
Abstract
Introduction
Malignancy is often associated with hematological disorders, but rarely is the diagnosis of malignancy secondary to the diagnosis of microangiopathic hemolytic anemia and thrombocytopenia.
Case reports
We report hereby two patients with metastatic gastric carcinoma presenting with microangiopathic hemolytic anemia and thrombocytopenia. Despite chemotherapy and repeated plasmapheresis in one patient, both patients succumbed shortly after the diagnosis of cancer was made.
Conclusion
A review of the literature regarding microangiopathic hemolytic anemia in cancer patients is discussed. In patients suffering from microangiopathic hemolytic anemia and thrombocytopenia, malignancy should be considered as a possible cause. Early diagnosis of malignancy may be critical for determining the patient’s prognosis and potentially avoiding unnecessary overtreatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Anemia, thrombocytopenia and elevated serum lactate dehydrogenase (LDH) levels are often seen in metastatic cancer patients; in most cases, bone marrow biopsy is not performed. In very rare cases, the diagnosis of malignancy is secondary to the diagnosis of microangiopathic hemolytic anemia (MAHA), caused by the related disorders of thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS) [1–3]. Although most cases of these syndromes are idiopathic, the diagnosis of underlying malignancy is important. Failure to diagnose disseminated malignancy exposes the patient to the major risks of unnecessary plasma exchange and delays chemotherapy. However, failure to urgently initiate plasma exchange treatment in a patient with TTP may result in death [4]. In this article, we report two cases of metastatic gastric carcinoma presenting with MAHA. Both patients died soon after the diagnosis of cancer was made.
Case 1
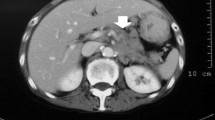
A 45-year-old male with a medical history of smoking for 20 years and peptic ulcer was hospitalized due to back pain and general weakness. Complete blood count showed anemia of 5.9 g/dl, low platelet count, high reticulocyte level, and high serum level of LDH. Blood smear showed MAHA. Spinal computerized tomography (CT) scan demonstrated lytic destruction of vertebrae D2–D3, destruction and collapse of L5 causing pressure on the nerve roots, and disseminated lytic lesions in all other vertebrae and sacrum. Chest and abdominal CT scans revealed fibrotic changes in both lungs, a liver enlarged to 18 cm, and abdominal lymphadenopathy. Bone marrow biopsy showed normal three lineage hematopoiesis; however, it was infiltrated with adenocarcinoma cells. The patient was transferred to our department for further evaluation and treatment.
Panendoscopy revealed an ulcerated tumor in the gastric cardia, with signs of recent non-active bleeding; biopsies from the lesion were consistent with poorly differentiated gastric adenocarcinoma, signet ring cell type. Radiotherapy was given due to impending spinal cord compression; 3,000 cGy was delivered to D1–D5, L3–S1. Despite thrombocytopenia (50,000/μl), chemotherapy was initiated a few days later and consisted of cisplatin 60 mg/m2 given IV on day 1 and 5-fluororouracil (5-FU) 600 mg/m2/day given by continuous infusion on days 1–4. This cycle was repeated 3 weeks later. During the first two chemotherapy courses, the patient was hemodynamically stable, without evidence of active bleeding. Repeated blood transfusions were given due to low hemoglobin (Hb) levels. The platelet count was low but stable, and the patient did not need platelet transfusion.
One day prior to the third chemotherapy course, the patient was admitted to hospital due to hypercalcemia of 14 mg/dl, Hb level of 6.7 g/dl and platelet count of 40,000/μl, with no clinical evidence of active bleeding. He was treated appropriately with normalization of the serum calcium level, but, despite repeated blood transfusions and plasma, blood tests showed low levels of Hb (7.2 g/dl), high levels of LDH and low platelet counts of 18,000/μl, with no clinical evidence of bleeding. Partial thromboplastin time (PTT) and international normalized ratio (INR) were within normal limits, and there was no evidence of renal dysfunction. However, his status continued to deteriorate and he died a week after this admission to hospital.
Case 2
A 32-year-old female was admitted due to fever and pneumonia and was treated with antibiotics. Because of coffee-ground vomiting, she underwent a gastroscopy which showed coffee-ground content with incomplete visualization. CT scans of her chest and abdomen showed pulmonary infiltrates that were interpreted as pneumonia and evidence of multiple lytic spine lesions. Blood tests showed evidence of hemolytic anemia with low Hb levels, low platelet counts, high LDH levels, and schystocyts in the blood smear. Microangiopathic hemolytic anemia was diagnosed. A test for ADAMTS 13 antibodies showed 17 U/ml (normal limits 0–15). Bone marrow biopsy was positive for adenocarcinoma. In spite of broad spectrum antibiotic treatment, septic shock developed and she was transferred to the intensive care unit (ICU).
The patient underwent repeated plasmapheresis (eight treatments performed on 8 days) and was given repeated blood transfusions and fresh frozen plasma. After stabilization, she was discharged from the ICU and admitted to the Oncology Department. Chemotherapy with a combination of IV cisplatin (20 mg/m2, days 1–5) and a reduced dose of etoposide (60 mg/m2, days 1–5) was initiated with granulocyte colony-stimulating factor (GCSF; Filgrastim) support. After the first cycle of chemotherapy, there was improvement in the platelet count and the LDH levels decreased. The patient’s clinical status improved, and the next chemotherapy cycle was given after 21 days with escalated doses of etoposide. During her hospitalization, a brain CT scan was performed due to worsening headaches, which demonstrated sinus vein thrombosis; subcutaneous enoxaparin 40 mg/day (due to low platelet counts) was started. After two cycles of chemotherapy, the patient underwent panendoscopy that showed evidence of linitis plastica, and the biopsy was positive for gastric signet ring cell carcinoma.
Two weeks after her second chemotherapy cycle, the patient was admitted to the hospital due to anemia with Hb level of 6 g/dl and platelet count of 8,000/μl. She received blood transfusions and underwent repeated plasmapheresis. She insisted on trying another line of chemotherapy, and because her clinical state suggested failure of the previous line of treatment, it was decided to treat her with a reduced dose (70%) of 5-FU (425 mg/m2, days 1–5) and leucovorin (20 mg/m2, days 1–5). Her clinical state improved, and it was decided to add a weekly dose of cetuximab (average dose 500 mg). After 21 days, the treatment was stopped after 1 day of chemotherapy due to clinical deterioration. The Hb levels continued to deteriorate and the patient went into a state of sepsis, dying several days later.
Discussion
TTP and HUS are rare, closely related disorders, characterized by MAHA and thrombocytopenia. In the clinical setting, it is often impossible to distinguish between TTP and HUS (TTP–HUS). TTP is classically described as a pentad of hemolytic anemia, thrombocytopenia, neurological symptoms and renal impairment, although only a minority of patients are presented with the complete [1, 3, 5, 6]. In the plasma of patients suffering from TTP, ultra large multimeres of von Willebrand’s factor (ULVWF) have been found. In idiopathic TTP, a deficiency of von Willebrand cleaving protease activity ADAMTS13 has been demonstrated. This protease has been identified as that which processes the ULVWF by proteolytic cleavage [7, 8]. Deficiency of protease can arise from either a congenital deficiency of ADAMTS13 or, more frequently, an autoantibody development. The high titer of antibodies to ADAMTS13 and low-enzyme activity set the rationale behind plasmapheresis that both removes the antibodies and supplies the enzyme in the plasma used for the apheresis process [9–11]. In cancer-associated TTP, there are often no antibodies to ADAMTS13. The absence of such correlation may explain the poor response to plasma exchange in these patients.
The majority of these cases are idiopathic; several etiologic causes have been identified including adverse reactions to drugs. Chemotherapeutic agents that have been described in association with these syndromes include cisplatin, mitomycin C, bleomycin, doxorubicin, 5-FU, interferon-alfa, carboplatin, daunorubicin, dacarbazine, oxaliplatin, lomustine, vinblastine, tamoxifen, carmustine, tretinoin, cytarabine and gemcitabine [12, 13]. Malignancy has been well described for many years as a cause of TTP–HUS [2]. Data from the Oklahoma TTP–HUS Registry of 376 consecutive patients treated with plasma exchange for a diagnosis of TTP or HUS for 17 years were analyzed for the presence of malignancy [4]. In 13 (3%) patients who were initially diagnosed with TTP and begun treatment with plasma exchange, systemic malignancy was subsequently discovered. Except for higher LDH levels (four had values exceeding 5,000 U/l), no difference was seen in the presenting symptoms or the laboratory tests, compared to other TTP–HUS patients. Among those 13 patients, ADAMTS13 activity was measured in 10; the median value was 50% (range 13–100%). One patient with disseminated malignancy had less than 10% ADAMTS13 activity, and none had less than 5%. The prognosis of malignancy-related TTP was much worse than idiopathic TTP, with 2/13 survivals compared to 118/148 survivors [14]. Yet other studies have suggested that ADAMTS13 deficiency may play a role in the pathogenesis of cancer-associated TTP, although the exact mechanism is not completely understood [4, 5].
In both cases presented in this report, the ADAMTS13 level was not studied. In the second case, there was evidence of mildly elevated levels of ADAMTS13 antibodies—15 units while up to 10 is the normal—and usually patients with idiopathic TTP are presented with more than 40 units. In the literature, TTP–HUS was associated with various cancers including breast carcinoma, non-small cell lung cancer, gastric carcinoma, prostate carcinoma, anal squamous cell carcinoma, colon carcinoma, multiple endocrine neoplasia type I, pancreatic carcinoma, renal carcinoma, acute lymphoblastic leukemia, diffuse large B-cell lymphoma and Kaposi’s sarcoma [4, 15–21]. Among the various malignancies, gastric carcinoma is the most common. According to the literature, 25–50% of the reported cases were diagnosed with gastric carcinoma [22, 23]. Our two presented cases also had the same diagnosis. The underlying cause for this finding is not known. Multiple mechanisms, such as tumor infiltration and endothelial damage due to disseminated carcinomatosis, have been suggested.
Levandovsky and colleagues [24] analyzed 178 consecutively treated patients with TTP–HUS, assessing whether clinical or laboratory characteristics could predict important short- and long-term outcome measures. They defined complete response to plasma exchange as a platelet count greater than 100,000/μl for two consecutive evaluations, declining LDH levels (if initially elevated), and no further neurological deficits or progression. A partial response was defined as stabilization of the platelet count below 100,000/μl, with no further neurological deficits or progression. According to these criteria, both patients in the current report had a short-lived partial response.
A significant difference in survival between those with co-morbidities, such as malignancy, and those without co-morbidities (p < 0.001) was found by Levandovsky et al. [24]. Malignancy-associated TTP–HUS had a grave prognosis; this finding agrees with the Oklahoma study [4], whose authors concluded that early recognition of cancer may not benefit many patients who are presented with MAHA and thrombocytopenia, since these patients often have widely disseminated cancer and a poor prognosis.
Without treatment, these syndromes are often fatal, with a mortality rate in excess of 95% [15]. Plasma exchange used to remove the ULVWF and autoantibodies in patients suffering from idiopathic TTP has been shown in several case series to produce response rates of approximately 80% and survival rates greater than 90% [25–29]. The role of plasma exchange in malignancy and chemotherapy-associated TTP is limited [1, 4]. Treatment of the underlying cancer was suggested to be important in the management of cancer-related TTP–HUS [4]. Still, despite early chemotherapy treatment, the survival of these patients remains poor, as in the current cases and other reports [4, 30].
Anemia, thrombocytopenia and elevated LDH levels are often seen in advanced cancer patients; in most cases, bone marrow biopsy or blood smear are not performed. These disturbances may be related to bone marrow suppression due to chemotherapy, radiation treatment and marrow infiltration by advanced cancer. Thus, TTP–HUS in these patient populations is probably underdiagnosed; however, it seems that there is no clinical benefit to performing clinical studies, since these patients usually have a poor prognosis regardless of evaluation and treatment. In cases of presenting TTP–HUS without known malignancy, early diagnosis of systemic malignancy may be crucial for determining the patient’s prognosis and potentially avoiding unnecessary overtreatment.
References
George JN. How I treat patients with thrombotic thrombocytopenic purpura–hemolytic uremic syndrome. Blood. 2000;96:1223–9.
George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006;354:1927–35.
Ruggenenti P, Remuzzi G. Thrombotic microangiopathies. Crit Rev Oncol Hematol. 1991;11:243–65.
Francis KK, Kalyanam N, Terrell DR, Vesely SK, George JN. Disseminated malignancy misdiagnosed as thrombotic thrombocytopenic purpura: a report of 10 patients and a systematic review of published cases. Oncologist. 2007;12:11–9.
Spoormans I, Altintas S, Van den Brande J, Luijks A, Vermorken JB. Purpura in a patient with disseminated breast cancer: a rapidly progressive cancer-related thrombotic thrombocytopenic purpura. Ann Oncol. 2008;19:1204–7.
Moake JL, Byrnes JJ, Troll JH, Rudy CK, Weinstein MJ, Colannino NM, et al. Abnormal VIII: von Willebrand factor patterns in the plasma of patients with the hemolytic–uremic syndrome. Blood. 1984;64:592–8.
Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–94.
Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127:834–9.
Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, et al. von Willebrand factor-cleaving protease in thromboticthrombocytopenic purpura and the hemolytic–uremic syndrome. N Engl J Med. 1998;339:1578–84.
Furlan M, Robles R, Solenthaler M, Lammle B. Acquired deficiency of von Willebrand factor-cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood. 1998;91:2839–46.
Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339:1585–94.
Zakarija A, Bennett C. Drug-induced thrombotic microangiopathy. Semin Thromb Hemost. 2005;31:681–90.
Zupancic M, Shah PC, Shah-Khan F. Gemcitabine-associated thrombotic thrombocytopenic purpura. Lancet Oncol. 2007;8:634–41.
KremerHovinga JA, Vesely SK, Terrell DR, Lammle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115:1500–11.
Aksoy M, Erdem S, Tahsinglu M, Dincol G, Debbag F. Microangiopathic hemolytic anemia due to adenocarcinoma of the stomach. New Istanbul Contrib Clin Sci. 1975;11:103–7.
Podzamczer D, Carreras L, Condom E, Baucells JM, Vidaller A. Microangiopathic hemolytic anemia associated with pulmonary adenocarcinoma. JAMA. 1985;254:2554–5.
Eugene M, Deray G, Cacoub P, Achour A, Baumelou A. Hemolytic uremic syndrome and prostatic adenocarcinoma. Clin Nephrol. 1987;27:46.
Sill H, Hofler G, Kaufmann P, Horina J, Spuller E, Kleinert R, et al. Angiotropic large cell lymphoma presenting as thrombotic microangiopathy (thrombotic thrombocytopenic purpura). Cancer. 1995;75:1167–9.
Kouides PA, Phatak PD, Cramer SF. Fatal thrombotic thrombocytopenia purpura (TTP) presenting concurrently with metastatic multiple endocrine neoplasia (MEN) type I. Hematopathol Mol Hematol. 1996;10:161–70.
Gonzalez N, Rios E, Martin-Noya A, Rodríguez JM. Thrombotic thrombocytopenic purpura and bone marrow necrosis as a complication of gastric neoplasm. Haematologia. 2002;87:ECR01.
Pirrotta MT, Bucalossi A, Forconi F, Bocchia M, Mazzotta S, Sammassimo S, et al. Thrombotic thrombocytopenic purpura secondary to an occult adenocarcinoma. Oncologist. 2005;10:299–300.
Murgo AJ. Thrombotic microangiopathy in the cancer patients including those induced by chemotherapeutic agents. Semin Hematol. 1987;24:161–77.
Lesesne JB, Rothschild N, Erickson B, Korec S, Sisk R, Keller J, et al. Cancer-Associated hemolytic–uremic syndrome: analysis of 85 cases from a national registry. J Clin Oncol. 1989;7:781–9.
Levandovsky M, Harvey D, Lara P, Wun T. Thrombotic thrombocytopenic purpura–hemolytic uremic syndrome (TTP–HUS): a 24-year clinical experience with 178 patients. J Hematol Oncol. 2008;1:23.
Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura–hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325:398–403.
Kwaan HC, Soff GA. Management of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Semin Hematol. 1997;34:159–66.
Lara Jr PN, Coe TL, Zhou H, Fernando L, Holland PV, Wun T. Improved survival with plasma exchange in patients with thrombotic thrombocytopenic purpura–hemolytic uremic syndrome. Am J Med. 1999;107:573–9.
Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325:393–7.
Vesely SK, George JN, Rath W. Long term clinical outcome of thrombotic thrombocytopenic purpura–hemolytic uremic syndrome (TTP–HUS) among different patient groups. Blood. 1999;94:15a.
Vasko R, Koziolek M, Fuzesi L, Konig F, Strutz F, Muller GA. Fulminant plasmapheresis–refractory microangiopathy associated with advanced gastric cancer. Ther Apher Dial. 2010;14:222–5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kaidar-Person, O., Nasrallah, H., Haim, N. et al. Disseminated Carcinoma Diagnosed by Bone Marrow Biopsy in Patients with Microangiopathic Hemolytic Anemia and Thrombocytopenia: A Report of Two Cases with Gastric Cancer and a Review of the Literature. J Gastrointest Canc 42, 123–126 (2011). https://doi.org/10.1007/s12029-010-9204-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12029-010-9204-6