
Abstract
Environment enrichment (EE) has been demonstrated to improve the cognitive impairment that is induced by chronic cerebral hypoperfusion (CCH), but the underlying mechanism has not yet been elucidated. This study aimed to investigate the role of endogenous neuroprotection in EE-induced cognitive improvement in rats with CCH. Permanent bilateral common carotid artery occlusions (2-vessel occlusions (2VOs)) were performed to induce CCH in male adult Wistar rats. Four weeks after the surgeries, the rats were exposed to enriched environments for 4 weeks (6 h/day). Subsequently, we assessed the effects of EE on cognitive function, brain histone acetylation levels, neuroprotection-related transcription factors (i.e., cAMP response element-binding protein (CREB), phospho-CREB (p-CREB), hypoxia-inducible factor 1 (HIF-1) α, and nuclear regulatory factor 2 (Nrf2)), and oxidative stress and histological changes in the brain. After 2VO, the rats exposed to the EE treatment exhibited increased acetylation of histone 4 and increased p-CREB and Nrf2 protein levels in the brain. HIF-1α levels were increased after 2VO and reduced after EE treatment. The oxidative damage, histopathological changes in the brain, and spatial learning and memory impairments induced by 2VO were subsequently restored after EE treatment. These data indicate that EE promotes the acetylation of histone 4, regulates some neuroprotection-related transcription factors, attenuates oxidative damage, and protects against the histopathological damage to the brain induced by CCH. Together, the effects of EE in CCH rats might contribute to the recovery of spatial learning and memory.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebral hypoperfusion has been demonstrated to be associated with the cognitive deficits of both Alzheimer’s disease (AD) and vascular dementia (VaD) patients (de la Torre 2002; Osawa et al. 2004). In addition to the clinical reports, an increasing number of experimental studies have also proven that reduced cerebral blood supply can cause progressive cognitive impairment (Farkas et al. 2007). Thus, chronic cerebral hypoperfusion (CCH) is thought to be one of the initial conditions that are critical to the development of cognitive dysfunction (Gorelick et al. 2011; Ruitenberg et al. 2005).
Cerebral hypoperfusion can cause deranged energy metabolism, protein synthesis abnormalities, glial activation, apoptosis, oxidative stress, neuronal damage, and white matter lesions; these factors might be the pathophysiological mechanisms that contribute to cognitive impairment (Farkas et al. 2007; Liu and Zhang 2012). Despite the detrimental effects of hypoperfusion, such as declines in cellular oxygen levels and energy supplies, hypoperfusion also induces compensatory or endogenous adaptive and regenerative mechanisms that defend against hypoperfusion injury and allow for the recovery of brain function (Farkas et al. 2007; Choy et al. 2006). The regulation of the endogenous adaptive and neuroprotective processes includes the concerted activation of various transcription factors that mediate the upregulation of neurotrophic factors, antioxidant enzymes, and proteins that are involved in cellular energy metabolism. These transcription factors include the nuclear regulatory factor 2 (Nrf2)–antioxidant response element (ARE) pathway, the cAMP response element-binding protein (CREB) pathway, and hypoxia-inducible factor 1 (HIF-1) (Stranahan and Mattson 2012; Kitagawa 2007). Because a large number of clinical trials that have sought to identify protective strategies against dementia have failed, the recruitment of endogenous neuroprotective pathways represents a potential strategy for the development of therapeutics for the cognitive impairment induced by CCH.
Environment enrichment (EE) is a housing manipulation that provides a complex combination of enhanced sensory, cognitive, and motor stimulation and has been extensively used to investigate the influence of the environment on behavior, particularly in terms of complex cognition (Nithianantharajah and Hannan 2006). Several experimental studies have reported beneficial effects of EE on cognitive function during normal ageing and in models of AD and VaD (Nithianantharajah and Hannan 2006; Gobbo and O'Mara 2004). We previously demonstrated that EE can ameliorate learning and memory impairments induced by CCH (Sun et al. 2010). Although the mechanisms by which EE preserves cognition remain obscure, numerous studies have shown that functional improvement related to EE is associated with the upregulation of neurotrophic factors, the enhancement of synaptic plasticity, the stimulation of neurogenesis (Nithianantharajah and Hannan 2006), and reductions of apoptosis and oxidative damage (Herring et al. 2010; Stranaha et al. 2010; Cechetti et al. 2012). Based on these beneficial effects of EE, we hypothesize that the activation of endogenous neuroprotective pathways might be implicated in the beneficial effects of EE on cognition.
The present study was therefore designed to validate the effects of EE on CCH-related cognitive impairment and to investigate whether EE-induced cognitive protection is associated with the activation of endogenous neuroprotection. Because permanent bilateral common carotid artery occlusion (2-vessel occlusion (2VO)) in rats is widely accepted as an adequate experimental model of CCH that successfully imitates the pathophysiological features of human AD and VaD (Farkas et al. 2007), 2VO was used to induce CCH in the present study. The nuclear expression of endogenous neuroprotection-related transcription factors and the corresponding protective effects were measured after 2VO rats were exposed to EE.
Materials and Methods
Animals and Housing
The experiments were performed on 7-week-old male Wistar rats (Laboratory Animal Research Center of Wuhan University, Wuhan, China) that weighed 220–250 g at the beginning of the study. Forty-eight rats (four per cage) were group-housed in polypropylene cages (35 cm × 29 cm × 21 cm) in a controlled environment at 22 ± 2 °C with 55 ± 5 % humidity and maintained on 12-h light/dark cycle with lights on at 07:00 and ad libitum access to water and food. All animal procedures were performed with protocols that were designed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Ethics Committee of the Medical School of Wuhan University.
Model Establishment and Experimental Design
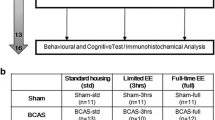
The animals were matched for body weight and randomly assigned to three groups in accordance with the random number table: (1) sham surgery plus a standard environment (sham + SE), n = 16; (2) 2VO surgery plus a standard environment (2VO + SE), n = 16; and (3) 2VO surgery plus an enriched environment (2VO + EE), n = 16.
2VO Animal Model
The 2VO surgeries were performed as previously described (Xu et al. 2010). Food and water were withheld for 1 day prior to surgery. The rats were anesthetized with 10 % chloral hydrate (350 mg/kg, intraperitoneal (i.p.)) and allowed to breathe spontaneously. The bilateral common carotid arteries of the rats were exposed via a midline ventral incision, carefully separated from the vagal nerves, and permanently ligated with silk sutures. The rats that underwent sham operations were treated in a similar manner with the exception that the common carotid arteries were not ligated. During surgery, body temperature was maintained at 37.5 ± 0.5 °C. After surgery, the wounds were sutured, and rats were allowed to recover from anesthesia before being returned to their cages.
Enriched Environment
The EE protocols that we employed were described by Fischer et al. (Fischer et al. 2007). Eight rats were housed together in a large specially designed cage (90 cm long × 75 cm wide × 45 cm high) that contained objects including toys, tunnels of different colors and shapes, running wheels, and wooden or metal stairs and platforms (Fig. 1). The objects were changed on a daily basis. For the SE groups, four animals were housed in each standard laboratory cage, and each cage contained sawdust bedding but no additional objects. Food and water were available ad libitum to both groups during the period of enrichment.

Enriched environment example in our experiment
After surgery, all rats were housed in the standard environment (SE) with free access to food and water for 4 weeks. During the subsequent 4 weeks, the 2VO + EE animals were exposed to an EE (6 h each day from 10:00 to 16:00), whereas the sham + SE and 2VO + SE animals remained in the SE.
The spatial learning and memory performances of the rats were evaluated on the days after the environmental treatment periods via the Morris water maze (MWM) test. The rats were subsequently sacrificed. Samples for Western blot assays and histopathological and immunohistochemical analyses were obtained from the brains of the rats.
MWM Test
The MWM consisted of a black circular pool of 2.0 m in diameter that was filled to a depth of 32 cm with opaque water at a temperature of 20 ± 1 °C. For the cued training (four trials per day for the first 5 days), the rats were released into the water facing the sidewalls at four different locations, and each rat was allowed 60 s to find the platform. If the rat did not find the platform, it was guided to the platform and allowed to rest there for at least 20 s. The escape latencies were recorded during the 5-day cued training. After the last cued trial of day 5, the platform was removed from the pool, and each rat received one 30-s swim probe trial on day 6; the time spent in the target quadrant in which the platform had previously been located during training was recorded. All data were acquired via a computerized video imaging analysis system.
Western Blotting
After behavioral testing, rats were anesthetized as described above (i.e., 10 % chloral hydrate solution, 350 mg/kg, i.p.), and the brains were rapidly removed and dissected to obtain the hippocampi. The tissue was immediately frozen in liquid nitrogen and stored at −80 °C until Western blotting. Nuclear proteins were extracted from the hippocampus using a kit from the Keygen Institute of Biotechnology according to the manufacturer’s instructions (Keygen, Nanjing, China). Protein samples (40 μg) were fractionated with 10 % or 12 % SDS-PAGE, transferred to 0.45-μm PVDF membranes at 200 mA for 1 h, and stained with Ponceau red to confirm equal loading. The blots were blocked with 5 % nonfat milk in Tris-buffered saline containing 0.1 % Tween-20 (TBST) at room temperature for 1 h. The membranes were then incubated with primary antibodies at 4 °C overnight. Anti-CREB (Millipore Chemicon International, Temecula, CA, USA, 1:1000 dilution), anti-phospho-CREB (Millipore, Billerica, MA, USA, 1:1000 dilution), anti-HIF-1α (BD Transduction Laboratories, San Diego, CA, USA, 1:1000 dilution), anti-Nrf2 antibody (Abcam, Cambridge, MA, USA, 1:1000 dilution), anti-acetyl histone H4 (Lys5/8/12/16, Millipore Corporation, Billerica, MA, USA, 1:2000 dilution), anti-acetyl Histone H3 (Lys14, Millipore Corporation, Billerica, MA, USA, 1:2000 dilution), and anti-β-actin(Santa Cruz, Dallas, TX, USA, 1:2000 dilution) were used. The blots were visualized via chemiluminescence (SuperSignal West Pico, Pierce, Rockford, IL, USA) after a 30-min incubation in the peroxidase-conjugated secondary antibody (anti-mouse: 1:3000 in TBST, KPL, Gaithersburg, MD, USA). Semiquantifications of the immunoreactive bands on the X-ray films were performed via optical density analyses (HPIAS 2000, Tongji Qianping Company, Wuhan, China). The densities were normalized to β-actin.
Histopathology and Immunohistochemistry
The rats were anesthetized and perfused transcardially with 4 % buffered PFA at pH 7.4. The brain was removed, stored in fixative for at least 24 h, embedded in paraffin wax, and coronally sectioned at a thickness of 4 μm. The sections were subjected to histologic examination using a standard procedure previously been described (Shibata et al. 2004). Klüver–Barrera (KB) staining was used to observe any histological changes. The white matter (WM) lesions were graded as normal (grade 0), containing disarranged nerve fibers (grade 1), exhibiting the formation of marked vacuoles (grade 2), and exhibiting the disappearance of myelinated fibers (grade 3) by an investigator who was blind to the experimental conditions. The primary antibodies used for immunohistochemistry included those directed against the following: the astrocyte marker glial fibrillary acidic protein (GFAP; GeneTex, Irvine, CA, USA, 1:250 dilution); the microglial marker ionized calcium-binding adapter molecule 1 (Iba-1; Abcam, Cambridge, MA, USA, 1:100 dilution), and the lipid peroxidation marker 4-hydroxynonenal (4-HNE; Millipore Chemicon International, Temecula, CA, USA, 1:500 dilution). After blocking, the sections were incubated with the primary antibodies overnight at 4 °C, and the brain slices were washed and incubated with the secondary antibodies for 1 h at room temperature. The reaction products were developed using a 50-μl diaminobenzidine solution. As a negative control, similar sets of sections were stained in the absence of primary antibody. In the sections that were immunostained for GFAP or Iba-1, the numbers of immunopositive cells were counted in three visual fields (cells/mm2) in the cerebral cortex; corpus callosum; and the CA1, CA2, CA3, and DG subregions of the hippocampus at 100× magnification using the Image-Pro Plus 6.0 image analysis software. Additionally, the mean integral optical density (IOD) values of the cells that were positively immunostained for 4-HNE were obtained. First, we selected five slices that contained the largest differences in staining intensity and then analyzed them according to the manual to select the most appropriate parameters to ensure the inclusion of the maximal number of hippocampal pyramidal cells and the minimal number of other background cells. Next, we used these specific parameters to reanalyze all of the stained slices. GraphPad Prism 5.0 software was used to analyze the differences between groups.
Statistical Analyses
All results are presented as the mean ± the SEM. The statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad Software, Inc., USA). Differences in escape latencies were analyzed with a two-way repeated measures ANOVA, and Bonferroni posttests were used for post hoc multiple treatment comparisons. Kruskal–Wallis test followed by Dunn’s test was used to compare the severity of WM lesions between the groups. All other measurements were analyzed using one-way ANOVAs followed by Bonferroni post hoc tests. Statistical significance was defined as p < 0.05.
Results
EE Ameliorated the Spatial Learning and Memory Deficits Induced by CCH
The rats were subjected to five consecutive days of training in the MWM to investigate their spatial learning abilities following 4 weeks of EE or SE treatment. The escape latency decreased significantly during training (F(4,113) = 42.88, p < 0.01), and there was a significant difference among the three groups (F(2,113) = 19.41, p < 0.01; Fig. 2a). The Bonferroni posttest indicated that beginning on day 2, rats in the CCH group (2VO + SE) took significantly longer to find the platform compare to the sham + SE rats (days 2 and 5, p < 0.05; days 3–4, p < 0.01). The hypoperfused rats treated with EE (2VO + EE rats) took significantly less time to find the platform than did their 2VO + SE counterparts on days 3–5 (days 3 and 5, p < 0.05; day 4, p < 0.01).

The effects of EE on CCH-induced deficits in spatial learning and memory as measured with the Morris water maze. a Mean daily escape latencies (i.e., times from the starting to the hidden platform). b Times spent in the target quadrant during the probe trials. c Time spent in each quadrant during the probe trials. All values are expressed as the mean ± the SEM (n = 10–11). *p < 0.05, **p < 0.01 compared to the sham + SE group; #p < 0.05 compared to the 2VO + SE group
As shown in Fig. 2b, in the probe trials after the platform was removed, memory was evaluated by measuring the time spent in the target quadrant. We observed a significant difference in time spent in the target quadrant among the three groups (F(2,18) = 10.02, p < 0.01), and Bonferroni posttest indicated that the CCH rats (2VO + SE) spent less time in the platform region than did the sham + SE rats (p < 0.01). EE significantly increased the amount of time that the hypoperfusion rats remained in the platform region relative to the SE group of hypoperfusion rats (2VO + SE rats; p < 0.05).
EE Regulated the Nuclear Protein Levels of Transcription Factors That Are Related to Endogenous Neuroprotection
Western blotting analyses were used to examine the nuclear protein levels of target transcription factors (CREB/phospho-CREB (p-CREB), Nrf2, HIF-1α) in the hippocampus (Fig. 3). As shown in Fig. 3b, the level of p-CREB in the nucleus decreased following 2VO surgery (p < 0.05), and this level was restored following EE treatment (p < 0.05). The expression of CREB exhibited a similar trend between these two groups, but this trend was not statistically significant (p > 0.05, Fig. 3a). No difference in Nrf2 was observed after 2VO surgery (p > 0.05), but EE treatment significantly increased Nrf2 nuclear translocation in the hippocampus (p < 0.05; Fig. 3c). Additionally, the CCH induced by 2VO led to increased levels of HIF-1α in the nuclei of the hippocampus relative to the levels observed in the sham group (p < 0.01; Fig. 3d), and this level was reduced after EE treatment, although the latter difference was not significant (p > 0.05; Fig. 3d).

The effect of EE on the nuclear protein levels of endogenous neuroprotection-related transcription factors in the hippocampus following 2VO surgery. The results of Western blot analyses of the transcription factors (i.e., CREB/p-CREB, Nrf2, and HIF-1α) are shown in a–d, respectively. The images are representative immunoblotting bands of the transcription factors and β-actin. The histograms represent the quantitative results for CREB, p-CREB, Nrf2, and HIF-1α. The densities were normalized to β-actin, and the bars in a–d represent the mean relative optical densities ± the SEMs (n = 6). *p < 0.05, **p < 0.01 compared to the sham + SE group; #p < 0.05 compared to the 2VO + SE group
The Effect of EE on Histone Acetylation
Histone acetylation results in the alteration of chromatin into a more open and relaxed conformation that facilitates the interaction of transcription factors with specific gene promoters and, thus, the activation of the expression of downstream genes. The levels of acetylation of histone 3 at lysine 14 (H3K14) and histone 4 at lysines 5, 8, 12, and 16 (H4K5, H4K8, H4K12, H4K16) in the hippocampi after 2VO and EE treatment were examined by nuclear protein Western blotting (Fig. 4).

The effect of EE on histone acetylation in the hippocampus. Western blot analyses of the levels of histone acetylation. The images are representative immunoblotting bands for Ac-H3 (H3K14), Ac-H4 (H4K5, H4K8, H4K12, and H4K16), and β-actin. The histograms represent the quantitative results for Ac-H3 (a) and Ac-H4 (b). The densities were normalized to β-actin, and the bars in a, b represent the mean relative optical densities ± the SEMs (n = 6). #p < 0.05 compared to the 2VO + SE group
The results indicated that 2VO led to reduced histone 3 (H3K14) and histone 4 (H4K5, H4K8, H4K12, H4K16) acetylation, but these differences were not significantly different (p > 0.05; Fig. 4a, b). EE restored the levels of H4K5, H4K8, H4K12, and H4K16 acetylation (p < 0.01; Fig. 4b) but did not significantly affect the acetylation of H3K14 (p > 0.05; Fig. 4a).
EE Attenuated the CCH-Induced Oxidative Damage
The 4-HNE-modified protein can be used as a marker of oxidative neuronal damage (Calkins et al. 2009). The results of immunohistochemical analyses of 4-HNE in the cerebral cortex, corpus callosum, and hippocampus are shown in Fig. 5. After 2VO, there were significant increases in the 4-HNE levels in the cortex and the four subregions of the hippocampus. EE treatment reduced the levels of 4-HNE in the CA1, CA2, and CA3 subregions of the hippocampus (p < 0.05; Fig. 5b). No significant change was observed in the corpus callosum following 2VO and EE treatment (p > 0.05; Fig. 5b).

The effects of EE on the lipid peroxidation induced by chronic cerebral hypoperfusion. a Representative photomicrographs of 4-HNE immunohistochemistry in the corpus callosum (a–c), cerebral cortex (d–e), and four subregions of the hippocampus (g–i; j–l; m–o; p–r) after the sham operation and the 2VO surgeries with and without EE treatment. The arrows show the positive cells (dyed brown). The images are 100×. b The histograms represent the quantitative results of 4-HNE immunohistochemistry. The bars represent the mean IODs ± the SEMs (n = 4). *p < 0.05, **p < 0.01 compared to the sham + SE group; #p < 0.05 compared to the 2VO + SE group
EE Treatment Protected Against the Damage to the WM, Cortex, and Hippocampus
WM lesions and glial activation were measured with KB and immunohistochemical staining.
KB staining revealed that 2VO led to WM lesion in the corpus callosum (p < 0.05; Fig. 6a, d). After EE treatment, the severities of the WM lesions were reduced, although this difference was not significant (p > 0.05; Fig. 6a, d).



The effects of EE on CCH-induced damage to the WM, cortex, and hippocampus. a Representative photomicrographs of Klüver–Barrera (KB) staining in the corpus callosum. The images are 100×. b, c Representative photomicrographs of GFAP and Iba-1 immunohistochemistry in the corpus callosum, cerebral cortex, and four subregions of hippocampus after sham surgery and the 2VO surgeries with and without EE treatment. The arrows show the positive cells (dyed brown). The images are 100×. d Histograms showing the grading scores for the corpus callosum after Klüver–Barrera staining. e, f Histograms showing the densities of the cells that were immunoreactive for GFAP and Iba-1 (/mm2). The bars represent the mean ± the SEM (n = 4). *p < 0.05, **p < 0.01 compared to the sham + SE group; #p < 0.05 compared to the 2VO + SE group
Immunohistochemical staining revealed increased densities of glial fibrillary acidic protein (GFAP)-positive astrocytes in the corpus callosum and hippocampus (CA1, CA3, and DG subregions) after 2VO (p < 0.05 and p < 0.01; Fig. 6b, e), and EE treatment significantly decreased the densities of astrocytes in the corpus callosum and hippocampus (CA1 and DG subregions; p < 0.05). Similarly, 2VO led to increased numbers of ionized calcium-binding adapter molecule 1 (Iba-1)-positive microglia in the cortex and hippocampus (CA1, CA3, and DG subregions; p < 0.05 and p < 0.01; Fig. 6c, f), and the densities of microglia were reduced after EE in the cortex and CA3 subregion (p < 0.05).
Thus, 2VO-induced WM lesions were restored following EE treatment, and this restoration was paralleled by glial activation in the brain.
Discussion
In this study, we confirmed that EE was capable of reversing the spatial learning and memory impairments induced by CCH in rats as we have demonstrated previously (Sun et al. 2010; Zhang et al. 2013). We also revealed that this reversal was accompanied by the activations of some transcription factors and that the induced downstream neuroprotective reactions included the preservation of WM integrity, alleviation of oxidative damage, and inhibition of glial activation in the brain.
CREB is expressed in all cells in the brain and is a member of a family of proteins that function as transcription factors. There is strong evidence that CREB and the CREB-mediated system are involved in learning, memory, and neuronal survival and differentiation (Kitagawa 2007). Activation of CREB leads to the expression of genes that promote cell survival and synaptic plasticity, such as the neurotrophic factor brain-derived neurotrophic factor (BDNF), which plays pivotal roles in synaptic plasticity and neurogenesis, and this activation is also involved in mechanisms that are protective against excitotoxic, oxidative, and metabolic stress (Cheng and Mattson 1994; Spina et al. 1992). Here, we found that neither CCH nor EE treatment altered the nuclear level of CREB protein, but these treatments did affect CREB activation. As shown in the results, CCH reduced the expression of p-CREB, and this effect was rescued by exposure to EE. Consistently, a previous study also reported that the level of the CREB-mediated gene BDNF is restored by EE treatment after CCH (Sun et al. 2010). Thus, we speculate that EE activates CREB and the CREB-mediated system and that this activation might contribute to the neuroprotection and cognitive improvement that are associated with EE.
Nrf2 is a key transcription factor that modulates the anti-oxidative ability of brain and mediates the production of a battery of endogenous anti-oxidative enzymes (Calkins et al. 2009). The Nrf2 antioxidant response pathway is known as the primary cellular defense against the cytotoxic effects of oxidative stress (Yang et al. 2014). Moreover, oxidative stress has been regarded as a common pathophysiological change and an essential factor in the development of CCH-related cognitive impairment (Liu and Zhang 2012). Consistently, in present research, increased lipid peroxidation was observed in the cortex and hippocampus after 2VO. EE reduced the level of 4-HNE and alleviated the oxidative damage in the brain, which might have been related to the activated Nrf2 after EE treatment.
The hypoxia-inducible factor 1 (HIF-1)-mediated signaling pathway, which is activated by hypoxia and ischemia, controls a diverse range of adaptive and protective cellular processes including angiogenesis, erythropoiesis, and cellular metabolism that is targeted at increasing oxygen delivery to tissues (Karuppagounder and Ratan 2012). HIF-1 is a transcriptional complex consisting of HIF-1α and HIF-1β subunits. Under normoxic conditions, HIF-1α is hydroxylated and rapidly degraded. Under hypoxic–ischemic conditions, hydroxylases are less active; thus, the HIF-1 complex accumulates and translocates to the nucleus, which results in the transcription of HIF-1 target genes, which, in turn, promotes neuronal adaptation for survival under hypoxic conditions (Benarroch 2009). We found that HIF-1α, the functional subunit of HIF-1, was increased in the hippocampus after 2VO. Our previous study showed that the CCH induced by 2VO triggers a steady increase in HIF-1α levels in the hippocampus that begins as early as 12 h after the operation and continues until at least 56 days postoperation (Yang et al. 2013). These findings indicate that the compensatory or endogenous adaptive response that defends against hypoperfusion might be activated after 2VO. However, no increase in HIF-1α level was observed after EE treatment. Therefore, whether EE affects the activation of HIF-1 is unclear. Four weeks of EE treatment might have alleviated the hypoxic–ischemic conditions in the brains of the 2VO rats, which might have caused the increases in HIF-1α degradation. This supposition might explain why no increases in HIF-1α were detected 4 weeks after EE compared to the 2VO + SE rats in this study. Measurements of the changes in HIF-1α levels at different time points after EE treatment might aid the identification of the exact role of EE in the HIF-1-mediated signaling pathway.
Additionally, histone acetylation, which has been implicated in the facilitation of the interaction of transcription factors with specific gene promoters and the activation of downstream gene expression via the induction of more open and relaxed chromatin conformation, has recently been implicated in synaptic plasticity and learning behavior (Chuang et al. 2009). Consistent with the changes observed in some transcription factors, such as p-CREB and Nrf2, we found that EE upregulated the acetylation of histone 4 (i.e., H4K5, H4K8, H4K12, H4K16) in the hippocampi of the 2VO rats. A previous study reported a similar result; EE was found to increase the levels of hippocampal and cortical acetylation of histones 3 and 4 as soon as 3 h after treatment in a mouse model of neurodegenerative disease and to induce recovery from impaired learning and recovery of lost memories (Fischer et al. 2007). Moreover, the increments in histone acetylation that are caused by inhibitors of histone deacetylases might reinstate learning and memory (Fischer et al. 2007). Because the alterations to chromatin structure that are induced by histone acetylation might favor transcriptional initiation of various transcription factors (Fig. 7), Vecsey et al. suggested that the enhancement of memory-mediated histone deacetylase inhibitors is due to the activation of key genes that are regulated by the CREB/CBP transcriptional complex (Vecsey et al. 2007). These results suggest that the role of histone acetylation in the promotion of the transcriptional initiation of transcription factors might also be related to the cognitive improvements and neuroprotection induced by EE in the 2VO rats.

Transcription factor pathways affected by histone acetylation. Histone acetylation could transform the condensed chromatin into a more relaxed structure that is associated with greater levels of gene transcription. The alterations to chromatin structure that are induced by histone acetylation might favor transcriptional initiation of various transcription factors (TFs), including neuroprotective-related TFs CREB, HIF-1α, and Nrf2. Furthermore, histone acetyltransferases (HATs), enzymes that acetylate lysine residues on nucleosomal histones, also regulate the activity of certain TFs
Thus, this study demonstrated that the oxidative damage and histopathological damage to the brain that are induced by 2VO were alleviated by EE treatment in parallel with the regulation of histone acetylation and some neuroprotection-related transcription factors. All of these neuroprotective effects of EE in CCH rats might contribute to the recovery of spatial learning and memory. Thus, we speculate that the cognitive improvement induced by EE in CCH rats might result from the upregulation of endogenous neuroprotection. The recruitment of endogenous neuroprotective pathways might represent a possible target for therapeutic interventions for CCH related diseases, such as AD and VaD.
Abbreviations
- EE:
-
Environment enrichment
- CCH:
-
Chronic cerebral hypoperfusion
- 2VO:
-
2-Vessel occlusion
- AD:
-
Alzheimer’s disease
- VaD:
-
Vascular dementia
- CREB:
-
cAMP response element-binding protein
- p-CREB:
-
Phospho-CREB
- Nrf2:
-
Nuclear regulatory factor 2
- HIF-1α:
-
Hypoxia-inducible factor 1α
- GFAP:
-
Glial fibrillary acidic protein
- Iba-1:
-
Ionized calcium binding adapter molecule 1 (a marker of microglia)
- 4-HNE:
-
4-Hydroxynonenal
References
Benarroch EE (2009) Hypoxia-induced mediators and neurologic disease. Neurology 73:560–565
Calkins MJ, Johnson DA, Townsend JA, Vargas MR, Dowell JA, Williamson TP, Kraft AD, Lee JM, Li J, Johnson JA (2009) The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid Redox Signal 11:497–508
Cechetti F, Worm PV, Lovatel G, Moysés F, Siqueira IR, Netto CA (2012) Environmental enrichment prevents behavioral deficits and oxidative stress caused by chronic cerebral hypoperfusion in the rat. Life Sci 91:29–36
Cheng B, Mattson MP (1994) NT-3 and BDNF protect CNS neurons against metabolic/excitotoxic insults. Brain Res 640:56–67
Choy M, Ganesan V, Thomas DL, Thornton JS, Proctor E, King MD, van derWeerd L, Gadian DG, Lythgoe MF (2006) The chronic vascular and haemodynamic response after permanent bilateral common carotid occlusion in newborn and adult rats. J Cereb Blood Flow Metab 26:1066–1075
Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci 32:591–601
de la Torre JC (2002) Vascular basis of Alzheimer’s pathogenesis. Ann N Y Acad Sci 977:196–215
Farkas E, Luiten PG, Bari F (2007) Permanent, bilateral common carotid artery occlusion in the rat: a model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res Rev 54:162–180
Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH (2007) Recovery of learning and memory is associated with chromatin remodelling. Nature 447:178–182
Gobbo OL, O’Mara SM (2004) Impact of enriched-environment housing on brain-derived neurotrophic factor and on cognitive performance after a transient global ischemia. Behav Brain Res 152:231–241
Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC et al (2011) Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42:2672–2713
Herring A, Blome M, Ambree O, Sachser N, Paulus W, Keyvani K (2010) Reduction of cerebral oxidative stress following environmental enrichment in mice with Alzheimer-like pathology. Brain Pathol 20:166–175
Karuppagounder SS, Ratan RR (2012) Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J Cereb Blood Flow Metab 32:1347–1361
Kitagawa K (2007) CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J 274:3210–3217
Liu H, Zhang J (2012) Cerebral hypoperfusion and cognitive impairment: the pathogenic role of vascular oxidative stress. Int J Neurosci 122:494–499
Nithianantharajah J, Hannan AJ (2006) Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci 7:697–709
Osawa A, Maeshima S, Shimamoto Y, Maeshima E, Sekiguchi E, Kakishita K, Ozaki F, Moriwaki H (2004) Relationship between cognitive function and regional cerebral blood low in different types of dementia. Disabil Rehabil 26:739–745
Ruitenberg A, denHeijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM (2005) Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam study. Ann Neurol 57:789–794
Shibata M, Ohtani R, Ihara M, Tomimoto H (2004) White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 35:2598–2603
Spina MB, Squinto SP, Miller J, Lindsay RM, Hyman C (1992) Brain-derived neurotrophic factor protects dopamine neurons against 6-hydroxydopamine and N-methyl-4- phenylpyridinium ion toxicity: involvement of the glutathione system. J Neurochem 59:99–106
Stranaha AM, Lee K, Becker KG, Zhang Y, Maudsley S, Martin B, Cutler RG, Mattson MP (2010) Hippocampal gene expression patterns underlying the enhancement of memory by running in aged mice. Neurobiol Aging 31:1937–1949
Stranahan AM, Mattson MP (2012) Recruiting adaptive cellular stress responses for successful brain ageing. Nature 13:209–216
Sun H, Zhang J, Zhang L, Liu H, Zhu H, Yang Y (2010) Environmental enrichment influences BDNF and NR1 levels in the hippocampus and restores cognitive impairment in chronic cerebral hypoperfused rats. Curr Neurovasc Res 7:268–280
Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA (2007) Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci 27:6128–6140
Xu Y, Zhang JJ, Xiong L, Zhang L, Sun D, Liu H (2010) Green tea polyphenols inhibit cognitive impairment induced by chronic cerebral hypoperfusion via modulating oxidative stress. J Nutr Biochem 21:741–748
Yang Y, Zhang J, Liu H, Wang J, Xin J, Deng M (2013) Changes in levels of hypoxia-induced mediators in rat hippocampus during chronic cerebral hypoperfusion. Neurochem Res 38:2433–2439
Yang Y, Zhang J, Liu H, Zhang L (2014) Change of Nrf2 expression in rat hippocampus in a model of chronic cerebral hypoperfusion. Int J Neurosci 124:577–584
Zhang L, Zhang J, Sun H, Zhu H, Liu H, Yang Y (2013) An enriched environment elevates corticosteroid receptor levels in the hippocampus and restores cognitive function in a rat model of chronic cerebral hypoperfusion. Pharmacol Biochem Behav 103:693–700
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant number: 81171029 and 81000471)
Conflict of Interest
The authors declared that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ying Yang and Li Xiong contributed equally to this work.
Rights and permissions
About this article

Cite this article
Yang, Y., Zhang, J., Xiong, L. et al. Cognitive Improvement Induced by Environment Enrichment in Chronic Cerebral Hypoperfusion Rats: a Result of Upregulated Endogenous Neuroprotection?. J Mol Neurosci 56, 278–289 (2015). https://doi.org/10.1007/s12031-015-0529-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-015-0529-2