data and relative expression value, respectively. The results showed combined COL2A1 and ATP4B as a classifier for classification of GC and normal samples with high sensitivity (100 % in training samples and 96.3 % in test samples) and specificity (100 % in training samples and 95.65 % in test samples) (Fig. 5; Table 6). These data indicate that combining COL2A1 and ATP4B as a classifier was sensitive and specific in classifying the GC and normal samples both in training and test sets.

Sensitivity and specificity of the candidate biomarkers using ROC curves analyses. a–c ROC curves of COL2A1, ATP4B and combination (COL2A1 and ATP4B as a classifier) in the training set, respectively. d–f ROC curves of COL2A1, ATP4B and combination (COL2A1 and ATP4B as a classifier) in the test set, respectively. Higher sensitivity and specificity were observed in combination (COL2A1 and ATP4B as a classifier) group compared to COL2A1 and ATP4B as single group
Discussion
High-throughput microarray technologies had generated a large amount of data, after which various statistical and machine learning methods were adopted to analyse these data for finding gene or protein expressed pattern investigation and search for new biomarkers of human diseases. Selecting the biomarkers which contain most useful information used in molecular classification of human diseases was a most emergent and essential work for microarray data analysis. In this article, we reported an optimized data-mining and prediction model for identification of biomarkers based on gene expression profiling data from a group of Chinese GC patients.
In this new strategy, we used an optimized method of combined multiple machine learning algorithms for data mining in small set of gene expression data. Three machine learning algorithms were used to select feature genes based on differentially expressed gene profiling of GC, which contains 1,519 genes. Our results showed that different feature genes were selected by different algorithms; there was a common set of 32 genes contained by all algorithms. DT determined the final classification rule set; COL2A1 and ATP4B were identified as an optimal classifier for distinguishing GC samples from normal ones. These two genes have been validated in 88 test GC cases compared with the matched normal samples, including 30 cases in PCR experiment and 58 cases in IHC staining. Our classification model used combination of two genes that classified GC and normal samples to a high accuracy. Some previous studies also used a two-gene classifier in the investigation of subclass of human diseases, including cancer [21, 22].
The function and clinical significance of COL2A1 and ATP4B were reported by some researchers. COL2A1 is located on human chromosome 12q13, encoding the alpha-1 chain of type II collagen, a fibrillar collagen found in cartilage and the vitreous humour of the eye. There are two transcripts identified for this gene. Most of the research paper reported that the COL2A1 gene polymorphism is related to some of the genetic diseases, including advanced stages of osteoarthritis [23, 24], skeletal dysplasia [25, 26], knee osteoarthritis [27], congenital toxoplasmosis [28] and stickler syndrome [29–32]. However, no research articles reported that the gene polymorphism and differential expression were associated with human cancer. We supposed that type II collagen family is an important protein family which participated in sustaining the stabilization of the physiological structure in normal cells, tissues and organs. ATP4B is located on human chromosome 13q34, encoding the member of the P-type cation-transporting ATPases. This enzyme is a proton pump that catalyses the hydrolysis of ATP coupled with the exchange of H+ and K+ ions across the plasma membrane and responsible for gastric acid secretion [33]. In the mouse model, it was reported that ATP4B was required for normal function, development and membrane structure of mouse parietal cells [34]. No previous research has reported that this gene was associated with the development of human cancer, although our microarray results showed that it was down-regulated in 20 training and 58 test GC specimens.
In this article, we have documented a systematic data-mining model for biomarkers identification based on a gene expression profiling data, and we identified COL2A1 and ATP4B as a signature for GC classification. The two-gene signature obtained by our predictive model was validated in validation samples by real-time PCR and IHC staining and might be useful o recognize the biological characteristics of GC.
References
Lau SK, Boutros PC, Pintilie M, Blackhall FH, Zhu CQ, Strumpf D, Johnston MR, Darling G, Keshavjee S, Waddell TK, Liu N, Lau D, Penn LZ, Shepherd FA, Jurisica I, Der SD, Tsao MS. Three-gene prognostic classifier for early-stage non small-cell lung cancer. J Clin Oncol. 2007;25(35):5562–9.
Yoshihara K, Tajima A, Komata D, Yamamoto T, Kodama S, Fujiwara H, Suzuki M, Onishi Y, Hatae M, Sueyoshi K, Fujiwara H, Kudo Y, Inoue I, Tanaka K. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009;100(8):1421–8.
Yan Z, Li J, Xiong Y, Xu W, Zheng G. Identification of candidate colon cancer biomarkers by applying a random forest approach on microarray data. Oncol Rep. 2012;28(3):1036–42.
Peyre M, Commo F, Dantas-Barbosa C, Andreiuolo F, Puget S, Lacroix L, Drusch F, Scott V, Varlet P, Mauguen A, Dessen P, Lazar V, Vassal G, Grill J. Portrait of ependymoma recurrence in children: biomarkers of tumor progression identified by dual-color microarray-based gene expression analysis. PLoS ONE. 2010;5(9):e12932.
Colombo J, Fachel AA, De Freitas Calmon M, Cury PM, Fukuyama EE, Tajara EH, Cordeiro JA, Verjovski-Almeida S, Reis EM, Rahal P. Gene expression profiling reveals molecular marker candidates of laryngeal squamous cell carcinoma. Oncol Rep. 2009;21(3):649–63.
Crispi S, Calogero RA, Santini M, Mellone P, Vincenzi B, Citro G, Vicidomini G, Fasano S, Meccariello R, Cobellis G, Menegozzo S, Pierantoni R, Facciolo F, Baldi A, Menegozzo M. Global gene expression profiling of human pleural mesotheliomas: identification of matrix metalloproteinase 14 (MMP-14) as potential tumour target. PLoS ONE. 2009;4(9):e7016.
Fèvre-Montange M, Champier J, Durand A, Wierinckx A, Honnorat J, Guyotat J, Jouvet A. Microarray gene expression profiling in meningiomas: differential expression according to grade or histopathological subtype. Int J Oncol. 2009;35(6):1395–407.
Li W, Wang R, Yan Z, Bai L, Sun Z. High accordance in prognosis prediction of colorectal cancer across independent datasets by multi-gene module expression profiles. PLoS ONE. 2012;7(3):e33653.
Yang S, Chen J, Guo Y, Lin H, Zhang Z, Feng G, Hao Y, Cheng J, Liang P, Chen K, Wu H, Li Y. Identification of prognostic biomarkers for response to radiotherapy by DNA microarray in nasopharyngeal carcinoma patients. Int J Oncol. 2012;40(5):1590–600.
Lahat G, Tuvin D, Wei C, Wang WL, Pollock RE, Anaya DA, Bekele BN, Corely L, Lazar AJ, Pisters PW, Lev D. Molecular prognosticators of complex karyotype soft tissue sarcoma outcome: a tissue microarray-based study. Ann Oncol. 2010;21(5):1112–20.
Yan Z, Xiong Y, Xu W, Gao J, Cheng Y, Wang Z, Chen F, Zheng G. Identification of hsa-miR-335 as a prognostic signature in gastric cancer. PLoS ONE. 2012;7(7):e40037.
Quackenbush J. Microarray analysis and tumour classification. N Engl J Med. 2006;354:2463–72.
Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7(1):55–65.
Yang L. Incidence and mortality of gastric cancer in China. World J Gastroenterol. 2006;12:17–20.
Zang SZ, Guo RF, Zhang L, Lu Y. Integration of statistical inference methods and a novel control measure to improve sensitivity and specificity of data analysis in expression profiling studies. J Biomed Inform. 2007;40:552–60.
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–7.
Li YX, Ruan XG. Feature selection for cancer classification based on support vector machine. J Comput Res Dev. 2005;42:1796–801.
Lee C, Lee G. Information gain and divergence-based feature selection for machine learning-based text categorization. Inf Process Manag. 2006;42:155–65.
Kingsford C, Salzberg SL. What are decision trees? Nat Biotechnol. 2008;26:1011–3.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) method. Methods. 2001;25:402–8.
Price ND, Trent J, El-Naggar AK, Cogdell D, Taylor E, Hunt KK, Pollock RE, Hood L, Shmulevich I, Zhang W. Highly accurate two-gene classifier for differentiating gastrointestinal stromal tumors and leiomyosarcomas. Proc Natl Acad Sci USA. 2007;104(9):3414–9.
Zhang X, Yan Z, Zhang J, Gong L, Li W, Cui J, Liu Y, Gao Z, Li J, Shen L, Lu Y. Combination of hsa-miR-375 and hsa-miR-142-5p as a predictor for recurrence risk in gastric cancer patients following surgical resection. Ann Oncol. 2011;22(10):2257–66.
Gálvez-Rosas A, González-Huerta C, Borgonio-Cuadra VM, Duarte-Salazár C, Lara-Alvarado L, de los Angeles Soria-Bastida M, Cortés-González S, Ramón-Gallegos E, Miranda-Duarte A. A COL2A1 gene polymorphism is related with advanced stages of osteoarthritis of the knee in Mexican Mestizo population. Rheumatol Int. 2010;30(8):1035–9.
Hämäläinen S, Solovieva S, Hirvonen A, Vehmas T, Takala EP, Riihimäki H, Leino-Arjas P. COL2A1 gene polymorphisms and susceptibility to osteoarthritis of the hand in Finnish women. Ann Rheum Dis. 2009;68(10):1633–7.
Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL. Identification of three novel mutations in the COL2A1 gene in four unrelated Chinese families with spondyloepiphyseal dysplasia congenita. Biochem Biophys Res Commun. 2011;413(4):504–8.
Mark PR, Torres-Martinez W, Lachman RS, Weaver DD. Association of a p.Pro786Leu variant in COL2A1 with mild spondyloepiphyseal dysplasia congenita in a three-generation family. Am J Med Genet A. 2011;155A(1):174–9.
Xu P, Yao J, Hou W. Relationships between COL2A1 gene polymorphisms and knee osteoarthritis in Han Chinese women. Mol Biol Rep. 2011;38:2377–81.
Jamieson SE, de Roubaix LA, Cortina-Borja M, Tan HK, Mui EJ, Cordell HJ, Kirisits MJ, Miller EN, Peacock CS, Hargrave AC, Coyne JJ, Boyer K, Bessieres MH, Buffolano W, Ferret N, Franck J, Kieffer F, Meier P, Nowakowska DE, Paul M, Peyron F, Stray-Pedersen B, Prusa AR, Thulliez P, Wallon M, Petersen E, McLeod R, Gilbert RE, Blackwell JM. Genetic and epigenetic factors at COL2A1 and ABCA4 influence clinical outcome in congenital toxoplasmosis. PLoS ONE. 2008;3(6):e2285.
Zechi-Ceide RM, Jesus Oliveira NA, Guion-Almeida ML, Antunes LF, Richieri-Costa A, Passos-Bueno MR. Clinical evaluation and COL2A1 gene analysis in 21 Brazilian families with Stickler syndrome: identification of novel mutations, further genotype/phenotype correlation, and its implications for the diagnosis. Eur J Med Genet. 2008;51(3):183–96.
Gerth-Kahlert C, Grisanti S, Berger E, Höhn R, Witt G, Jung U. Bilateral vitreous hemorrhage in a newborn with Stickler syndrome associated with a novel COL2A1 mutation. J AAPOS. 2011;15(3):311–3.
Yaguchi H, Ikeda T, Osada H, Yoshitake Y, Sasaki H, Yonekura H. Identification of the COL2A1 mutation in patients with type I Stickler syndrome using RNA from freshly isolated peripheral white blood cells. Genet Test Mol Biomarkers. 2011;15(4):231–7.
Richards AJ, McNinch A, Martin H, Oakhill K, Rai H, Waller S, Treacy B, Whittaker J, Meredith S, Poulson A, Snead MP. Stickler syndrome and the vitreous phenotype: mutations in COL2A1 and COL11A1. Hum Mutat. 2010;31(6):E1461–71.
Göõz M, Hammond CE, Larsen K, Mukhin YV, Smolka AJ. Inhibition of human gastric H(+)-K(+)-ATPase alpha-subunit gene expression by Helicobacter pylori. Am J Physiol Gastrointest Liver Physiol. 2000;278(6):G981–91.
Scarff KL, Judd LM, Toh BH, Gleeson PA, Van Driel IR. Gastric H(+), K(+)-adenosine triphosphatase beta subunit is required for normal function, development, and membrane structure of mouse parietal cells. Gastroenterology. 1999;117(3):605–18.
Acknowledgments
We thank Prof. Jiangeng Li (Academy of Electronic Information & control Engineering, Beijing University of Technology, Beijing, China.) for the great help in processing the gene expression data using machine learning algorithms.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yan, Z., Xu, W., Xiong, Y. et al. Highly accurate two-gene signature for gastric cancer. Med Oncol 30, 584 (2013). https://doi.org/10.1007/s12032-013-0584-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-013-0584-x