
Abstract
Background
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancerrelated mortality in the USA, and the overall incidence of the disease is increasing such that it is expected to be the third leading cause of cancer-related deaths in the next decade. Minimal improvements in therapy have not changed the overall mortality rate over the past decade for patients with PDAC. The purpose of this review is to identify new data regardign the role of Transforming growth factor beta (TGF-β) based therapeuics in patients with PDAC.
Methods
The literature was searched for peer reviewed manuscripts regarding the use of TGF-β inhibitors in PDAC therapy and the mechanism in which TGF-β intracellular signaling effects patient survival.
Results
TGF-β plays a vital, context-dependent role as both a tumor suppressor and promoter of PDAC. The downstream effects of this duality play a significant role in the immunologic response of the tumor microenvironment (TME), epithelial-mesenchymal transformation (EMT), and the development of metastatic disease. Immunologic pathways have been shown to be successful targets in the treatment of other diseases, though they have not been shown efficacious in PDAC. TGF-β-mediated EMT does play a critical role in PDAC progression in the development of metastases. The use of anti-TGF-β-based therapies in phase I and II clinical trials for metastatic PDAC demonstrate the importance of understanding the role of TGF-β in PDAC progression.
Conclusion
This review clarifies the recent literature investigating the role of anti-TGF-β-based therapy in PDAC and areas ripe for targeted investigations and therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a malignancy whose treatment continues to present significant challenges for clinicians. When diagnosed, it tends to present itself as advanced stage, metastatic disease, and with a poor prognosis. It is projected to become the third leading cause of cancer-related deaths in the next decade, and the death toll from PDAC is expected to continue to rise [1]. Among the leading causes of cancer-related deaths, it is the only one whose overall mortality rate is not decreasing [2]. The optimization of operative resection over the past 2 decades has significantly reduced perioperative morbidity and mortality, and it has resulted in increased stage-specific survival rates [3]. However, the overall mortality has remained stagnant, with an overall 5-year survival at less than 8%, and death primarily due to metastatic disease [2, 4].
The overwhelming use of immunotherapy to successfully treat several other malignancies continues to spur investigation for similar therapies to treat PDAC [5,6,7,8,9]. Transforming growth factor beta (TGF-β) signaling pathways play a central role in the immunogenic response to cancer. [7, 10] While we have shown that patients with increased systemic inflammation during neoadjuvant PDAC therapy have lower overall survival [11], it appears that immunotherapy is failing in PDAC patients due to local peri-tumoral immunosuppression. Two mechanisms by which PDAC induces local immunosuppression are through tumor-associated macrophages [12,13,14] and suppression of cytotoxic CD8+ T cells [9, 15]. The PDAC tumor microenvironment (TME) is a desmoplastic-rich environment which provides for physical barriers to immunosurveillance, in addition to the aforementioned immunosuppression, which is primarily due to TGF-β expression from cancer cells [13, 16,17,18].
Our group and others have demonstrated that TGF-β acts as a tumor suppressor in normal pancreatic cells and stages I and II PDAC through significant blockade of cell proliferation; when disrupted, it has been shown to have tumorigenic activity in many late-stage malignancies, including PDAC [19,20,21]. This duality, termed the TGF-β paradox, is evident as upregulated and overexpressed TGF-β has been shown to induce stromal proliferation in the PDAC TME, as well as promote the epithelial-mesenchymal transformation (EMT) that leads to metastases [14, 20, 22, 23]. The full characterization of the effects of these pathways has yet to be established, but the paradox of TGF-β is well described [24, 25].
This review is aimed at delineating the potential key role of TGF-β in the development of metastatic PDAC through changes in the TME and EMT. Key challenges that remain will be discussed in the context of the utility of anti-TGF-β-based therapy. TGF-β inhibitors, such as galunisertib and AP12009, are in phase I and II clinical trials for metastatic PDAC but will likely only benefit patients with PDAC that is TGF-β dependent, a phenomenon that may not be universal. Previous work by Heldin et al. and others have been critical to our understanding of the complexity of TGF-β signaling [26]. The purpose of this review is to build on that work because of the growing interest in the use of TGF-β inhibitors in clinical trials to treat PDAC.
Canonical and Non-canonical TGF-β Signaling
The TGF-β signaling pathway is a complex pathway of interrelated proteins that has a wide range of effects including angiogenesis, apoptosis, embryogenesis, cell differentiation, immune response, and immune suppression [27]. Depending on the cell type and cellular context, TGF-β will activate different subsets of genes, leading to the diverse effects seen with TGF-β signaling [28]. There are three TGF-β ligands in humans termed TGF-β1–3, with β1 being the most commonly expressed. It also has significant activity in the exocrine pancreas [27]. There are over 30 other ligands that bind to TGF-β receptors which include activins, bone morphogenetic proteins, and growth and differentiation factors [26].
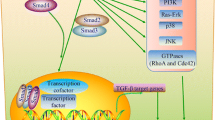
The stable TGF-β dimer in the TME binds to and acts on one of three TGF-β receptors (TGF-βR1–3). TGF-βR1 is a transmembrane receptor with an intracellular serine-threonine-rich GS domain, which after activation, phosphorylates downstream targets, primarily the SMAD proteins [29]. The SMAD proteins carry out a variety of intracellular and transcriptional actions through what is termed as a canonical TGF-β pathway, and these lead to primarily tumor suppressive activity and normal cellular function [30, 31].
Receptor-regulated SMADs in the canonical pathway include SMAD2 and SMAD3, which bind with a co-regulator, SMAD4, allowing activation and transcription of specific target genes (Fig. 1). Inhibitor SMAD proteins, such as SMAD7, are produced as a result of the SMAD2/3/4 induction, and they act to suppress the initial activation of SMAD 2/3, and provide negative feedback control of the TGF-β-regulated pathway [29]. Molecular variability due to mutations in SMAD4 is generally associated with worse overall survival in both primary and metastatic PDAC demonstrating the importance of the canonical TGF-β/SMAD signaling cascade [32,33,34].

The canonical TGF-β results in controlled cell growth, apoptosis, and overall tumor suppression. Any variations to this, such as mutation or loss of SMAD4, and an inability to bind to SMAD2/3, or non-SMAD signaling results in cancer cell growth, increased migration, and overall tumor promotion
The non-canonical TGF-β signaling pathway includes activation of Akt and direct phosphorylation of PAR6 [30]. Through Akt, it appears that EMT proteins are upregulated and normal cellular polarization is lost [13, 30]. Some of the most notable downstream effects of non-canonical TGF-β signaling include inhibition of the cell cycle as well as pro-apoptotic effects [13]. Inhibition of cyclins and cyclin-dependent kinases is achieved by an increased expression of p15 and p21 among others [13, 29, 30].
Other signaling proteins that can be activated by TGF-β include the mitogen-activated protein kinases (MAPKs), the Rho family of small GTPases, phosphatidylinositide 3-kinase (PI3K), and TNF receptor-associated factor 4/6 (TRAF 4/6) (Fig. 2) [34, 35]. Overall, the downstream effects of activating these pathways are the regulation of cell proliferation, differentiation, and apoptosis. These pathways are implicated in the tumorigenesis function of TGF-β, promoting angiogenesis, metastasis, and EMT [34].

The non-canonical TGF-β signaling pathways. This pathway, also known as the TGF-beta/SMAD4 independent pathway, transmits its signal through factors such as the Rho family of small GTPases, TNF receptor-associated factor 4/6 (TRAF 4/6), the mitogen-activated protein kinases (MAPKs), and phosphatidylinositide 3-kinase (PI3K). Activation of RhoA/ROCK leads to the induction of actin stress fiber formation during EMT via a non-transcriptional mechanism. Activated TRAF 4/6 can initiate the nuclear factor kappaB (NF-kB) signaling pathway, which results in the inflammation and other cell survival processes. TGF-β activation of PI3K and AKT leads to a physical interaction between the PI3K p85 subunit and the receptor complex, resulting in translational responses via mTOR/S6kinase pathways. Activation of MAPKs leads to transcriptional regulation of target genes, either through direct interaction with the nuclear SMAD protein complex, or through other downstream proteins. Meanwhile, activated JNK/p38/ERK work together with SMADs to regulate cellular apoptosis and proliferation, mediating metastasis, angiogenesis, and cellular growth via transcriptional regulation. In the case of pancreatic cancer, the overexpression of TGF-β causes an increase in the signaling in the pathways depicted below, thus promoting tumorigenesis rather than tumor suppression
Molecular Variations in Metastatic Pancreatic Ductal Adenocarcinoma and Potential Targets
PDAC is thought to result from accumulating DNA mutations and molecular alterations in ductal cells which follow a dysplastic transformation of pancreatic intraepithelial neoplasms (PanIN) from PanIN-1 to PanIN-3 before becoming invasive cancer [10]. These changes coincide with several known mutations that are found at increasing rates as the histologic grade of the tissue approaches cancer. KRAS mutations, being the most common, are followed by CDK2, P53, SMAD4, and SWI/SWF [36]. Of interest regarding TGF-β is the SMAD4 mutation, as it is a well-described downstream mediator of TGF-β activity, and its loss coincides with the loss of TGF-β’s tumor suppression [34]. Additionally, pathologic SMAD family molecular alterations, especially SMAD4 mutations, have an important role in activating several non-canonical TGF-β downstream pathways including Sox4, MAPK, ERK, JNK, P38, PI3K/AKT, and WNT/β-catenin [34].
The most common site of metastatic PDAC is the liver due to the portal venous blood flow. However, it is clear that while PDAC cells escape the pancreas, other events are needed to allow for metastatic implantation in the extra-pancreatic microenvironment [37]. The liver niche (or less commonly the lung niche) that is required to allow for PDAC cell growth is not entirely understood but recent work by Giovannetti et al. suggests that pre-metastatic niche formation is a requisite for clinically identifiable metastatic disease [38].
We have investigated this with a slightly different approach. By investigating molecular variations in metastatic PDAC cells by the location of disease, we may be able to describe therapeutic resistance and outcomes by the site as a marker for specific molecular variations. To this end, we have found that the specific pattern of molecular variations in PDAC metastases is associated with the location of metastatic development [39]. This data favors the seed theory of metastatic development that PDAC cells may circulate but cannot develop into metastases for a given pre-metastatic niche (the soil) until the “correct” molecular variations occur for that given niche. In effect, this becomes a stochastic problem that explains the often very late presentation of these patients—so many molecular variations and pre-metastatic niche events are needed that it takes decades for metastatic PDAC development but is not identifiable until those events occur [37].
TGF-β as a PDAC Tumor Suppressor
TGF-β has a well-established role as a tumor suppressor in early stage PDAC [27]. Recent clinical data has shown the critical nature of the tumor suppressor function of the canonical TGF-β signaling. TGF-β exerts its tumor suppression via its role as a regulator of cell proliferation [28]. Specifically, TGF-β stimulation is known to inhibit cell cycle progression into the G1 phase via the induction of cyclin-dependent kinase inhibitors (CDKIs), INK4B, and p21. Simultaneously, it inhibits the expression of MYC, which is known to promote cell proliferation [28].
Shugang et al. have demonstrated that the loss of SMAD4 expression, a critical protein in canonical TGF-β signal cascade, confers a nearly twofold worse outcome [40]. Other groups have shown that STAT3 can commandeer SMAD3, through protein-protein interactions, and limit the canonical signal through the lack of a substrate for the TGF-β receptor kinase [41].
Other work has shown that the tumor suppressor role of TGF-β is only effective when there are no defects in the canonical signal cascade, suggesting an explanation for the apparent TGF-β paradox [42]. Similarly, groups have demonstrated that a variety proto-oncogenes, such as BCL6 and human T cell leukemia virus type I (HTLV-1) tax, showcase their tumorigenic activity via the suppression of TGF-β signaling, further supporting TGF-β’s tumor suppressor role [28].
TGF-β as Tumor Promoter in the Tumor Microenvironment
One of the key features of the local growth of PDAC is the significant stromal proliferation. TGF-β has many functions that are involved in the normal physiologic function of mesenchymal stem cells (MSC) within the bone marrow, controlling their differentiation and activity into fibroblasts, among other things [6]. The relationship, however, is not solely one-sided, as the MSCs are major producers of TGF-β. When combined with the downstream effects of SMAD4 mutations, TGF-β induces transformation of epithelial cells to more primitive forms [20]. The plasticity process involves a loss of epithelial features, such as expression of E-cadherin, and a gain in mesenchymal features, thus promoting invasiveness and stem-cell-like features in cancer cells [20].
In tumors, the presence of fibroblasts has clearly been shown to promote tumor growth, dedifferentiation, and angiogenesis, creating an environment ripe for EMT and lymphovascular invasion [23]. The process of EMT leads to the loss of polarity and cell-cell contact in carcinoma cells, as well as the acquisition of fibroblast-like characteristics [28]. During EMT, claudins, occludins, and scaffold proteins are downregulated via PAR6 and RhoA pathways, resulting in the degradation of tight junctions. Simultaneously, the cell surface protein complex structure is altered independently of nuclear gene regulation [43].
The Ras pathway has also been implicated in the induction of EMT together with TGF-β signaling [44]. Ras, together with PI3K, have been shown to activate Src family tyrosine kinases, which cause the destabilization of E-cadherin-beta-catenin complexes and the disruption of adherens junctions. The induction of SNAI1 by TGF-β is also highly dependent on active Ras signals [28, 44].
Recent studies have also shown that a mutation of p53, an established tumor suppressor, is involved in the switching of TGF-β from a tumor suppressor to a tumor promoter [45]. The mutant p53, together with an activated Smad complex, abrogate the ability of p63 to downregulate sharp-1, leading to an inability to suppress metastasis.
Additionally, the production of fibroblasts in the TME further increases TGF-β production, inducing more desmoplasia, the attraction of more fibroblasts, and further increases in the TGF-β production, worsening the problem as the tumor is no longer suppressed in this environment. Instead, the tumor now finds itself in a place well suited for growth and invasion.
TGF-β and β-Catenin in PDAC
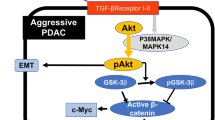
The duality of TGF-β and the paradox itself relates to the evidence that TGF-β is tumor promotive in late-stage PDAC. Our group and others have demonstrated that TGF-β increases EMT, inactivates cytotoxic T cells, neoangiogenesis, and increases fibrosis [14, 15, 42, 46]. The majority of the evidence suggests that non-canonical signaling is responsible for EMT with RAS/RAF signaling playing an important role, as well as snail expression [47, 48]. Interestingly, JNK1 expression has an integral role in the canonical pathway that allows for tumor suppression, whereas lack of JNK1 abrogates the SMAD2/3/4 complex from activating tumor suppressor transcription activity and resulting in tumor promotive EMT [26].
A key aspect of non-canonical TGF-β signaling is the crosstalk with other non-SMAD-based pathways and eventual induction of cancer stem cells and EMT. Previously, our group and others had suggested a relationship between TGF-β and β-catenin in PDAC [21, 24, 49]. Now, others have demonstrated a more precise relationship between these two pathways [50, 51].
This crosstalk between TGF-β and β-catenin is responsible for the increased fibrosis, via the canonical Wnt pathway [52]. However, it is not exclusive to the Wnt pathway. TGF-β is capable of interacting with other factors that are independent of Wnt leading to the activation and accumulation of β-catenin [53]. Given the importance of β-catenin signaling in cancer [54], and the clear evidence that Wnt ligands are not needed for downstream β-catenin effects [55], the use of TGF-β may have untoward effects through inadvertent off-target activation of β-catenin pathways.
TGF-β Inhibitors in Advanced PDAC
Advanced PDAC—a metastatic or locally advanced disease that is not resectable—represents the greatest challenge for patients as the vast majority of patients will die from metastatic disease. Antibodies, inhibitors, and other drugs to target the TGF-β pathways have undergone phase I and phase II clinical trials with most of the data only published in the abstract form. There has been some data published after peer review demonstrating the safety of anti-TGF-β-based therapies.
Recent work by Javle et al. out of the University of Texas M.D. Anderson Cancer Center has suggested that different levels of SMAD4 and TGF-β1 protein expression in tumors from patients with advanced PDAC correlates well with overall survival [56]. This clinical data helps explain the TGF-β paradox seen in both clinical settings and in models of PDAC [24, 30, 42]. While TGF-β inhibitors are undergoing clinical trials, the TGF-β paradox should give pause as to the efficacy of these therapies.
Recently, phase I clinical trial data has demonstrated that galunisertib, a small molecule TGF-β signal inhibitor that works by preventing phosphorylation of SMAD2, is safe and well tolerated when combined with gemcitabine in the treatment of advanced PDAC [57, 58]. Currently, there are three clinical trials investigating galunisertib in the setting of PDAC and an additional trial AP12009 for patients with tumors overexpressing TGF-β2 (http://clinicaltrials.gov). Multiple phase 2 clinical trials have recently been published in the abstract form suggesting an increase in survival when TGF-β inhibitors are added to gemcitabine for advanced PDAC [59]. There are ongoing phase 2 and plans for phase 3 clinical trials investigating this combination but the data is not yet fully matured.
While the preliminary data is welcoming, phase III studies are absolutely critical to not only prove effectiveness but almost as important to understand the off-target effects of anti-TGF-β therapy in a Wnt-independent manner. Our group and others are actively investigating this interaction with hopes of developing rationale combinational therapies to treat PDAC patients.
Conclusion
Pancreatic ductal adenocarcinoma remains a difficult disease to fully contain. The dichotomy of function that TGF-β demonstrates as both a tumor suppressor and promoter adds a significant amount of difficulty to its use as a molecular target for immunotherapy. Further work must be done aimed at isolating key downstream regulators that can be attacked without disrupting the canonical function of TGF-β as an immune modulator and tumor regulator.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–89.
Luberice K, Downs D, Sadowitz B, Ross S, Rosemurgy A. Has survival improved following resection for pancreatic adenocarcinoma? Am J Surg. 2017;214(2):341–6.
Huttner FJ, Fitzmaurice C, Schwarzer G, et al. Pylorus-preserving pancreaticoduodenectomy (pp Whipple) versus pancreaticoduodenectomy (classic Whipple) for surgical treatment of periampullary and pancreatic carcinoma. The Cochrane database of systematic reviews. 2016;2:Cd006053.
Gao J, Wu Y, Su Z, Amoah Barnie P, Jiao Z, Bie Q, et al. Infiltration of alternatively activated macrophages in cancer tissue is associated with MDSC and Th2 polarization in patients with esophageal cancer. PLoS One. 2014;9(8):e104453.
Kerkar SP, Leonardi AJ, van Panhuys N, Zhang L, Yu Z, Crompton JG, et al. Collapse of the tumor stroma is triggered by IL-12 induction of Fas. Mol Ther. 2013;21(7):1369–77.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–90.
Qi W, Huang X, Wang J. Correlation between Th17 cells and tumor microenvironment. Cell Immunol. 2013;285(1–2):18–22.
Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol. 2016;28(8):401–9.
Seo YD, Pillarisetty VG. T-cell programming in pancreatic adenocarcinoma: a review. Cancer Gene Ther. 2017;24(3):106–13.
Glazer ES, Rashid OM, Pimiento JM, Hodul PJ, Malafa MP. Increased neutrophil-to-lymphocyte ratio after neoadjuvant therapy is associated with worse survival after resection of borderline resectable pancreatic ductal adenocarcinoma. Surgery. 2016;160(5):1288–93.
Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098.
Pickup M, Novitskiy S, Moses HL. The roles of TGF beta in the tumour microenvironment. Nat Rev Cancer. 2013;13(11):788–99.
Hussain SM, Reed LF, Krasnick BA, Miranda-Carboni G, Fields RC, Bi Y, et al. IL23 and TGF-ss diminish macrophage associated metastasis in pancreatic carcinoma. Sci Rep. 2018;8(1):5808.
Principe DR, DeCant B, Mascarinas E, Wayne EA, Diaz AM, Akagi N, et al. TGFbeta signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016;76(9):2525–39.
Stylianou A, Gkretsi V, Stylianopoulos T. Transforming growth factor-beta modulates pancreatic cancer associated fibroblasts cell shape, stiffness and invasion. Biochim Biophys Acta. 2018;1862:1537–46.
Moir JA, Mann J, White SA. The role of pancreatic stellate cells in pancreatic cancer. Surg Oncol. 2015;24(3):232–8.
Lohr M, Schmidt C, Ringel J, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001;61(2):550–5.
Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20(10):1218–49.
David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, et al. TGF-beta tumor suppression through a lethal EMT. Cell. 2016;164(5):1015–30.
Glazer ES, Welsh E, Pimiento JM, Teer JK, Malafa MP. TGFbeta1 overexpression is associated with improved survival and low tumor cell proliferation in patients with early-stage pancreatic ductal adenocarcinoma. Oncotarget. 2017;8(1):999–1006.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13(11):788–99.
Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S. TGF-beta - an excellent servant but a bad master. J Transl Med. 2012;10:183.
Lebrun JJ. The dual role of TGFbeta in human cancer: from tumor suppression to cancer metastasis. ISRN molecular biology. 2012;2012:381428.
Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21(2):166–76.
Truty MJ, Urrutia R. Basics of TGF-beta and pancreatic cancer. Pancreatology. 2007;7(5–6):423–35.
Ikushima HM, K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–24.
Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700.
Massague J. TGF beta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30.
Heldin C-H, Moustakas A. Role of Smads in TGF beta signaling. Cell Tissue Res. 2012;347(1):21–36.
Singh P, Srinivasan R, Wig JD, Radotra BD. A study of Smad4, Smad6 and Smad7 in surgically resected samples of pancreatic ductal adenocarcinoma and their correlation with clinicopathological parameters and patient survival. BMC Res Notes. 2011;4:560.
Yamada S, Fujii T, Shimoyama Y, Kanda M, Nakayama G, Sugimoto H, et al. SMAD4 expression predicts local spread and treatment failure in resected pancreatic cancer. Pancreas. 2015;44(4):660–4.
Zhao M, Mishra L, Deng CX. The role of TGF-beta/SMAD4 signaling in cancer. Int J Biol Sci. 2018;14(2):111–23.
Xia X, Wu W, Huang C, Cen G, Jiang T, Cao J, et al. SMAD4 and its role in pancreatic cancer. Tumor Biol. 2015;36:111–9.
Tanaka S. Molecular pathogenesis and targeted therapy of pancreatic cancer. Ann Surg Oncol. 2016;23(Suppl 2):S197–205.
Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16(9):553–65.
Giovannetti E, van der Borden CL, Frampton AE, Ali A, Firuzi O, Peters GJ. Never let it go: stopping key mechanisms underlying metastasis to fight pancreatic cancer. Semin Cancer Biol. 2017;44:43–59.
Ferguson MD, Dong L, Wan J, Deneve JL, Dickson PV, Behrman SW, et al. Molecular alterations associated with DNA repair in pancreatic adenocarcinoma are associated with sites of recurrence. J Gastrointest Cancer. 2018.
Shugang X, Hongfa Y, Jianpeng L, Xu Z, Jingqi F, Xiangxiang L, et al. Prognostic value of SMAD4 in pancreatic cancer: A meta-analysis. Transl Oncol. 2016;9(1):1–7.
Wang G, Yu Y, Sun C, Liu T, Liang T, Zhan L, et al. STAT3 selectively interacts with Smad3 to antagonize TGF-beta. Oncogene. 2016;35(33):4388–98.
Principe DR, Doll JA, Bauer J, et al. TGF-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst. 2014;106(2):djt369.
Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307(5715):1603–9.
Vogelmann R, Nguyen-Tat MD, Giehl K, Adler G, Wedlich D, Menke A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J Cell Sci. 2005;118(20):4901–12.
Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A mutant p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137(1):87–98.
Craven KE, Gore J, Wilson JL, Korc M. Angiogenic gene signature in human pancreatic cancer correlates with TGF-beta and inflammatory transcriptomes. Oncotarget. 2016;7(1):323–41.
Shields MA, Ebine K, Sahai V, Kumar K, Siddiqui K, Hwang RF, et al. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol Cancer Res. 2013;11(9):1078–87.
Kim H, Choi JA, Kim JH. Ras promotes transforming growth factor-beta (TGF-beta)-induced epithelial-mesenchymal transition via a leukotriene B4 receptor-2-linked cascade in mammary epithelial cells. J Biol Chem. 2014;289(32):22151–60.
Kwak HJ, Park DW, Seo JY, Moon JY, Kim TH, Sohn JW, et al. The Wnt/beta-catenin signaling pathway regulates the development of airway remodeling in patients with asthma. Exp Mol Med. 2015;47:e198.
Chen S, Huang J, Liu Z, Liang Q, Zhang N, Jin Y. FAM83A is amplified and promotes cancer stem cell-like traits and chemoresistance in pancreatic cancer. Oncogene. 2017;6(3):e300.
Sannino G, Armbruster N, Bodenhofer M, et al. Role of BCL9L in transforming growth factor-beta (TGF-beta)-induced epithelial-to-mesenchymal-transition (EMT) and metastasis of pancreatic cancer. Oncotarget. 2016;7(45):73725–38.
Akhmetshina AP, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; Schneider, H.; Sadowski, A.; Riener, M.; MacDougald, O. A.; Distler, O.; Schett, G.; Distler, J. H. W. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun 2012;3:1–12.
Zhou BL, Y.; Kahn, M.; Ann, D. K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M. S.; Liebler, J. M.; Minoo, P.; Crandalll, E.D.; Borok, Z. Interactions between β-catenin and transforming growth factor-β signalling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J Biol Chem 2012;287(10):7026–7038.
Shang S, Hua F, Hu ZW. The regulation of beta-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8(20):33972–89.
Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31(12):2714–36.
Javle M, Li Y, Tan D, Dong X, Chang P, Kar S, et al. Biomarkers of TGF-beta signaling pathway and prognosis of pancreatic cancer. PLoS One. 2014;9(1):e85942.
Ikeda M, Takahashi H, Kondo S, Lahn MMF, Ogasawara K, Benhadji KA, et al. Phase 1b study of galunisertib in combination with gemcitabine in Japanese patients with metastatic or locally advanced pancreatic cancer. Cancer Chemother Pharmacol. 2017;79(6):1169–77.
Fujiwara Y, Nokihara H, Yamada Y, Yamamoto N, Sunami K, Utsumi H, et al. Phase 1 study of galunisertib, a TGF-beta receptor I kinase inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015;76(6):1143–52.
de Gramont A, Faivre S, Raymond E. Novel TGF-beta inhibitors ready for prime time in onco-immunology. Oncoimmunology. 2017;6(1):e1257453.
Funding
This research is supported by UTHSC Chancellor’s Oncology Fund (ESG).
Author information
Authors and Affiliations
Contributions
Conception and Design: SMH and ESG.
Acquisition and review of data: MAA, JPF, and ESG.
Analysis and interpretation of data, critical discussion: MAA, SMH, and ESG.
Writing, review, and/or manuscript revision: MAA, JPD, SMH, and ESG.
Study supervision: ESG.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Alvarez, M.A., Freitas, J.P., Mazher Hussain, S. et al. TGF-β Inhibitors in Metastatic Pancreatic Ductal Adenocarcinoma. J Gastrointest Canc 50, 207–213 (2019). https://doi.org/10.1007/s12029-018-00195-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12029-018-00195-5