
Abstract
Astrocyte glucose metabolism functions to maintain brain activity in both normal and stress conditions. Dysregulation of astrocyte glucose metabolism relates to development of neuronal disease, such as multiple sclerosis and Alzheimer’s disease. In response to acute stress, beta2-adrenergic receptor is activated and initiates multiple signaling events mediated by Gs, Gi, arrestin, or other effectors depending on specific cellular contexts. In astrocytes, beta2-adrenergic receptor promotes glucose uptake through GLUT1 and accelerates glycogen degradation via coupling to Gs and second messenger cAMP-dependent pathway. Beta2-adrenergic receptor may regulate other steps in astrocyte glucose metabolism, such as lactate production or transduction. Inappropriate regulation of beta2-adrenergic receptor activity can disrupt normal glucose metabolism, and leads to accelerate neuronal disease development. It was demonstrated that the absence of beta2-adrenergic receptor in astrocytes occurred in multiple sclerosis patients, and the increased beta2-adrenergic receptor activity relates to Alzheimer’s disease. A clear view of beta2-adrenergic receptor-mediated signaling pathways in regulating astrocyte glucose metabolism could help us to develop neuronal diseases treatment by targeting to the beta2-adrenergic receptor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Astrocytes are star-shaped brain glial cells with important functions including physical structuring of the brain, blood–brain barrier formation, modulation of synaptic transmission, and nutrient support to adjacent neurons. Residing close to microvasculature and neuronal axons, astrocytes are poised ideally to supply axons with metabolic intermediates, such as glutamate, GABA, and probably lactates, which are essential for neuron function under normal or stress conditions. In resting state, the brain consumes 20 % of total body oxygen in contrast to its 2 % relative weight, suggesting the importance of oxidative phosphorylation in glucose metabolism of both astrocytes and neurons (Genc et al. 2011). However, in response to detrimental factor or urgent situation, such as hypoxia condition with reduced oxygen in blood, astrocytes can also switch to anaerobic pathway and produce then transfer lactate to around axons. Both oxidative and aerobic glucose metabolism in astrocytes are tightly regulated by concerted actions of neuronal transmitter, nutrient, and other factors, and dysfunction of glucose metabolism regulation may lead to development or acceleration of many neuronal diseases, such as multiple sclerosis, Parkinson’s disease, and Alzheimer’s disease (AD, Hunt et al. 2007; Laureys et al. 2010).
Beta2-adrenergic receptor (β2AR) is one of the major receptors of endogenous “fight or flight response hormone,” norepinepherine, which regulates multiple steps of glucose metabolism in astrocytes (Cohen et al. 1997; Carnevale et al. 2007). Numerous studies have shown expression of beta-adrenergic receptor in both gray and white matters by radioligand binding and immunohistochemistry, in which β2AR is the dominant component of human white matter astrocytes (Aoki 1992; Sutin and Shao 1992; Zeinstra et al. 2000). A recent research with specific receptor subtype knockout mice, with radio ligand binding together with RT-PCR, also demonstrated expression of all three types of beta-adrenergic receptors in astrocytes (Liu et al. 1992; Salm and McCarthy 1992; Shao and Sutin 1992; Mantyh et al. 1995; Zeinstra et al. 2000; Catus et al. 2011).
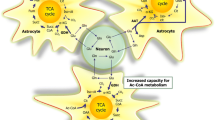
Being the first radiolabeled and cloned other than rhodopsin, β2AR is the best characterized G protein-coupled receptor in signaling and biochemical studies, which is highlighted by recent advanced works such as crystal structure of β2AR–Gs protein complexes, multiple conformations of β2AR with different ligands, and its numerous downstream signaling at cellular level (Robishaw et al. 1986; Daaka et al. 1997; Hall et al. 1998; Xiao et al. 2010; Hara et al. 2011; Kahsai et al. 2011; Kobilka 2011; Rasmussen et al. 2011). Generally, with endogenous ligand norepinepherine stimulation, β2AR primarily couples to Gs proteins, successively activates adenylyl cyclase, and produces cAMP (Robishaw et al. 1986). The increased cellular concentration of cAMP functions as the “second messenger”; activates cascade connection, such as PKA and CREB; and results in a “first wave” of biological effects (Fig. 1). The activated PKA conversely phosphorylates β2AR, uncoupled it from Gs then switches the receptor to couple to Gi (Daaka et al. 1997; Xiao et al. 1999). The switching mechanism of β2AR from Gs to Gi during persistent agonist stimulation has important anti-apoptotic effects in cardioprotection (Zhu et al. 2001; Xiao et al. 2006).

Signaling cascade by beta2 adrenergic receptor and its regulation on astrocyte glucose metabolism
Concurrent with its phosphorylation by PKA, β2AR is also phosphorylated by one or two specific GRKs on its c-terminal or intracellular third loop depending on its specific cellular contexts (Wilkins and Scolding 2008). The phosphorylated receptor recruits beta-arrestins, terminates Gs signaling at one side, and initiates a “second wave” of signaling by scaffolding different downstream molecules, such as Src, ERK, and PDE (Lin et al. 1999; Luttrell et al. 1999; Perry et al. 2002; Nelson et al. 2008). The arrestin-mediated β2AR signaling regulates multiple cellular activities, which is signified by recent research in β2AR regulation of DNA damage in stress response pathway (Hara et al. 2011). Unless general G or arrestin mediated pathway for all GPCRs, it is also reported that specific downstream signals are initiated by β2AR in specific physiological conditions. β2AR can directly interact with NHERF through its c-terminal, regulating Na+/H+ exchange (Hall et al. 1998). In recent research of Alzheimer’s disease, activated β2AR interacts with presenilin-1, stimulates γ-secretase activity, and accelerates amyloid plaque formation (Ni et al. 2006). Thus, as the most important endocrine receptor, β2AR may regulate distinct cellular functions due to variation in the abundance of its downstream effectors in specific organs or cell types. In this minireview, we will examine current knowledge of β2AR function and in regulating astrocyte glucose metabolism and its relevance to neuronal disease, which may shed light for future studies.
β2AR Regulate Glucose Transport Through GLUT1
Glucose is regarded as the main carbon source of brain energy metabolism. During rest conditions astrocytes and neuron uptake glucose from the blood with similar rate. After being taken up by astrocytes, glucose can be metabolized through glycolysis or be stored via glycogen synthesis. Once there is intense neuronal activity or stress signal, astrocytes increase glucose uptake rate while the neuron does not change, suggesting a tight regulation of glucose transport in astrocytes (Pellerin et al. 2007; Chuquet et al. 2010).
β2AR first regulates glucose transport from the blood vessel cells into astrocytes (Fig. 1). As early as 1990, Hsu et al. observe a marked increase of 14C-labeled glucose transport induced by beta2 and beta3 adrenergic receptor agonist isoproterenol after 30 min incubation, accompanied with an activation of adenylyl cyclase (Hsu and Hsu 1990). Further studies with specific β2AR agonist zinterol and beta3 adrenergic receptor agonist CL316243 revealed that beta3 adrenergic receptor regulates early time points while β2AR functions at later time of glucose transport (Sato et al. 2007; Gibbs et al. 2008). The fact that two adenylate cyclase inhibitors DDA and SQ22536 block β2AR-mediated glucose transport suggests that β2AR regulates glucose transport through coupling to Gs and activation of adenylate cyclase pathway. Just recently Catus et al. did a thorough research on three beta-adrenergic receptor functions in regulation of glucose transport with subtype specific receptor knockout animals. They demonstrated that activation of beta2 receptor upregulated glucose transport (Catus et al. 2011). Application of GLUT inhibitor cytochalasin B blocks beta-adrenergic receptor-mediated glucose transport, suggesting that β2AR likely regulate glucose transport through directly affecting GLUT1 activity. Neurons express GLUT3, while astrocytes mostly express GLUT1 (Pellerin et al. 2007; Genc et al. 2011). This glucose subtype specificity determines their different regulatory property, which may explain the glucose uptake rate change occurring in astrocytes but not neurons under stress. Whether do β2AR regulate GLUT1 function through phosphorylation, membrane anchoring, or protein expression needs further scrutinized inspection.
β2AR in Astrocyte Glycogenesis and Lactate Metabolism
Astrocytes are the primary glycogen stores in the brain, and 40 % glucose taken up by astrocytes enters into glycogen synthesis (Prebil et al. 2011), which is majorly promoted by alpha adrenergic receptor (Hertz and Gibbs 2009; Hutchinson et al. 2011) in physiological conditions. Neurons can also synthesize glycogen while this activity is inhibited to prevent the harmful effects of glycogen to these cells. Thus, astrocyte glycogen is the only glycogen source in the brain and is necessary to maintain neuronal activity in case of energy deprivation, hypoxia, ischemia, or too much neuronal activity. With a low blood glucose concentration or stress condition, glycogen stored in astrocytes undergoes glycogenlysis in response to glucagon or epinephrine, not only providing energy for it own consumption but also generating glucose or lactate to supply peripheral axon demand. Binding of β2AR in astrocytes either by norepinephrine or isoproterenol leads to coupling the receptor to Gs and activation of adenylyl cyclase (Fig. 1). The activated adenylyl cyclase increases intracellular cAMP and promotes glycogenlysis through a PKA-mediated phosphorylation cascade (Brown et al. 2003, 2005; Dienel et al. 2007). The PKA activates phosphorylase kinase, which, in turn, phosphorylates glycogen phosphorylase b at Ser14, rearranges residues region 10-22 into a stable alpha helical conformation, increases phosphorylase activity up to 25 %, and enhances further AMP activation. The activated phosphorylase cleaves glycogen at α-1-4 position and substitutes with a phosphoryl group, generating glucose-1-phosphate (G1P). The conversion of phosphorylase b to phosphorylase a by PKA controls the rate-limiting step of glycogen degradation to monomers (Johnson et al. 1978; Sorg and Magistretti 1991; Fillenz et al. 1999; Magistretti and Pellerin 1999; Allaman et al. 2000; Wender et al. 2000; Zaccolo et al. 2006; Brown and Ransom 2007; Walls et al. 2009).
G1P is isomerized to glucose-6 phosphate (G6P) by phosphoglucomutase. Most of G6P are converted to pyruvate via glycolysis as the concentration of glucose-6-phosphatase is quite low in astrocytes (Dringen and Hamprecht 1993; Magistretti and Pellerin 1999). Pyruvate can either go through oxidative phosphorylation in the mitochondria to provide ATP for astrocytes’ usage or be converted to lactate through anaerobic metabolism. The significance of anaerobic metabolism as energy supply is highlighted by the fact that 50 % glucose versus 5 % oxygen increase is taken up by astrocytes under stress condition (Fox et al. 1988). Astrocytes express specific LDH5 which prefers converting pyruvate to acetate whereas neurons only have LDH1 that favors the reverse reaction. There is an astrocyte–neuron lactate shuttle hypothesis (ANLSH model) that lactate generated by astrocytes can be provided to adjacent neurons for emergent demands. Although several computational studies questioned the efficiency of the glucose utilization in the process, experimental and computational evidence supports the functional importance of the hypothesis (Jolivet et al. 2010; Mangia et al. 2011; Bouzier et al. 1998; Genc et al. 2011; Newman et al. 2011). For example, exogenous lactate serves as the main substrate for C6 glioma cell oxidative metabolism monitored by NMR (Bouzier et al. 1998); the MCT inhibitor that blocks the lactate transport impairs learning memory and in silico preference of ANLSH model in hypoxia-induced condition (Bouzier et al. 1998; Genc et al. 2011; Newman et al. 2011).
β2AR has been identified to regulate anaerobic metabolism and lactate production. With human exercise test, it is found that infusion of epinephrine increases blood lactate concentration. The nonselective beta-adrenergic receptor blocker propranolol attenuates oxygen–carbohydrate index while beta1 adrenergic receptor antagonist metoprolol has no effect. These results suggest that β2AR not beta1 adrenergic receptor regulates this nonoxidative metabolism (Seifert et al. 2009). β2AR probably regulates lactate production by accelerating glycogenlysis, while it is also possible that it can directly regulate LDH or lactate transport. It is reported that both of β2AR agonists isoproterenol and clenbuterol elevate LDH4 and LDH5 expression in ventricular myocytes (Kaundal et al. 2007). Whether or not a similar effect of beta2 adrenergic receptor functions in regulation of astrocyte specific LDH expression is never examined. It is also not known whether LDH can be phosphorylated downstream of β2AR activation, which provide another level of potential β2AR regulatory mechanism in lactate production.
The lactate produced by astrocytes will be transported outside of the astrocytes through monocarboxylate transporters (MCTs). Astrocytes are abundant with MCT1 and MCT4 while neurons are enriched with MCT2. It is known that MCT1 and MCT4 prefer to releasing lactate outside while MCT2 prefer uptaking lactate, which supports the ANLSH model (Pellerin et al. 1998; Bergersen 2007; Genc et al. 2011). In neurons, norepinepherine stimulates MCT2 expression through PI3K/Akt pathway. Conversely, the same stimulation may also increase the expression of MCT1 or MCT4 in astrocytes through β2AR. Such hypothesis is waiting for further evidences (Chenal and Pellerin 2007).
Dysregulation of β2AR in Astrocyte Glucose Metabolism Relates to Development of Multiple Sclerosis and Alzheimer’s Disease
There are signs indicating the association of astrocyte glucose metabolism dysfunction with neuronal diseases, such as multiple sclerosis, Alzheimer’s disease and Parkinson’s disease(Steele and Robinson 2012; Alexander 2002; Freemantle et al. 2006; Maragakis and Rothstein 2006). As perhaps the most important neuronal transmitter receptor, β2AR regulates the transition of astrocytes from rest to active state to respond to acute stress, and to accomplish the fine-tuning metabolic interactions between astrocytes and around neurons. In most cases, astrocyte β2AR expression is upregulated in areas of the CNS or optic nerve injury (Mantyh et al. 1995; Hodges-Savola et al. 1996). However, clinical studies revealed that β2AR is absent in plaques and alba of multiple sclerosis patients’ postmortem brain sections compared with nonneurologic disease patients, through both immunohistochemistry and quantitative autoradiography with [3H]-labeled dihydroalprenolol (De Keyser et al. 1999; Zeinstra et al. 2000). This observation indicates that β2AR may involve in a rescuing mechanism during neuronal injury, and the lack of β2AR in astrocyte cells may cause or accelerate multiple sclerosis development.
Inflammatory cell infiltration, glial cell hyperplasia plaque formation, axon damage and loss are all involved in multiple sclerosis development (Frohman et al. 2005; Wilkins and Scolding 2008). β2AR has been extensively reviewed by several recent papers for its importance in immune inflammatory astrocyte responses (Laureys et al. 2010). Yet, recent research found that drugs suppressing the inflammatory response, such as IFN-beta and CD52, were unable to prevent chronic neurological damage in multiple sclerosis (Coles et al. 1999; Kidd et al. 1999; Confavreux et al. 2000). These results suggest that other mechanisms, such as glucose metabolism disorder, also play important roles in multiple sclerosis development. When hypoglycemia or nerve activity becomes strong, glycogenolysis in astrocytes plays as an important energy supplier for axons (Fillenz et al. 1999). Extending away from their cell bodies, axons depend on local production of ATP to maintain ion gradients and sustain energy supply. In the white matter, primary axonal energy metabolism takes place at abut of astrocytes and Ranvier nodes (Brown 2007). Lack of β2AR in multiple sclerosis patients causes disorder of astrocyte glucose transportation and glycogenlysis, and decreases the energy supply to axons. The energy deprivation of axons will decrease its ATP synthesis, yielding insufficient energy to fulfill Na+–K+ pump requirements. The failure of Na+–K+ pump finally leads to excessive Na+ influx and uncontrolled depolarizations, followed by the opening of voltage-sensitive Ca2+channels and reverse operation of the Na+–Ca2+ exchanger, namely intake Ca2+ and the discharge Na+. The rise in axonal Ca2+ then leads to microtubule breakdown and unwanted apoptosis (Gaskin et al. 1975; Ransom and Fern 1997; Stys and Jiang 2002). Thus deficiency of β2AR in astrocytes causes dysregulation of astrocyte glucose metabolism, and finally contributes to progressive axonal degeneration and multiple sclerosis development.
Dysregulation of glucose metabolism may also contribute to neuron degeneration of Alzheimer’s disease. Positron emission tomography studies demonstrate that glucose uptake is impaired in Alzheimer’s disease patients (Minoshima et al. 1994; Freemantle et al. 2006). The observation that disruption of glucose metabolism precedes the amyloid plaque formation suggests a relation of astrocytes glucose metabolism in AD development (Small et al. 2000).
As environmental factors such as acute stress are important risk factors of Alzheimer’s disease, there is no doubt that dysregulation of β2AR has important impact on Alzheimer’s disease development. It was initially identified that both β2AR and β1AR showed smaller but significant (25 %) increases in aggressive Alzheimer’s disease subjects versus both nonaggressive Alzheimer’s disease patients and controls (Russo-Neustadt and Cotman 1997). In neurons, activation of β2AR associates with presenilin-1, enhances γ-secretase activity, and promotes amyloid plaque formation (Ni et al. 2006). Conversely, amyloid beta can directly interact with β2AR, induces PKA-dependent AMPA receptor hyperactivity, and promotes β2AR internalization and degradation (Wang et al. 2010, 2011). All these results indicate that hyperreactive β2AR relates to AD and β2AR selective blockers have therapeutic potential for Alzheimer’s disease treatment. The concept is supported by a recent study that beneficial effects are seen from β2AR selective antagonist application with an induced acute stress mouse model (Yu et al. 2010).
The hyperreactive β2AR in neurons seems to contradict to the observation of hypometabolic glucose state in the brain of Alzheimer’s disease patients (Wang et al. 2011). A possible explanation is that acute stress induces significant β2AR downregulation in neurons and astrocytes. After activation, β2AR should be phosphorylated by GRKs or PKA, binding to beta arrestin and internalized. Some internalized receptor will be recycled to the cell membrane, while some will be targeted to lysozyme for degradation. A superactive receptor may also have rapid inactivation kinetic, and a long-term stress may induce more receptor degradation that can account for lowered glucose activity in the brain of Alzheimer’s disease patients. Till now, two polymorphisms of β2AR are identified to associate with sporadic late onset of Alzheimer’s disease. One is G16R, and the other is Q27E (Yu et al. 2008). The R16 polymorphism has enhanced agonist-mediated desensitization, and E27 polymorphism displays increased agonists signaling in the vasculature. As β2AR signaling and desensitization are cell specific and also depend on intracellular regulators’ abundance, such as PKA, GRK, and arrestin, a thorough study of the effects of these mutations on β2AR degradation and desensitization in astrocytes is in need. The impact of β2AR internalization on glucose metabolism and its relation to Alzheimer’s disease should also be examined.
Conclusions and Perspective
The “fight or flight hormone receptor” β2AR play important roles in regulating astrocyte glucose metabolism in acute stress. Together with other factors, dysregulation of β2AR will disrupt fine-tuning glucose metabolism in astrocytes and may contribute to the development of neuronal disease such as multiple sclerosis and Alzheimer’s disease. In astrocytes, β2AR is demonstrated to be an important regulator of glucose uptake and glycogen degradation. Lack of β2AR in multiple sclerosis patients may destroy energy supply from astrocytes to axons, contributing to neuronal degeneration and multiple sclerosis development. While in Alzheimer’s disease, hyperactivity of β2AR or desensitized β2AR polymorphism is identified in promoting amyloid beta formation and neuronal degeneration. Studies of β2AR and its polymorphism in desensitization and downregulation in astrocytes likely elucidate its function relevance, which may reconcile the contradiction of observed less glucose metabolism with higher β2AR activity in the brain of Alzheimer’s disease patients.
β2AR has been demonstrated to regulate cellular activity through coupling to different effectors, such as Gs, Gi, beta arrestin, NHERF, and presenilin, depending on specific cellular contexts. Compared with the heart and skeletal muscle systems, the combination signaling and function of β2AR in astrocyte cell glucose metabolism are less investigated. The β2AR molecular regulatory target of glucose transport in astrocytes and whether or not β2AR involves in lactate transport and aerobic oxidation through regulation of LDH expression and activity of MCTs await further investigation. Current available tools of B2AR accumulated in the past three decades of research, including specific receptor knockout and trans-gene models, multiple ligands with different receptor activation properties, and knowledge of the fine-tuning downstream regulatory mechanisms, provide good opportunities for a better understanding of its function in astrocytes glucose metabolism and relation to neuronal disease. Such understanding will lay out the foundation for further development and usage of specific beta2 receptor ligands in preventing or therapeutic treatment of neuronal disease, such as multiple sclerosis and Alzheimer’s disease.
References
Alexander GE, Chen K et al (2002) Longitudinal pet evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer’s disease treatment studies. Am J Psychiatry 159(5):738–745
Allaman I, Pellerin L et al (2000) Protein targeting to glycogen mRNA expression is stimulated by noradrenaline in mouse cortical astrocytes. Glia 30(4):382–391
Aoki C (1992) Beta-adrenergic receptors: astrocytic localization in the adult visual cortex and their relation to catecholamine axon terminals as revealed by electron microscopic immunocytochemistry. J Neurosci 12(3):781–792
Bergersen LH (2007) Is lactate food for neurons? Comparison of monocarboxylate transporter subtypes in brain and muscle. Neuroscience 145(1):11–19
Bouzier AK, Voisin P et al (1998) Glucose and lactate metabolism in C6 glioma cells: evidence for the preferential utilization of lactate for cell oxidative metabolism. Dev Neurosci 20(4–5):331–338
Brown GC (2007) Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans 35(Pt 5):1119–1121
Brown AM, Ransom BR (2007) Astrocyte glycogen and brain energy metabolism. Glia 55(12):1263–1271
Brown AM, Tekkok SB et al (2003) Glycogen regulation and functional role in mouse white matter. J Physiol 549(Pt 2):501–512
Brown AM, Sickmann HM et al (2005) Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res 79(1–2):74–80
Carnevale D, De Simone R et al (2007) Microglia–neuron interaction in inflammatory and degenerative diseases: role of cholinergic and noradrenergic systems. CNS Neurol Disord Drug Targets 6(6):388–397
Catus SL, Gibbs ME et al (2011) Role of beta-adrenoceptors in glucose uptake in astrocytes using beta-adrenoceptor knockout mice. Br J Pharmacol 162(8):1700–1715
Chenal J, Pellerin L (2007) Noradrenaline enhances the expression of the neuronal monocarboxylate transporter MCT2 by translational activation via stimulation of PI3K/Akt and the mTOR/S6K pathway. J Neurochem 102(2):389–397
Chuquet J, Quilichini P et al (2010) Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J Neurosci 30(45):15298–15303
Cohen Z, Molinatti G et al (1997) Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab 17(8):894–904
Coles AJ, Wing MG et al (1999) Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol 46(3):296–304
Confavreux C, Vukusic S et al (2000) Relapses and progression of disability in multiple sclerosis. N Engl J Med 343(20):1430–1438
Daaka Y, Luttrell LM et al (1997) Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 390(6655):88–91
De Keyser J, Wilczak N et al (1999) Astrocytes in multiple sclerosis lack beta-2 adrenergic receptors. Neurology 53(8):1628–1633
Dienel GA, Ball KK et al (2007) A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: implications for glycogen turnover. J Neurochem 102(2):466–478
Dringen R, Hamprecht B (1993) Differences in glycogen metabolism in astroglia-rich primary cultures and sorbitol-selected astroglial cultures derived from mouse brain. Glia 8(3):143–149
Fillenz M, Lowry JP et al (1999) The role of astrocytes and noradrenaline in neuronal glucose metabolism. Acta Physiol Scand 167(4):275–284
Fox PT, Raichle ME et al (1988) Nonoxidative glucose consumption during focal physiologic neural activity. Science 241(4864):462–464
Freemantle E, Vandal M et al (2006) Omega-3 fatty acids, energy substrates, and brain function during aging. Prostaglandins Leukot Essent Fatty Acids 75(3):213–220
Frohman EM, Filippi M et al (2005) Characterizing the mechanisms of progression in multiple sclerosis: evidence and new hypotheses for future directions. Arch Neurol 62(9):1345–1356
Gaskin F, Cantor CR et al (1975) Biochemical studies on the in vitro assembly and disassembly of microtubules. Ann N Y Acad Sci 253:133–146
Genc S, Kurnaz IA et al (2011) Astrocyte-neuron lactate shuttle may boost more ATP supply to the neuron under hypoxic conditions–in silico study supported by in vitro expression data. BMC Syst Biol 5:162
Gibbs ME, Hutchinson DS et al (2008) Role of beta-adrenoceptors in memory consolidation: beta3-adrenoceptors act on glucose uptake and beta2-adrenoceptors on glycogenolysis. Neuropsychopharmacology 33(10):2384–2397
Hall RA, Premont RT et al (1998) The beta2-adrenergic receptor interacts with the Na+/H+-exchanger regulatory factor to control Na+/H+ exchange. Nature 392(6676):626–630
Hara MR, Kovacs JJ et al (2011) A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature 477(7364):349–353
Hertz L, Gibbs ME (2009) What learning in day-old chickens can teach a neurochemist: focus on astrocyte metabolism. J Neurochem 109(Suppl 1):10–16
Hodges-Savola C, Rogers SD et al (1996) Beta-adrenergic receptors regulate astrogliosis and cell proliferation in the central nervous system in vivo. Glia 17(1):52–62
Hsu CC, Hsu CS (1990) Effect of isoproterenol on the uptake of [14C]glucose into glial cells. Neurosci Res 9(1):54–58
Hunt A, Schonknecht P et al (2007) Reduced cerebral glucose metabolism in patients at risk for Alzheimer's disease. Psychiatry Res 155(2):147–154
Hutchinson DS, Catus SL et al (2011) Alpha-adrenoceptors activate noradrenaline-mediated glycogen turnover in chick astrocytes. J Neurochem 117(5):915–926
Johnson LN, Wilson KS et al (1978) Crystallographic studies on the structure and function of glycogen phosphorylase b. Biochem Soc Trans 6(6):1108–1111
Jolivet R, Allaman I et al (2010) Comment on recent modeling studies of astrocyte–neuron metabolic interactions. J Cereb Blood Flow Metab 30(12):1982–1986
Kahsai AW, Xiao K et al (2011) Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat Chem Biol 7(10):692–700
Kaundal M, Katoch SS et al (2007) Beta-agonists enhance the lactic dehydrogenase (LDH) expression in serum and ventricular myocytes of mice. Acta Physiol Hung 94(3):249–259
Kidd D, Barkhof F et al (1999) Cortical lesions in multiple sclerosis. Brain 122(Pt 1):17–26
Kobilka BK (2011) Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci 32(4):213–218
Laureys G, Clinckers R et al (2010) Astrocytic beta(2)-adrenergic receptors: from physiology to pathology. Prog Neurobiol 91(3):189–199
Lin FT, Miller WE et al (1999) Feedback regulation of beta-arrestin1 function by extracellular signal-regulated kinases. J Biol Chem 274(23):15971–15974
Liu Y, Jia WG et al (1992) Morphology and distribution of neurons and glial cells expressing beta-adrenergic receptors in developing kitten visual cortex. Brain Res Dev Brain Res 65(2):269–273
Luttrell LM, Ferguson SS et al (1999) Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283(5402):655–661
Magistretti PJ, Pellerin L (1999) Astrocytes couple synaptic activity to glucose utilization in the brain. News Physiol Sci 14:177–182
Mangia S, DiNuzzo M et al (2011) Response to ‘comment on recent modeling studies of astrocyte-neuron metabolic interactions’: much ado about nothing. J Cereb Blood Flow Metab 31(6):1346–1353
Mantyh PW, Rogers SD et al (1995) Beta 2-adrenergic receptors are expressed by glia in vivo in the normal and injured central nervous system in the rat, rabbit, and human. J Neurosci 15(1 Pt 1):152–164
Maragakis NJ, Rothstein JD (2006) Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2(12):679–689
Minoshima S, Foster NL et al (1994) Posterior cingulate cortex in Alzheimer's disease. Lancet 344(8926):895
Nelson CD, Kovacs JJ et al (2008) Beta-arrestin scaffolding of phosphatidylinositol 4-phosphate 5-kinase Ialpha promotes agonist-stimulated sequestration of the beta2-adrenergic receptor. J Biol Chem 283(30):21093–21101
Newman LA, Korol DL et al (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6(12):e28427
Ni Y, Zhao X et al (2006) Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat Med 12(12):1390–1396
Pellerin L, Pellegri G et al (1998) Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci 20(4–5):291–299
Pellerin L, Bouzier-Sore AK et al (2007) Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55(12):1251–1262
Perry SJ, Baillie GS et al (2002) Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 298(5594):834–836
Prebil M, Jensen J et al (2011) Astrocytes and energy metabolism. Arch Physiol Biochem 117(2):64–69
Ransom BR, Fern R (1997) Does astrocytic glycogen benefit axon function and survival in CNS white matter during glucose deprivation? Glia 21(1):134–141
Rasmussen SG, DeVree BT et al (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477(7366):549–555
Robishaw JD, Smigel MD et al (1986) Molecular basis for two forms of the G protein that stimulates adenylate cyclase. J Biol Chem 261(21):9587–9590
Russo-Neustadt A, Cotman CW (1997) Adrenergic receptors in Alzheimer’s disease brain: selective increases in the cerebella of aggressive patients. J Neurosci 17(14):5573–5580
Salm AK, McCarthy KD (1992) The evidence for astrocytes as a target for central noradrenergic activity: expression of adrenergic receptors. Brain Res Bull 29(3–4):265–275
Sato M, Hutchinson DS et al (2007) Functional domains of the mouse beta(3)-adrenoceptor associated with differential G-protein coupling. Biochem Soc Trans 35(Pt 5):1035–1037
Seifert TS, Brassard P et al (2009) Cerebral non-oxidative carbohydrate consumption in humans driven by adrenaline. J Physiol 587(Pt 1):285–293
Shao Y, Sutin J (1992) Expression of adrenergic receptors in individual astrocytes and motor neurons isolated from the adult rat brain. Glia 6(2):108–117
Small GW, Ercoli LM et al (2000) Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer's disease. Proc Natl Acad Sci USA 97(11):6037–6042
Sorg O, Magistretti PJ (1991) Characterization of the glycogenolysis elicited by vasoactive intestinal peptide, noradrenaline and adenosine in primary cultures of mouse cerebral cortical astrocytes. Brain Res 563(1–2):227–233
Steele ML, Robinson SR (2012) Reactive astrocytes give neurons less support: implications for Alzheimer’s disease. Neurobiol Aging 33(2):423.e1–423.e13
Stys PK, Jiang Q (2002) Calpain-dependent neurofilament breakdown in anoxic and ischemic rat central axons. Neurosci Lett 328(2):150–154
Sutin J, Shao Y (1992) Resting and reactive astrocytes express adrenergic receptors in the adult rat brain. Brain Res Bull 29(3–4):277–284
Walls AB, Heimburger CM et al (2009) Robust glycogen shunt activity in astrocytes: effects of glutamatergic and adrenergic agents. Neuroscience 158(1):284–292
Wang D, Govindaiah G et al (2010) Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J 24(9):3511–3521
Wang D, Yuen EY et al (2011) Amyloid beta peptide-(1-42) induces internalization and degradation of beta2 adrenergic receptors in prefrontal cortical neurons. J Biol Chem 286(36):31852–31863
Wender R, Brown AM et al (2000) Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J Neurosci 20(18):6804–6810
Wilkins A, Scolding N (2008) Protecting axons in multiple sclerosis. Mult Scler 14(8):1013–1025
Xiao RP, Cheng H et al (1999) Recent advances in cardiac beta(2)-adrenergic signal transduction. Circ Res 85(11):1092–1100
Xiao RP, Zhu W et al (2006) Subtype-specific alpha1- and beta-adrenoceptor signaling in the heart. Trends Pharmacol Sci 27(6):330–337
Xiao K, Sun J et al (2010) Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc Natl Acad Sci USA 107(34):15299–15304
Yu JT, Tan L et al (2008) Polymorphisms at the beta2-adrenergic receptor gene influence Alzheimer’s disease susceptibility. Brain Res 1210:216–222
Yu NN, Wang XX et al (2010) Blocking beta2-adrenergic receptor attenuates acute stress-induced amyloid beta peptides production. Brain Res 1317:305–310
Zaccolo M, Di Benedetto G et al (2006) Restricted diffusion of a freely diffusible second messenger: mechanisms underlying compartmentalized cAMP signalling. Biochem Soc Trans 34(Pt 4):495–497
Zeinstra E, Wilczak N et al (2000) [3H]dihydroalprenolol binding to beta adrenergic receptors in multiple sclerosis brain. Neurosci Lett 289(1):75–77
Zhu WZ, Zheng M et al (2001) Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci USA 98(4):1607–1612
Acknowledgments
This work was supported by grants from the Ministry of Science and major basic research project (grant number 2012CB910402), National Nature Science foundation of China (grant numbers: No. 31100580 and No. 81171062), and the foundation for excellent Young and Middle-Aged Scientists of Shandong Province, China, No.BS2011SW020.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dong, Jh., Chen, X., Cui, M. et al. Beta2-Adrenergic Receptor and Astrocyte Glucose Metabolism. J Mol Neurosci 48, 456–463 (2012). https://doi.org/10.1007/s12031-012-9742-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-012-9742-4