
Abstract
Gamma-Aminobutyric acid (GABA) receptors are present in peripheral and central glia and modulate important physiological parameters of glial cells. Schwann cells (SC), the peripheral nervous system glial cells, play essential roles in nerve regeneration, but they are unsuitable for bioengineering of nerve repair. Increasing interest has been focused on adult stem cells derived from bone marrow (BM-MSC) or adipose tissue (ASC), which can be differentiated into SC-like phenotype and used as SC replacements. SC-like adult stem cells express GABA-B receptors that can modulate their proliferation. The aim of this study was to investigate GABA-A receptors functional expression in differentiated stem cells. BM-MSC and ASC were found to express GABA-A α2 and β3, but not β1 mRNA transcripts. Protein expression levels of GABA-A α2 and β3 receptors were upregulated following SC-like differentiation as shown by Western blot studies. GABA-A receptor stimulation with muscimol increased the proliferation rate of SC, differentiated BM-MSC and differentiated ASC. In conclusion, GABA-A α2 and β3 receptor subunits are present in BM-MSC and ASC and upregulated following glial differentiation. GABA-A subunits in differentiated stem cells and SC assemble in functional receptors modulating cell proliferation. Functional GABA-A and GABA-B receptors represent a possible pharmacological target to modulate SC-like stem cells physiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurotransmitters, such as γ-aminobutyric acid (GABA), are key molecules in neuron–glia interactions and represent a novel and promising approach for the pharmacological management of nerve regeneration (Magnaghi et al. 2009). GABA is the main inhibitory neurotransmitter of the mammalian adult central nervous system (CNS), although GABA is also shown to determine depolarisation, thus yielding an excitatory action in early developing neurons (Ben-Ari 2002). GABA exerts its action through different receptor classes: the metabotropic GABA-B receptor, which is coupled with inhibitory G proteins, and the ionotropic GABA-A receptor, which gates Cl− ions. GABA-A receptors are members of the Cys-loop ligand-gated ion channel superfamily (Olsen and Sieghart 2008), and they are assembled from a pool of 19 homologous subunits (that is α1–6, β1–3, γ1–3, δ, ε, π, θ and ρ1–3) (Simon et al. 2004), forming numerous, mostly hetero-oligomeric, pentamers (Olsen and Sieghart 2008). Although GABA-A receptor pharmacology is highly heterogeneous and depends upon subunit composition, most of GABA-A receptors are activated by GABA and the selective agonist muscimol, blocked by bicuculline and picrotoxin and modulated by benzodiazepines, barbiturates and neurosteroids (Olsen and Sieghart 2009). Immunocytochemical and electron microscopic studies indicate that the majority of GABA-A receptors present in the brain are composed of α-, β- and γ-subunits (Olsen and Sieghart 2009), with a subunit stoichiometry of 2α, 2β and 1γ (or 1ρ) and with the GABA binding site located at the α/β interface (Olsen and Sieghart 2009). The majority of GABA-A receptors contain a single type of α- and β-subunit variant, with the α1β2γ2 combination being the most abundant population, followed by α2β3γ2 and α3β3γ2 (Fritschy and Brunig 2003).
It is now clear that the actions of GABA are not only limited to the CNS and to its role as a neurotransmitter (von Blankenfeld and Kettenmann 1991; Pastor et al. 1995; Watanabe et al. 2002). The first evidence of the presence of GABA and its receptors in the peripheral nervous system (PNS) date back to the 1970–1980s (Brown and Marsh 1978; Brown et al. 1979; Jessen et al. 1979; Morris et al. 1983; Olsen et al. 1984; Bhisitkul et al. 1987); however, it was not until more recently that GABA-A receptors were identified in rat sciatic nerves and in cultured Schwann cells (SC) (Melcangi et al. 1999; Magnaghi et al. 2001, 2006). Interestingly, SC have been recently shown to express also functional GABA-B receptors (Magnaghi et al. 2004, 2006). Moreover, pharmacological stimulation of glial GABA-A and GABA-B receptors modulates key physiological parameters such as SC proliferation and myelin protein expression (Magnaghi et al. 2001, 2004). In particular, stimulation of GABA-A receptors with specific agonists and with allosteric modulators increases the expression levels of the peripheral myelin protein PMP22 in cultured neonatal SC (Magnaghi et al. 2001; Melcangi et al. 2005). Conversely, treatments with baclofen, a specific agonist of GABA-B receptors, decrease PMP22 and glycoprotein zero (P0) expression levels of cultured SC and rat sciatic nerves (Magnaghi et al. 2004, 2008). Moreover, stimulation with baclofen decreases forskolin(fsk)-induced SC proliferation (Magnaghi et al. 2004), whereas treatments with the allosteric GABA-A receptor modulator allopregnanolone have been recently reported to increase SC proliferation (Perego et al. 2011). Finally, SC have been also shown to synthesise GABA, which in turn activates GABA-A receptors, with an autocrine mechanism, and determine the phosphorylation of cAMP responsive element binding protein (Magnaghi et al. 2010). Altogether these data emphasise the importance of the GABAergic system in modulating SC biology, suggesting a role of GABA as a gliotransmitter in regulating neuron–glia interactions in the PNS (Faroni and Magnaghi 2011; Magnaghi 2009).
Adult stem cells derived from the bone marrow (BM-MSC) and from the adipose tissue (ASC) are easily harvested and rapidly expandable in culture, and they can be differentiated in multiple lineages (Pittenger et al. 1999; Zuk et al. 2002). BM-MSC and ASC have been shown to express general mesenchymal stem cell markers (that is CD29, CD44, CD54, CD90, CD105, alkaline phosphatase and integrin β1) and to lack expression of the hematopoietic antigens CD14 and CD45 (Caddick et al. 2006; Kingham et al. 2007; Brohlin et al. 2009; Kalbermatten et al. 2011; Novikova et al. 2011; Reid et al. 2011; Tse et al. 2011). Moreover, a sub-population of BM-MSC and ASC has been shown to express the stem cell marker stro-1 and the neuronal precursor marker nestin (Caddick et al. 2006; Kingham et al. 2007). Interestingly, BM-MSC and ASC have been shown to successfully differentiate into a SC-like phenotype (dBM-MSC and dASC, respectively) with molecular, functional and morphological similarities with SC (Caddick et al. 2006; Kingham et al. 2007). In a recent study, we have demonstrated the functional expression of the metabotropic GABA-B receptors in SC-like BM-MSC and ASC, suggesting the potential role of the GABAergic system in regulating adult stem cell physiology (Faroni et al. 2011). It is thus of interest to investigate the presence of the ionotropic GABA-A receptors on SC-like stem cells and their putative role in modulating stem cell physiological processes and regenerative potential.
Materials and Methods
Animals and Tissue Handling
All the experiments requiring animals were performed in accordance with the UK Animals (Scientific Procedures) Act, 1986. Following terminal anaesthesia with CO2 and cervical dislocation, tissues were harvested from the animal and processed as required. For gene and protein expression studies, total RNA or whole tissue lysates were obtained from fresh frozen brain and sciatic nerves (SN) of adult male Sprague-Dawley rats. For immunohistochemical analyses, SN were harvested from adult rats and rapidly fixed in paraformaldehyde.
Adult (aSC) and Neonatal (nSC) Schwann Cells Cultures
SC were harvested from the sciatic nerves of neonatal or adult Sprague-Dawley rats using previously established protocols (di Summa et al. 2009; Faroni et al. 2011). Cultures were maintained in low glucose Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, Poole, UK) supplemented with 10% (v/v) of foetal bovine serum (FBS; Biosera, Ringmer, UK), 1% (v/v) of penicillin–streptomycin solution (P–S; PAA, Yeovil, UK), 14 μM forskolin (Sigma-Aldrich, Poole, UK) and 63 ng ml-1 of glial growth factor-2 (GGF-2; Acorda Therapeutics Inc., Hawthorne, NY, USA). Cells were incubated in 5% CO2 at 37°C and maintained at sub-confluent levels onto poly-d-Lysine (PDL; Sigma-Aldrich, Poole, UK)-coated 75-cm2 flasks, with medium changes every 72 h. SC identity was confirmed as previously described (Faroni et al. 2011).
Bone Marrow and Adipose-Derived Stem Cells Harvesting
BM-MSC and ASC were isolated, respectively, from bone marrow aspirates and fat pads of male adult Sprague-Dawley rats as described previously (Caddick et al. 2006; Kingham et al. 2007). Briefly, the cavities of femoral and tibial bones were exposed and flushed with alpha modified Eagle’s medium (αMEM; Sigma-Aldrich, Poole, UK), containing 1% (v/v) P–S and 10% FBS (stem cell growth media, SCGM), triturated using a 21-gauge needle and finally filtered through a 100-μm BD Falcon™ Cell strainer (BD Bioscience, Oxford, UK). Similarly, subcutaneous, visceral and inguinal fat pads were passed through a 100-μm cell strainer after mechanical and enzymatic dissociation with collagenase type I (Life technologies, Paisley, UK). The resulting cell suspensions were pelleted by 5 min of centrifugation at 900 rpm, resuspended in SCGM and plated onto 75-cm2 flasks. When confluent, cells were detached with trypsin–EDTA (Life technologies, Paisley, UK), split and re-plated.
Stem Cell Characterisation and Pluripotency
Undifferentiated BM-MSC and ASC cultures were assessed for stem cell markers expression by immunocytochemical means. Specific antibodies for the stem cell surface marker Stro-1 and for the neural precursor marker nestin were used to stain passage 2 (p2) cultures as described below. In order to confirm stem cell pluripotency, BM-MSC and ASC were incubated with specific media to induce differentiation into the three mesodermal-derived lineages as previously reported (Pittenger et al. 1999). In particular, for adipogenic differentiation, cells were incubated in adipogenic induction media consisting in DMEM low glucose containing 10% FBS, 1% P-S, 1 μM dexamethasone (DX), 10 μg/ml insulin, 0.05 mM 3-isobutyl-1-methylxanthine and 100 μM indomethacin (all from Sigma-Aldrich, Poole, UK). After 1 week, cells were incubated for 2 days in a maintenance media consisting in DMEM containing 10% FBS, 1% P-S and 10 μg/ml insulin. This cycle was repeated three times and after 21 days cells were fixed in 10% formalin and fat droplets were stained with Oil Red O staining (Sigma-Aldrich, Poole, UK). For osteogenic differentiation, cells were incubated for 3 weeks in a specific media inducing calcification, that is, αMEM containing 10% FBS, 1% P–S, 0.1 μM DX, 100 μg/ml ascorbate and 10 mM β-glycerolphosphate (all Sigma-Aldrich, Poole, UK). After fixation of the cultures in 4% paraformaldehyde, Alizarin Red S staining was carried out to stain calcium deposits. Finally, for chondrogenic differentiation, cells were incubated in high-glucose DMEM containing 10% FBS, 1% P-S, 0.1 μM DX, 50 μg/ml ascorbate, ITS-Premix, 50 μg/ml transforming growth factor β and 50 μg/ml proline (all from Sigma-Aldrich, Poole, UK except from ITS-Premix, BD Bioscience, Oxford, UK). After 3 weeks of differentiation, cells were fixed in 10% formalin and cartilage proteoglycan deposition was detected with toluidine blue staining (Sigma-Aldrich, Poole, UK).
SC-Like Differentiation of Stem Cells
As previously described (Caddick et al. 2006; Kingham et al. 2007), p1–p2 ASC and BM-MSC cultures were initially incubated for 24 h in SCGM containing 1 mM β-mercaptoethanol to synchronise cell cycle (Sigma-Aldrich, Poole, UK), and this was followed by 3 days of further cell-preconditioning in SCGM supplemented with 35 ng ml−1 all-trans-retinoic acid (Sigma-Aldrich, Poole, UK). The medium was then replaced with stem cell differentiation medium containing 5 ng ml−1 platelet-derived growth factor (Sera Laboratories International, Haywards Heath, UK), 10 ng ml−1 basic fibroblast growth factor (Sera Laboratories International, Haywards Heath, UK), 14 μM fsk and 126 ng ml−1 GGF-2 (Acorda Therapeutics Inc., Hawthorne, NY, USA). The cells were incubated for 2 weeks under these conditions and passaged with trypsin/EDTA when needed, and fresh medium was added approximately every 72 h. Successful differentiation into a glial phenotype and molecular similarities with SC were confirmed by immunocytochemical assessment of glial markers (i.e. S100 and myelin protein zero, P0), see below for immunocytochemistry methods.
Reverse Transcription Polymerase Chain Reaction
Sub-confluent flasks of each experimental group (aSC, nSC, BM-MSC and ASC before and after glial differentiation) were trypsinised by treatment with trypsin/EDTA to collect the cells. Frozen brain and sciatic nerves and the trypsinised cells were homogenised and total RNA was extracted using RNeasy™ Mini Kit (Qiagen, Crawley, UK), according to the manufacturer protocol. Extracted RNA was treated with DNAse (Qiagen, Crawley, UK) to eliminate genomic contamination and finally eluted in water. After the measure of the concentrations by UV spectrophotometry, 1–10 ng of each RNA sample was reverse-transcribed for 30 min at 50°C, and cDNAs were amplified using One-Step reverse transcription polymerase chain reaction (RT-PCR) kit (Qiagen, Crawley, UK) with the following PCR cycling protocol: 30 s of denaturation at 95°C for 30 s, annealing for 30 s (number of cycles and optimal temperatures are indicated in Table 1), primer extension at 72°C for 1 min and a final extension step of 5 min at 72°C. The primer sequences and product size for all RT-PCRs are described in Table 1. For GABA-A β1 and β3, the products of the first amplification reaction were re-amplified with 30 cycles at 60°C using a standard PCR kit (Bioline, London, UK), performing a biphasic PCR methodology to detect the low-level transcripts. Specific primers for the housekeeping gene β-actin were used to confirm reverse transcription and ensure equal loading (see Table 1). A reaction omitting the template was carried out as a negative control. Brain and SN total mRNA (1 ng) were used as a positive controls, together with adult and neonatal SC whole cells lysates (10 ng). In addition, all PCR products were sequenced using Big Dye™ Terminator Sequencing Kit (Applied Biosystems Inc., Paisley, UK) to prove identity with published sequences (GABA-A α2 GenBank ID: NM_001135779.1; GABA-A β3 GenBank ID: AY742860.1).
Western Blot Analysis
Tissue and whole cell lysates were obtained by homogenising fresh frozen tissues and scraping cells from confluent flasks using a lysis buffer containing 100 mM 1,4-piperazinediethanesulfonic acid, 5 mM MgCl2, 20% (v/v) glycerol, 0.5% (v/v) Triton X-100, 5 mM ethylene glycol tetraacetic acid and a cocktail of protease and phosphatase inhibitors (Sigma-Aldrich, Poole, UK). Lysates were incubated for 30 min on ice and, after freeze–thaw, protein concentrations were determined using the Bio-Rad detergent-compatible protein assay (Bio-Rad Laboratories, Hemel Hempstead, UK). Before loading, proteins were denaturated for 5 min at 95°C in reducing Laemmli’s buffer, and 40 μg of proteins was separated at 120 V on 8% (w/v) sodium dodecyl sulphate–polyacrylamide gels. Following electrophoretic separation, the proteins were transferred to nitrocellulose membranes (GE Healthcare, Little Chalfont, UK) for 1 h at 80 V. In order to confirm successful protein transfer, membranes were stained with Pounceau red (Sigma-Aldrich, Poole, UK), before being blocked for 1 h in a Tris-buffered saline (TBS)-Tween solution (10 mM Tris pH 7.5, 100 mM NaCl, 0.1% (v/v) Tween) containing 5% (w/v) of non-fat dry milk. The membranes were thus incubated at 4°C in the following primary antibodies diluted in blocking buffer: rabbit polyclonal anti-GABA-A α2 (1:500; Abcam, Cambridge, UK) and rabbit polyclonal anti-GABA-β3 (1:250; Abcam, Cambridge, UK). After overnight incubation, membranes were washed with TBS-Tween and thus incubated for 1 h with an anti-rabbit horseradish peroxidase-conjugated (HRP) secondary antibody (1:2,000; Cell Signalling Technologies, Hitchin, UK) for chemiluminescence detection. Finally, blots were stripped for 30 min at room temperature with a glycine solution (100 mM, pH 2.9; Sigma-Aldrich, Poole, UK) to remove the antibodies bound to the nitrocellulose. After further blocking in TBS-milk, membranes were reprobed, using a rabbit primary antiserum directed against β-tubulin (1:2,000, Abcam, Cambridge, UK), followed by antigen detection with an anti-rabbit HRP-conjugated secondary antibody (1:2,000; Cell Signalling Technologies, Hitchin, UK). After digital acquisition of the films, the intensity of the signals was analysed by densitometry using ImageJ 64 imaging software (National Institutes of Health NIH, Bethesda, MD, USA).
Immunocytochemistry
Cells from each experimental group were plated on chamber slides (Nunc-Fisher Scientific, Loughborough, UK) or glass coverslips at a density of 5 × 103 cells/cm2 for all immunocytochemical studies. Twenty-four hours after seeding, cells were fixed for 20 min in 4% (w/v) paraformaldehyde (Sigma-Aldrich, Poole, UK) and washed in phosphate buffer solution (PBS). After 30 min of permeabilisation with 0.2% (v/v) Triton X-100 at room temperature, slides and coverslips were blocked for 1 h with normal goat or donkey serum solution (both 1:100; Sigma-Aldrich, Poole, UK). The following primary antibodies were applied overnight at 4°C for stro-1 (mouse monoclonal 1:50; R&D Systems, Abingdon, UK), nestin (mouse monoclonal 1:200; Millipore, Watford, UK), S100 (rabbit polyclonal 1:500; Dako, Ely, UK), myelin glycoprotein zero (P0, rabbit policlonal 1:200; Sigma-Genosys, Poole, UK), GABA-A α2 (rabbit polyclonal 1:500; Abcam, Cambridge, UK) and GABA-Α β3 (rabbit polyclonal 1:250; Abcam, Cambridge, UK). After 24 h, slides were rinsed in PBS before incubation in goat anti-rabbit FITC- or horse anti-mouse FITC-conjugated secondary antibodies (both from Vector Laboratories, Peterborough, UK), depending on the host species of the primary antibodies. After final washing with PBS, slides were mounted using Vectashield™ mounting medium for fluorescence containing 4′,6-diamidino-2-phenylindole (DAPI) for nuclear staining (Vector Laboratories, Peterborough, UK). For colocalisation studies of GABA-A α2 receptor subunits with the myelin structures, fixed and permeabilised sciatic nerves cross-sections were incubated for 20 min in green FluoroMyelin™ (1:1,000; Molecular Probes, Paisley, UK) prior to staining. The dye stains specifically myelin structures and allows further immunostaining with the same protocol described above, with the sole exception than a goat anti-rabbit Texas Red® (Vector Laboratories, Peterborough, UK) secondary antibody was used to stain GABA-A α2-subunits. Finally, images were acquired using a fluorescence microscope (Olympus BX60, Southend-on-Sea, UK) and processed with ImageJ 64 imaging software (National Institutes of Health NIH, Bethesda, MD, USA). In the case of GABA-A α2 receptor subunit, confocal analysis was performed with a Nikon C1 upright microscope. Images were collected every 0.5 μm, and Z-stacks were processed with ImageJ 64 (NIH, Bethesda, MD, USA).
Proliferation Assay
In order to assess cell proliferation, a CellTiter 96® Cell Proliferation assay (Promega, Southampton, UK) was performed. The assay is based on a tetrazolium compound 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS), whose conversion into a formazan product is proportional to cell number and detectable with spectrophotometric reading. Neonatal SC, dBM-MSC and dASC were seeded at density of 5 × 103 cells/well onto 24-well plates (Corning Inc., Corning, NY, USA) and incubated in their respective growth media (GM) containing growth factors. Plates were previously PDL-coated for assessment of SC proliferation. After overnight incubation at 37°C and 5% CO2, cells were rinsed with HBSS and the medium was replaced with fresh GM supplemented with 10 μM fsk (Sigma-Aldrich, Poole, UK) in the presence or absence of the selective GABA-A receptor agonist muscimol (100 μM; Sigma-Aldrich, Poole, UK). Fresh drugs were added every 24 h, and after 4 days of treatments, cells were rinsed with sterile PBS and incubated at 37°C with the MTS solution diluted in high-glucose DMEM without phenol red (Sigma-Aldrich, Poole, UK). After 4 h incubation, absorbances of the dyed media were recorded at 490 nm with an Asys UVM-340 microplate reader/spectrophotometer (Biochrom Ltd., Cambridge, UK). Data were expressed as a percentage of the values detected in controls (cells grown with only fsk). Three separate experiments were performed in triplicate, for nine total determinations for each experimental group.
Statistical Analysis
Statistical significance for the western blot and proliferation studies was estimated by unpaired two-tailed Student’s t test using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA). In western blot studies, densitometry data of GABA-A subunits were normalised for the levels of β-tubulin used as a loading control, and the data were expressed as mean ± standard error of the mean (SEM). In proliferation studies, data were normalised vs. controls grown only in fsk and expressed as percentage vs. fsk ± SEM. In both cases, levels of significance were expressed as p values versus controls (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
Adult Stem Cell Characterisation and Pluripotency
BM-MSC and ASC were successfully harvested from the bone marrow and from the adipose tissue (subcutaneous, visceral and inguinal fat pads) of adult Sprague-Dawley rats. After plating, undifferentiated stem cells (uBM-MSC and uASC) showed flattened fibroblast like morphology (Fig. 1 A, D). To confirm stem cells identity and neurogenic potential, cultures of uBM-MSC and uASC were stained for the stem cell marker stro-1 and for the neural precursor marker nestin. Both uBM-MSC and uASC showed immunopositivity for stro-1 (Fig. 1 B, E) and for nestin (Fig. 1 C, F). To assess stem cells multi-potency, undifferentiated cultures were incubated in differentiation media specific for the three mesodermal lineages (chondrogenic, adipogenic and osteogenic). After 3 weeks, differentiated stem cells showed staining for fat droplets (adipogenic lineage, Fig. 1 G, J), proteoglycans (chondrogenic lineage, Fig. 1 H, K) and calcium deposition (osteogenic lineage, Fig. 1 I, L), respectively. Following glial differentiation, SC-like BM-MSC and ASC (dBM-MSC and dASC, respectively) showed the typical SC spindle-shaped morphology (Fig. 1 M, P), and the immunocytochemical staining for glial markers showed immunopositivity for S100 and P0 (Fig. 1 N, Q, O, R).

Adult stem cell pluripotency and glial differentiation: Stem cells obtained from bone marrow (A) and fat pads (D) of adult rats showed a flattened fibroblast-like morphology. Undifferentiated stem cells were immunopositive for the stem cell marker stro-1 (arrowheads in B and E), and for the neural precursor marker nestin (C and F arrowheads), nuclei were stained in blue with DAPI. Both BM-MSC and ASC were successfully differentiated in three mesodermal lineages: adipogenic (Oil Red O staining of fat droplets, G and J), chondrogenic (toluidine blue staining of proteoglycans, H and K) and osteogenic (Alizarin Red S staining for calcium deposition, I and L). dBM-MSC (M) and dASC (P) were successfully transdifferentiated into a glial phenotype showing spindle-like morphology typical of SC. Differentiated stem cells showed immunopositivity for the glial markers S100 (N and Q) and P0 (O and R). Scale bars 50 μm
Adult Stem Cells Express GABA-A Receptor mRNAs
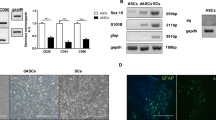
The presence of specific mRNA transcripts for GABA-A receptor subunits was assessed by qualitative RT-PCR. Using specific primers, amplicons sized 333 bp, corresponding to mRNAs encoding for GABA-A α2-subunit, were found in all the experimental groups analysed (uBM-MSC, dBM-MSC, uASC and dASC), as well as in brain, SN, adult and neonatal SC used as positive controls (Fig. 2). Interestingly, GABA-A α2 mRNA levels seemed to be upregulated in dBM-MSC and dASC if compared with the undifferentiated counterparts. In addition, uBM-MSC, dBM-MSC, uASC, dASC as well as all the positive controls were found to express specific mRNAs (355 bp) for GABA-A β3 receptor subunit (Fig. 2). Interestingly, differentiation of both BM-MSC and ASC seems to downregulate GABA-A β3 mRNAs expression levels. Following two rounds of PCR amplification, mRNA amplicons specific for GABA-A β1 (341 bp) were not detected in undifferentiated and differentiated stem cells, although they were present in the positive controls brain, SN, aSC and nSC (Fig. 2). To ensure equal loading and for quality assessment of the extracted mRNAs, specific primers for the housekeeping gene β-actin (product size 510 bp) were used in parallel amplifications in all PCR studies. A reaction with the omission of the template was carried out as a negative control, and no amplicons were detected. Moreover, PCR products were extracted and purified from the gel at the end of electrophoretic separation, and they were sequenced to verify their identity by comparison with published sequences (GABA-A α2 GenBank ID: NM_001135779.1; GABA-A β3 GenBank ID: AY742860.1). The sequences obtained were input in The National Center for Biotechnology (http://blast.ncbi.nlm.nih.gov/) website and subjected to Basic Local Alignment Search Tool with blastn and Megablast algorithms in order to find similarities with available nucleotide databases. At least 97% of identity with published sequences of GABA-A α2 and GABA-A β3 was found for all the amplicons sequenced.

Qualitative RT-PCR for GABA-A receptors: mRNA transcripts for GABA-A α2 were found in all the stem cells groups analysed that is undifferentiated stem cells (uBM-MSC and uASC) and SC-like stem cells (dBM-MSC and dASC) as well as in the positive controls (brain, sciatic nerve (SN), adult and neonatal SC). SC-like differentiation of BM-MSC and ASC seems to upregulate GABA-A α2 mRNA levels. Moreover, mRNA transcripts for GABA-A β3 were found in the positive controls and in all the experimental groups with expression levels that seem to be downregulated in dBM-MSC and dASC if compared to the undifferentiated cells. GABA-A β1 mRNA transcripts were not detected in stem cells samples in spite of two rounds of PCR amplification. A reaction omitting the template was carried out as a negative control. Specific primers for the housekeeping gene β-actin were used to check RNA quality and equal loading
GABA-A Receptor Proteins Are Expressed in Adult Stem Cells and Upregulated Following Glial Differentiation
Western blot analyses were performed to investigate the presence of GABA-A α2 and GABA-A β3 receptor subunits at the protein level. Using an antibody specifically raised against the GABA-A α2 isoform, a band of 51 kDa was detected in all stem cell groups and in the positive controls brain, SN, aSC and nSC (Fig. 3a). Interestingly, GABA-A α2 protein expression levels were significantly upregulated in ASC following glial differentiation (*p < 0.05, Fig. 3b). Similarly, dBM-MSC showed higher expression levels of GABA-A α2 subunit compared to uBM-MSC, although the difference did not reach statistical significance. A specific antibody directed against the GABA-A β3 subunit revealed a specific band of 51–56 kDa in uBM-MSC, dBM-MSC, uASC, dASC as well as in brain, SN, aSC and nSC used as positive controls (Fig. 3a). As seen for GABA-A α2, the densitometric analysis of the blots also revealed a statistically significant upregulation of GABA-A β3 protein levels in both BM-MSC and ASC after SC-like differentiation (*p < 0.05, Fig. 3b). Western blots for the housekeeping protein β-tubulin, giving a specific band of 50 kDa, were used to ensure equal loading and to normalise GABA-A subunits expression data.

Western blot analysis for GABA-A receptors: A specific band for GABA-A α2 subunit (51 kDa) was found in both adipose and bone marrow stem cells, as well as in the positive controls brain, sciatic nerve (SN) and SC (adult and neonatal) (a). GABA-A α2 was significantly upregulated in dASC following glial differentiation (b, *p < 0.05). Bone marrow SC-like stem cells showed higher expression levels of GABA-A α2; however, the difference with the undifferentiated counterpart was not statistically significant. A specific signal for GABA-A β3 (51–56 kDa) was found in all experimental groups analysed (a). GABA-A β3 protein expression levels were significantly upregulated in both dASC and dBM-MSC, compared to the respective undifferentiated cells (b, *p < 0.05). All expression data were normalised for the levels of β-tubulin used as loading control. Data are expressed as ratio between GABA subunit/β-tubulin ± SEM. Statistical significance was estimated by unpaired two-tailed Student’s t test (n = 3, *p < 0.05)
GABA-A Receptor Immunolocalisation in Adult Stem Cells
GABA-A α2 receptor subunit localisation was further investigated by immunocytochemical analysis. Undifferentiated and differentiated BM-MSC and ASC were stained for a specific antibody directed against GABA-A α2. Both uBM-MSC and uASC showed GABA-A α2 immunopositivity localised mainly in the intracellular compartment (Fig. 4A, C). Following glial differentiation, immunostaining for GABA-A α2 in both dBM-MSC and dASC was stronger and diffused throughout the cytoplasm (Fig. 4B, D). Confocal microscopy analysis was performed to confirm the differences in GABA-A α2 intracellular localisation following SC-like differentiation. As shown in the representative Z-stacks in columns h and i (Fig. 4), GABA-A α2 is localised intracellularly in undifferentiated stem cells (column h in Fig. 4) in structures resembling the endoplasmic reticulum (stacks were collected every 0.5 μm). After SC differentiation, GABA-A α2 staining was diffused in the cytoplasm and cell extensions (column i in Fig. 4), with a similar pattern observed in cultured SC used as positive controls (Fig. 4E). Moreover, SN cross sections were stained for GABA-A α2 showing intense staining in SC wrapping the axons (Fig. 4F). Finally, using a counterstain for myelin structures (that is green Fluoromyelin™), GABA-A α2 receptor was localised in myelinating SC ensheating the axons (Fig. 4G). As negative controls, stainings omitting the primary antibody were performed on fixed cells and tissues without showing immunopositivity (data not shown).

Immunolocalisation of GABA-A subunits: uBM-MSC (A), dBM-MSC (B), uASC (C) and dASC (D) showed immunopositivity for GABA-A α2 subunit. The cytoplasmic distribution of differentiated stem cells was similar to SC (E) used as positive controls (GABA subunit in green, DAPI stained nuclei in blue). Sciatic nerves cross sections showed GABA-A α2 immunopositivity in the cytoplasm of SC myelinating the axons. In F, GABA-A α2 in green, SC nuclei in blue. In G, GABA-A α2 staining of SC cytoplasm in red and myelin structures stained in green. In undifferentiated stem cells, GABA-A α2 was localised mainly intracellularly as shown in the representative Z-stack montage in column H, whereas subunit distribution in differentiated stem cells was more diffused throughout the cytoplasm and cell extensions (I column). Z-stack images were acquired with a confocal Nikon C1 upright microscope; images were collected every 0.5 μm. dBM-MSC (J) and dASC (K) were immunopositive for GABA-A β3 subunit, and staining pattern was similar to SC (L), used as positive controls. Scale bars 20 μm
Immunocytochemistry analysis was also performed to investigate GABA-A β3 receptor subunit. Confirming western blot studies, both dBM-MSC and dASC showed immunopositivity for GABA-A β3 subunit (Fig. 4J, K), with a similar pattern of cytoplasmic distribution observed in the positive controls nSC (Fig. 4L). Staining was performed also in undifferentiated culture, but immunopositivity was below the detection limit of immunocytochemistry techniques (data not shown).
GABA-A Receptor Stimulation Increases Cell Proliferation
The simultaneous expression of two of the most abundant GABA-A receptor subunits suggests the formation of functional receptors in SC and SC-like adult stem cells. Hence, we assessed whether the pharmacological stimulation with the specific GABA-A agonist muscimol was able to modulate the fsk-induced proliferation of nSC, dBM-MSC and dASC, using a MTS-based proliferation assay. Following 4 days of muscimol treatments, there was a significantly increased nSC proliferation compared to the controls grown only in presence of 10 μM forskolin (Fig. 5a, increase of 18.23 ± 3.87% vs. forskolin, ***p < 0.001). Similarly, muscimol exerted the same effect on the proliferation of dBM-MSC (Fig. 5b, increase of 11.60 ± 3.71% vs. forskolin, **p < 0.01) and dASC (Fig. 5c, increase of 10.00 ± 1.83% vs. forskolin, ***p < 0.001), suggesting that the assembly of functional GABA-A receptors on differentiated stem cells is able to modulate proliferation.

Functional GABA-A receptors modulate SC and stem cell proliferation: To assess GABA-A receptor influence on cell proliferation, a MTS-based assay was performed. Four days of stimulation with the specific GABA-A agonist muscimol significantly increased the proliferation rate of fsk-treated nSC (a, increase of 18.23 ± 3.87% vs. forskolin, ***p < 0.001), dBM-MSC (b, increase of 11.60 ± 3.71% vs. forskolin, **p < 0.01) and dASC (c, increase of 10.00 ± 1.83% vs. forskolin, ***p < 0.001). Data are expressed as percent of variation vs. controls grown only in the presence of forskolin ± SEM. Three independent experiments were performed in triplicate for a total of nine determination for each experimental condition (fsk forskolin, mus muscimol). Statistical significance was evaluated with a two-tailed Students’ t test (**p < 0.01, ***p < 0.001).
Discussion
In the present study, we investigated the functional expression of GABA-A receptors in adult stem cells differentiated into a SC-like phenotype. In the adult CNS, GABA mediates synaptic inhibition and generates a rapid phasic inhibitory synaptic transmission and also a tonic inhibition by producing currents in extrasynaptic and perisynaptic locations (Olsen and Sieghart 2009). However, GABA, its receptors and the enzymes required to synthetise GABA are present well before the formation of synapses, and they play key roles in the development of neural tissues acting as an autocrine/paracrine signalling system (Varju et al. 2001; Ben-Ari 2002). GABA plays important roles also in the glia compartment of the CNS, modulating the physiology of glial cells, and represents a pivotal molecule in neuron–glia interactions (Velez-Fort et al. 2011). Our previous studies have demonstrated the function of GABA-A and GABA-B receptors in the glial cells of PNS (Magnaghi et al. 2001, 2004, 2008, 2010). Interestingly, SC have been shown to express GABA-A α2, α3 and β1–3 receptors (Melcangi et al. 1999), as well as GABA-B-1a, GABA-B-1b, GABA-B-1c and GABA-B-2 receptors (Magnaghi et al. 2004). Moreover, the functional GABA-A and B receptors on SC were shown to be important modulators of peripheral glia, regulating SC proliferation and myelin protein expression (Magnaghi et al. 2001, 2004, 2008; Perego et al. 2011). All these evidence suggest that GABA receptors on SC are potential pharmacological targets to modulate SC physiology in regenerative processes (Magnaghi et al. 2009).
We have shown the potential of BM-MSC and ASC to differentiate into a SC-like phenotype (Caddick et al. 2006; Kingham et al. 2007). Importantly, differentiated BM-MSC and ASC have morphological, molecular and functional similarities with SC (Kingham et al. 2007; Mahay et al. 2008a, b; Brohlin et al. 2009; Kaewkhaw et al. 2011), they are able to form myelin (Keilhoff et al. 2006; Xu et al. 2008; Mantovani et al. 2010) and they improve nerve regeneration in vivo (Tohill et al. 2004; di Summa et al. 2011). Recently, we have demonstrated that BM-MSC and ASC express both the subunits required to assemble functional metabotropic GABA-B receptors and that treatments with GABA-B agonist decrease the proliferation rate of dASC (Faroni et al. 2011), as previously shown in SC (Magnaghi et al. 2004). There are also evidence that GABA is present in murine ASC after neuronal–glial differentiation (Safford et al. 2004) and that GABA-A receptors are expressed in human ASC differentiated into a neuronal phenotype (Anghileri et al. 2008). In a recent study, the presence of GABA-A (α1, β3 and ε) and GABA-B2 receptors has been shown in BM-MSC differentiated along a neuronal-like phenotype (Marcoli et al. 2008). Furthermore, in vivo studies have shown increased expression of GABA-A receptors after transplantation of BM-MSC in mice after cerebral infarct (Shichinohe et al. 2006) and in rats after spinal cord injury (Yano et al. 2006), suggesting in vivo neuronal differentiation of transplanted BM-MSC in the region of the lesion (Shichinohe et al. 2006; Yano et al. 2006).
In this study, we showed the presence of the ionotropic GABA-A receptor subunits in undifferentiated BM-MSC and ASC at mRNA and protein level and GABA-A α2 and β3 proteins upregulation following glial differentiation. In particular, both BM-MSC and ASC were found to express GABA-A α2 and β3 subunits, which are known to form one of the most abundant native GABA-A receptor (Fritschy and Brunig 2003; Olsen and Sieghart 2009). GABA-A β3 protein upregulation following SC-like differentiation seems to be in contrast with the apparent downregulation observed with the RT-PCR; however, this discrepancy could be ascribed to a different mRNA/protein turnover and to the increased amplification required to detect β3 amplicons. The two steps of PCR amplification required to detect GABA-A β3 transcripts in undifferentiated BM-MSC is consistent with previous real-time PCR studies showing low expression levels of GABA-A β3 receptor subunit (Marcoli et al. 2008). Moreover, the GABA-A β3 protein upregulation following glial differentiation of BM-MSC and ASC detected by western blot analyses is comparable to previously reported mRNA upregulation in BM-MSC differentiated into a neuronal phenotype (Marcoli et al. 2008). The presence of GABA-A receptors in SC-like stem cells further confirms the molecular similarities between dBM-MSC, dASC and SC, the latter being reported to express α2 and β3 subunits (Melcangi et al. 1999).
We report here that GABA-A stimulation with a specific agonist increases the proliferation of SC-like stem cells and SC. These data are consistent with recent observations showing that allopregnanolone, a GABA-A allosteric modulator, increases the proliferation of SC (Perego et al. 2011) and of neonatal glial progenitors (Gago et al. 2004). Interestingly, we have previously demonstrated that stimulation with a specific GABA-B receptor ligand decreases SC and SC-like stem cell proliferation (Magnaghi et al. 2004; Faroni et al. 2011). This is not surprising since although GABA-A and GABA-B receptors respond to the same GABA neurotransmitter, they belong to two separate class of receptors with different roles and mechanisms of actions (Bowery et al. 1984; Bowery and Smart 2006). Moreover, the stimulation of GABA-A or GABA-B receptors has been already reported to effect oppositely the proliferation of mouse embryonic stem cells (Schwirtlich et al. 2010). In SC, the two classes of GABA receptors exert different and cross-interacting effects on SC proliferation and myelin protein expression, playing important roles in neuron–glia interactions and suggesting the importance of GABAergic system in the control of SC differentiation/proliferation (Faroni and Magnaghi 2011; Magnaghi 2007; Magnaghi et al. 2009).
More studies will be needed to clarify the physiological role of GABA receptors in SC-like stem cells, but the presence of both GABA-A and GABA-B receptors and their opposite effects on regulating cell proliferation suggests that the GABAergic signalling could be a potential player in the mechanisms regulating stem cell differentiation and proliferation as reported in SC (Magnaghi et al. 2009). In this scenario, we might speculate that the interplay between the two GABAergic systems could prompt stem cells differentiation with a reduction of proliferation (via GABA-B receptors), whereas GABA-A stimulation increases cell proliferation and thus could be a signal of stem cells de-differentiation.
References
Anghileri E, Marconi S, Pignatelli A et al (2008) Neuronal differentiation potential of human adipose-derived mesenchymal stem cells. Stem Cells Dev 175:909–916
Ben-Ari Y (2002) Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci 39:728–739
Bhisitkul RB, Villa JE, Kocsis JD (1987) Axonal GABA receptors are selectively present on normal and regenerated sensory fibers in rat peripheral nerve. Exp Brain Res 663:659–663
Bowery NG, Smart TG (2006) Gaba and glycine as neurotransmitters: a brief history. Br J Pharmacol 147(Suppl 1):S109–S119
Bowery NG, Price GW, Hudson AL et al (1984) Gaba receptor multiplicity. Visualization of different receptor types in the mammalian CNS. Neuropharmacology 232B:219–231
Brohlin M, Mahay D, Novikov LN et al (2009) Characterisation of human mesenchymal stem cells following differentiation into Schwann cell-like cells. Neurosci Res 641:41–49
Brown DA, Marsh S (1978) Axonal GABA-receptors in mammalian peripheral nerve trunks. Brain Res 1561:187–191
Brown DA, Adams PR, Higgins AJ, Marsh S (1979) Distribution of GABA-receptors and GABA-carriers in the mammalian nervous system. J Physiol (Paris) 756:667–671
Caddick J, Kingham PJ, Gardiner NJ, Wiberg M, Terenghi G (2006) Phenotypic and functional characteristics of mesenchymal stem cells differentiated along a Schwann cell lineage. Glia 548:840–849
di Summa PG, Kingham PJ, Raffoul W et al (2009) Adipose-derived stem cells enhance peripheral nerve regeneration. J Plast Reconstr Aesthet Surg 63:1544–1552
di Summa PG, Kalbermatten DF, Pralong E et al (2011) Long-term in vivo regeneration of peripheral nerves through bioengineered nerve grafts. Neuroscience 181:278–291
Faroni A, Magnaghi V (2011) The neurosteroid allopregnanolone modulates specific functions in central and peripheral glial cells. Front Endocrin 2:103. doi:10.3389/fendo.2011.00103
Faroni A, Mantovani C, Shawcross SG et al (2011) Schwann-like adult stem cells derived from bone marrow and adipose tissue express gamma-aminobutyric acid type B receptors. J Neurosci Res 89:1351–1362
Fritschy JM, Brunig I (2003) Formation and plasticity of GABAergic synapses: physiological mechanisms and pathophysiological implications. Pharmacol Ther 983:299–323
Gago N, El-Etr M, Sananes N et al (2004) 3alpha,5alpha-Tetrahydroprogesterone (allopregnanolone) and gamma-aminobutyric acid: autocrine/paracrine interactions in the control of neonatal Psa-Ncam+ progenitor proliferation. J Neurosci Res 786:770–783
Jessen KR, Mirsky R, Dennison ME, Burnstock G (1979) GABA may be a neurotransmitter in the vertebrate peripheral nervous system. Nature 281(5726):71–74
Kaewkhaw R, Scutt AM, Haycock JW (2011) Anatomical site influences the differentiation of adipose-derived stem cells for Schwann-cell phenotype and function. Glia 595:734–749
Kalbermatten DF, Schaakxs D, Kingham PJ, Wiberg M (2011) Neurotrophic activity of human adipose stem cells isolated from deep and superficial layers of abdominal fat. Cell Tissue Res 3442:251–260
Keilhoff G, Goihl A, Langnase K, Fansa H, Wolf G (2006) Transdifferentiation of mesenchymal stem cells into Schwann cell-like myelinating cells. Eur J Cell Biol 851:11–24
Kingham PJ, Kalbermatten DF, Mahay D et al (2007) Adipose-derived stem cells differentiate into a Schwann cell phenotype and promote neurite outgrowth in vitro. Exp Neurol 2072:267–274
Magnaghi V (2007) Gaba and neuroactive steroid interactions in glia: new roles for old players? Curr Neuropharmacol 51:47–64
Magnaghi V, Cavarretta I, Galbiati M, Martini L, Melcangi RC (2001) Neuroactive steroids and peripheral myelin proteins. Brain Res Brain Res Rev 371–3:360–371
Magnaghi V, Ballabio M, Cavarretta IT et al (2004) GABAB receptors in Schwann cells influence proliferation and myelin protein expression. Eur J Neurosci 1910:2641–2649
Magnaghi V, Ballabio M, Consoli A et al (2006) GABA receptor-mediated effects in the peripheral nervous system: a cross-interaction with neuroactive steroids. J Mol Neurosci 281:89–102
Magnaghi V, Ballabio M, Camozzi F et al (2008) Altered peripheral myelination in mice lacking GABAB receptors. Mol Cell Neurosci 373:599–609
Magnaghi V, Procacci P, Tata AM (2009) Chapter 15: novel pharmacological approaches to Schwann cells as neuroprotective agents for peripheral nerve regeneration. Int Rev Neurobiol 87:295–315
Magnaghi V, Parducz A, Frasca A et al (2010) Gaba synthesis in Schwann cells is induced by the neuroactive steroid allopregnanolone. J Neurochem 1124:980–990
Mahay D, Terenghi G, Shawcross SG (2008a) Growth factors in mesenchymal stem cells following glial–cell differentiation. Biotechnol Appl Biochem 51(Pt 4):167–176
Mahay D, Terenghi G, Shawcross SG (2008b) Schwann cell mediated trophic effects by differentiated mesenchymal stem cells. Exp Cell Res 314(14):2692–2701
Mantovani C, Mahay D, Kingham M et al (2010) Bone marrow- and adipose-derived stem cells show expression of myelin mRNAs and proteins. Regen Med 53:403–410
Marcoli M, Candiani S, Tonachini L et al (2008) In vitro modulation of gamma amino butyric acid (GABA) receptor expression by bone marrow stromal cells. Pharmacol Res 575:374–382
Melcangi RC, Magnaghi V, Cavarretta I et al (1999) Progesterone derivatives are able to influence peripheral myelin protein 22 and p0 gene expression: possible mechanisms of action. J Neurosci Res 564:349–357
Melcangi RC, Cavarretta IT, Ballabio M et al (2005) Peripheral nerves: a target for the action of neuroactive steroids. Brain Res Brain Res Rev 482:328–338
Morris ME, Di Costanzo GA, Fox S, Werman R (1983) Depolarizing action of GABA (gamma-aminobutyric acid) on myelinated fibers of peripheral nerves. Brain Res 2781–2:117–126
Novikova LN, Brohlin M, Kingham PJ, Novikov LN, Wiberg M (2011) Neuroprotective and growth-promoting effects of bone marrow stromal cells after cervical spinal cord injury in adult rats. Cytotherapy 137:873–887
Olsen RW, Sieghart W (2008) International Union of Pharmacology. Lxx. Subtypes of gamma-aminobutyric acid(a) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev 603:243–260
Olsen RW, Sieghart W (2009) GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 561:141–148
Olsen RW, Snowhill EW, Wamsley JK (1984) Autoradiographic localization of low affinity GABA receptors with [3 h]bicuculline methochloride. Eur J Pharmacol 992–3:247–248
Pastor A, Chvatal A, Sykova E, Kettenmann H (1995) Glycine- and GABA-activated currents in identified glial cells of the developing rat spinal cord slice. Eur J Neurosci 76:1188–1198
Perego C, Cairano ES, Ballabio M, Magnaghi V (2011) Neurosteroid allopregnanolone regulates EAAC1-mediated glutamate uptake and triggers actin changes in Schwann cells. J Cell Physiol. doi:10.1002/jcp.22898
Pittenger MF, Mackay AM, Beck SC et al (1999) Multilineage potential of adult human mesenchymal stem cells. Science 2845411:143–147
Reid AJ, Sun M, Wiberg M et al (2011) Nerve repair with adipose-derived stem cells protects dorsal root ganglia neurons from apoptosis. Neuroscience 199:515–522
Safford KM, Safford SD, Gimble JM, Shetty AK, Rice HE (2004) Characterization of neuronal/glial differentiation of murine adipose-derived adult stromal cells. Exp Neurol 1872:319–328
Schwirtlich M, Emri Z, Antal K et al (2010) GABA(A) and GABA(B) receptors of distinct properties affect oppositely the proliferation of mouse embryonic stem cells through synergistic elevation of intracellular Ca(2+). FASEB J 244:1218–1228
Shichinohe H, Kuroda S, Yano S et al (2006) Improved expression of gamma-aminobutyric acid receptor in mice with cerebral infarct and transplanted bone marrow stromal cells: an autoradiographic and histologic analysis. J Nucl Med 473:486–491
Simon J, Wakimoto H, Fujita N, Lalande M, Barnard EA (2004) Analysis of the set of GABA(A) receptor genes in the human genome. J Biol Chem 27940:41422–41435
Tohill M, Mantovani C, Wiberg M, Terenghi G (2004) Rat bone marrow mesenchymal stem cells express glial markers and stimulate nerve regeneration. Neurosci Lett 3623:200–203
Tse KH, Kingham PJ, Novikov LN, Wiberg M (2011) Adipose tissue and bone marrow-derived stem cells react similarly in an ischaemia-like microenvironment. J Tissue Eng Regen Med. doi:10.1002/term.452
Varju P, Katarova Z, Madarasz E, Szabo G (2001) GABA signalling during development: new data and old questions. Cell Tissue Res 3052:239–246
Velez-Fort M, Audinat E, Angulo MC (2011) Central role of GABA in neuron–glia interactions. Neuroscientist. doi:10.1177/1073858411403317
von Blankenfeld G, Kettenmann H (1991) Glutamate and GABA receptors in vertebrate glial cells. Mol Neurobiol 51:31–43
Watanabe M, Maemura K, Kanbara K, Tamayama T, Hayasaki H (2002) GABA and GABA receptors in the central nervous system and other organs. Int Rev Cytol 213:1–47
Xu Y, Liu L, Li Y et al (2008) Myelin-forming ability of Schwann cell-like cells induced from rat adipose-derived stem cells in vitro. Brain Res 1239:49–55
Yano S, Kuroda S, Shichinohe H et al (2006) Bone marrow stromal cell transplantation preserves gammaaminobutyric acid receptor function in the injured spinal cord. J Neurotrauma 2311:1682–1692
Zuk PA, Zhu M, Ashjian P et al (2002) Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 1312:4279–4295
Acknowledgments
The authors are grateful to Acorda Therapeutics, USA, for kindly supplying GGF-2 for the continuation of this work. We also thank Dr Pietro di Summa for precious methodological discussions and The University of Manchester DNA sequencing and Bioimaging facilities for sequence data and assistance with confocal microscopy analysis. This work was funded by Fondazione San Paolo di Torino (grant no. 2229/2008 to V.M.). Alessandro Faroni was funded by a BBSRC DTG fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Faroni, A., Terenghi, G. & Magnaghi, V. Expression of Functional γ-Aminobutyric Acid Type A Receptors in Schwann-Like Adult Stem Cells. J Mol Neurosci 47, 619–630 (2012). https://doi.org/10.1007/s12031-011-9698-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9698-9