
Abstract
Anemia is very common in aneurysmal subarachnoid hemorrhage (aSAH), with approximately half of the aSAH patient population developing moderate anemia during their hospital stay. The available evidence (both physiologic and clinical) generally supports an association of anemia with unfavorable outcomes. Although aSAH shares a number of common mechanisms of secondary insult with other forms of acute brain injury, aSAH also has specific features that make it unique: an early phase (in which early brain injury predominates) and a delayed phase (in which delayed cerebral ischemia and vasospasm predominate). The effects of both anemia and transfusion are potentially variable between these phases, which may have unique considerations and possibly different risk–benefit profiles. Data on transfusion in this population are almost exclusively limited to observational studies, which suffer from significant heterogeneity and risk of bias. Overall, the results are conflicting, with the balance of the studies suggesting that transfusion is associated with unfavorable outcomes. The transfusion targets that are well established in other critically ill populations should not be automatically applied to patients with aSAH because of the unique disease characteristics of this population and the limited representation of aSAH in the clinical trials that established these targets. There are two upcoming clinical trials evaluating transfusion in aSAH that should help clarify specific transfusion targets. Until then, it is reasonable to base transfusion decisions on the current guidelines and use an individualized approach incorporating physiologic and clinical data when available.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Anemia is ubiquitous in the critically ill population, affecting close to 100% of patients with admissions longer than 7 days [1]. Although it may be intuitive that moderate anemia (hemoglobin [Hb] level < 10 g/dL) is well tolerated in healthy individuals, there is now substantive evidence that it is also tolerated in most critical care populations as well [2, 3]. Further, transfusion to maintain higher Hb levels has not proven to be beneficial and, in fact, may be associated with harm in specific subgroups of critically ill patients, specifically those who are younger and less sick [2]. Unfortunately, the neurocritically ill population has been largely underrepresented in these trials, and specific data regarding both anemia and red blood cell (RBC) transfusion are lacking in this population. A systematic review of comparative studies in neurocritically ill patients from 2012 concluded that because of a general paucity of evidence for “clinically significant outcomes” and the overall risk of bias of the included studies, no specific recommendations could be made regarding whether a liberal or restrictive RBC transfusion strategy should be used [4]. In the case of aneurysmal subarachnoid hemorrhage (aSAH), the unique disease course and pathophysiology may make patients particularly vulnerable to the negative effects of anemia [5]. These unique aspects of aSAH should give us pause in applying what we know about managing anemia from other critically ill populations while we seek evidence specifically in this patient population. In this review, we provide an overview of anemia in aSAH, the physiologic considerations for RBC transfusion, and a review of the evidence on which these considerations are based.
Normal Cerebral Oxygenation
The brain is highly metabolically active, with the third highest metabolic rate of all human organs and tissues [6]. The brain relies almost exclusively on oxygen-coupled glucose metabolism to maintain normal function, so it requires a steady flow (delivery) of oxygenated arterial blood [7]. Despite its small contribution to total body mass (2%), the brain receives a large proportion of the resting cardiac output (10–20%) and accounts for 20% of the total basal oxygen consumption at rest (approximately 50 mL O2/min or 3.5 mL/100 g/min) [8].
Delivery of oxygen to the brain (CDO2) is determined by cerebral blood flow (CBF) and the arterial oxygen content (CaO2) of the blood, as follows: CDO2 = CBF × CaO2 [9]. CBF in normal adults is tightly maintained at 50 mL/100 g/min [10]. It can be conceptualized as laminar flow through a tube and therefore be described using both Ohm’s and Hagen-Poiseuille’s equations [11]. As such, the determinants of CBF are arterial blood pressure, venous pressure or intracranial pressure (ICP) (whichever is greater), the radius of the cerebral blood vessels, and blood viscosity [11]. The CaO2 is almost completely determined by the concentration of Hb and the oxygen saturation (SaO2), with a very small contribution from dissolved oxygen: CaO2 = Hb × 1.36 × (%SaO2/100) + (0.003 × PaO2) [9]. Under normal conditions in the resting brain, the ratio of oxygen consumption to delivery (known as the oxygen extraction ratio or fraction) is around 40–44% [12]. This provides a degree of buffer capacity against tissue hypoxia in the event of reduced CDO2.
Anemia and the “Normal Brain”
Multiple human studies have explored the impact of a reduced Hb concentration (and therefore a reduced CaO2) on cerebral oxygen physiology using healthy volunteers who underwent isovolemic hemodilution as a model for anemia. CaO2 has an inverse linear relationship with CBF: as CaO2 is decreased, there is a corresponding increase in CBF [13,14,15,16]. Animal studies suggest that in response to hemodilution, the regulatory mechanism leading to increased CBF is both complex and multifactorial but predominantly occurs via vasodilation and is primarily mediated by nitric oxide metabolites [9, 17, 18]. This increase in CBF attempts to maintain CDO2 and ultimately preserve tissue oxygenation. However, studies in both animals and humans show that the increase in CBF is not sufficient to completely maintain CDO2, especially at lower Hb concentrations [14, 15, 19,20,21]. Importantly, despite this drop in CDO2, the cerebral metabolic rate of oxygen (CMRO2) remains unchanged because of an increase in cerebral O2 extraction [14, 15, 19]. Therefore, to maintain the CMRO2 in the setting of isovolemic hemodilution (anemia), cerebral O2 extraction increases to compensate for the reduced CDO2.
Although the compensatory increase in CBF may not always be sufficient to maintain CDO2 in the setting of severe anemia (as previously illustrated), it is nevertheless a key component of the response that attempts to preserve tissue oxygenation. Both animal and human studies show that vasodilation is an important contributor to increased CBF in the setting of isovolemic hemodilution [22, 23]. Although hemodilution-induced vasodilation increased CBF, it also caused a reduction in the vasodilator reserve, which could limit the autoregulatory response to additional insults [23]. This vasodilation is part of the cerebrovascular autoregulation response to variation in pressure [24].
Pressure, in this case cerebral perfusion pressure (CPP), is another potentially important factor in the determination of CBF. Any substantial reduction in mean arterial pressure or increase in ICP could reduce CBF. However, because of autoregulation of the cerebral vasculature, the radius of the vessels changes in response to changes in CPP to maintain a constant CBF across a wide range of CPPs or mean arterial pressures (50–150 mmHg) [25]. In the normal brain, autoregulation is preserved, but in the injured brain, autoregulatory capacity can be altered or abolished entirely, leading to impaired CBF and delivery of oxygen [26, 27].
Conversely, although blood viscosity is a factor in CBF, its clinical significance is questioned. Both animal and human studies have found that viscosity likely has a limited to nonexistent role in the control of CBF [19, 28].
Anemia and the Injured Brain
In the injured brain, the negative effects of anemia can be exacerbated by both the features of the injury itself (e.g., elevated ICP or vasospasm) and the compensatory mechanisms that attempt to normalize tissue perfusion and oxygenation (vasodilation via autoregulation). Indeed, animal models of brain injury suggest that the injured brain is highly susceptible to anemia. For example, a canine model of global cerebral ischemia, using isovolemic hemodilution to simulate anemia, demonstrated that at a hematocrit level of 26% there was uncoupling of CaO2, CDO2, and the CMRO2 [29]. Although CBF increased, both CDO2 and CMRO2 fell, and the oxygen extraction fraction increased above the critical level for irreversible ischemia [29]. This may reflect a decrease in the cerebral perfusion reserve and the occurrence of irreversible ischemia.
In a rat model of traumatic brain injury, injured regions of the brain suffered significant secondary injury in the setting of anemia from isovolemic hemodilution [30]. This is compared with noninjured regions, which maintained normal cerebral oxygen tension and did not suffer injury [30]. Finally, In a mathematical modeling study of the effect of anemia on focal ischemic stroke in rabbits, when Hb concentrations were < 10 g/dL, the oxygen uptake in the ischemic penumbra “decreased progressively,” converting potentially salvageable tissue into a permanent infarct [31].
These findings appear to translate to the human population with aSAH. A study of eight patients with transcranial doppler ultrasound-confirmed vasospasm in aSAH assessed cerebral oxygen physiology before and after isovolemic hemodilution, from a hematocrit level of 36–28% (Hb 11.9–9.2 g/dL) [21]. By measuring global and regional CBF with radiolabelled tracers (133Xenon and 99mTc-HMPAO (technetium-99m-labeled hexamethylpropyleneamine oxime)), they demonstrated that anemia leads to disordered cerebral oxygen physiology and increased ischemic brain volume, which could ultimately lead to worse clinical outcomes [21].
Anemia in aSAH is Common and Problematic
Anemia is a common occurrence in aSAH, with approximately 50% of patients developing moderate anemia during their hospital stay [32]. Despite the fact that few patients (~ 5%) have anemia on presentation to the hospital, moderate anemia tends to occur early (day 4 post ictus) in the majority who develop it [32].
Multiple independent risk factors have been associated with the development of anemia in aSAH, including female sex, aSAH clinical severity (modified Fisher grade of 3 or 4 and/or a poor clinical grade), surgical aneurysm treatment, a baseline hematocrit level of < 36%, a history of hypertension or oral anticoagulant use, and the presence of ≥ 3 Systemic Inflammatory Response Syndrome criteria on admission [32, 33]. Inflammation is thought to be a key factor leading to increased consumption and decreased RBC production through myelosuppression as well as abnormalities with erythropoietin and iron metabolism that occur in critically ill patients [34]. Additionally, anemia can be exacerbated by a variety of factors associated with treatment, including hemodilution from fluid resuscitation, intraoperative loss during aneurysm repair, or blood sampling for laboratory testing [34].
Anemia and Adverse Clinical Outcomes in aSAH
Multiple studies have examined the association of anemia with clinical outcomes in aSAH (Table 1). Anemia appears to be associated with unfavorable outcome, though there are conflicting findings across studies [32, 35,36,37,38,39,40,41]. It is difficult to draw firm conclusions because these studies are generally single-center retrospective observational cohort studies with relatively small sample sizes. Further, there is significant heterogeneity across the studies with respect to a number of features, including the definition of anemia used, the time course of anemia studied, the number and type of adjusted confounders, and the specific outcome measures assessed. It thus remains unclear whether anemia is simply a marker of comorbidity and severity of illness or truly an independent prognostic factor.
Physiologic Effects of Anemia in aSAH
Physiologic data strongly support the association of anemia with harm in aSAH. A prospective observational cohort study of 20 patients with poor-grade (Hunt and Hess grade 4 or 5) aSAH used multimodal monitoring to evaluate the association of anemia with brain tissue hypoxia (using brain tissue oxygen tension [PbtO2]) and cellular energy dysfunction (using cerebral microdialysis lactate/pyruvate ratio) [42]. An Hb level < 9 g/dL was a strong independent predictor of both brain tissue hypoxia and cellular energy dysfunction, adjusted for CPP, central venous pressure, the ratio of partial pressure of oxygen in arterial blood to the fraction of inspiratory oxygen (P/F ratio), and presence of symptomatic vasospasm [42].
A retrospective cohort study of 34 patients with subarachnoid hemorrhage used multimodal monitoring data (PbtO2 and cerebral microdialysis) from a total of 259 h of monitoring to evaluate the association of low Hb levels with brain tissue hypoxia and cellular energy dysfunction [43]. The magnitude of anemia impacted brain hypoxia; for every 1-g/dL decrease in the Hb level, there was a 70% increase in brain hypoxia [43]. Compared to an Hb level of 10.1–11 g/dL, an Hb level of ≤ 9 g/dL was associated with a 280% increase in cellular energy dysfunction.
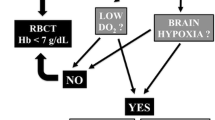
Although aSAH shares the basic physiological principles of cerebral oxygenation and metabolism with other forms of brain injury, there are unique features that may contribute to a particular vulnerability to anemia of this population (Fig. 1). The postbleed period of aSAH is thought to be complicated by two distinct phases: a period of early brain injury (typically during the first 72 h) and a period of delayed cerebral ischemia (DCI)/vasospasm (beyond 72 h until 21 days) [44].

Adapted from Okazaki T, Kuroda Y. Aneurysmal subarachnoid hemorrhage: intensive care for improving neurological outcome. J Intensive Care. 2018;6:28. https://doi.org/10.1186/s40560-018-0297-5 and from Dodd WS, Laurent D, Dumont AS, et al. Pathophysiology of delayed cerebral ischemia after subarachnoid hemorrhage: a review. J Am Heart Assoc. 2021;10:e021845.
Impact of anemia and RBC transfusion in aSAH: a conceptual model. aSAH aneurysmal subarachnoid hemorrhage, CPP cerebral perfusion pressure, DO2 xxx, ICP intracranial pressure, PbtO2 brain tissue oxygen tension.
Anemia During the Early aSAH Phase (0–72 h): Early Brain Injury
Early brain injury is a relatively new concept in aSAH, and as a result, the pathophysiologic processes are not well understood [44]. There is a variety of proposed mechanisms believed to contribute to injury during this phase, including the following [45]:
-
Sudden increase in ICP leading to decreased CPP, impaired autoregulation, and possibly transient or persistent ischemia
-
Neuronal cell death and endothelial damage leading to cytotoxic edema and breakdown of the blood–brain barrier potentiating vasogenic edema
-
Further cell death via microcirculatory failure, microthrombosis, altered ionic hemostasis, excitotoxicity, oxidative stress, and neuronal swelling
-
Energy dysfunction due to cortical spreading depolarizations or mitochondrial dysfunction
-
Subarachnoid blood or intraventricular blood may cause microglial activation and inflammation
During this early phase, CDO2 is at particular risk of being reduced because of elevated ICP (from edema or hydrocephalus), intentionally lowered blood pressure (to prevent aneurysmal rebleeding), and possible cardiac dysfunction (neurogenic stress cardiomyopathy). Meanwhile, in the setting of impaired autoregulatory mechanisms, liberalized blood pressure targets post aneurysm securement could lead to hyperemia, edema, and elevated ICP, further worsening CDO2, particularly to regional areas of injury and ischemia.
In general, most of the studies of anemia in aSAH do not discriminate between the early and the delayed phases of aSAH. However, a secondary analysis on 413 patients from the CONSCIOUS-1 (Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage) study was able to achieve some distinction between these phases. The CONSCIOUS-1 study was a phase 2 randomized, double blind, placebo-controlled dose-finding trial in patients with aSAH that compared clozosentan (an endothelin receptor antagonist) to a placebo, with the primary end point of angiographic vasospasm [36]. The secondary analysis found that anemia (Hb level < 10 g/dL) was present in 29% of patients during days 1–3, and it was associated with poor neurologic outcome and increased risk of death [36]. Interestingly, anemia that occurred during the “peak DCI risk period” (days 5–9) was only associated with poor neurologic outcome but not mortality [36].
Anemia During the Delayed Phase (72 h to 21 Days): DCI/Vasospasm
The delayed phase of aSAH is classically the period from 72 h up to 21 days post rupture and is typified by both vasospasm and DCI. Previously, it was thought that macrovascular vasospasm led directly to DCI; however, contemporary evidence shows that this is not the case: approximately 70% of patients develop angiographic vasospasm, whereas only 30% develop DCI [44]. Other mechanisms are clearly contributing, of which CDO2 and Hb concentration may be one important element.
Based on the earlier discussion of the physiology of cerebral oxygen delivery, there is a significant possibility that anemia will reduce the CDO2, leading to increased risk of ischemia. In a study of eight patients with aSAH and vasospasm, isovolemic hemodilution to a hematocrit level of 28% produced a 12% decrease in global cerebral oxygen delivery and a 7% increase in the volume of ischemic brain tissue, despite an overall increase in CBF [21].
Additional evidence suggesting the importance of CaO2 and CDO2 in the time period of vasospasm and DCI comes from a retrospective cohort study of 245 patients with aSAH [38]. Patients with unfavorable outcomes had a lower Hb level between days 6 and 11 [38]. However, the notion that a higher Hb level is associated with better outcomes is challenged by conflicting data which suggests that an elevated Hb level on admission is associated with a higher incidence of DCI and deep vein thrombosis [46].
Transfusion in aSAH: Available Evidence
Transfusion and Physiologic Outcomes
Studies evaluating physiologic outcome measures support the intuitive notion that transfusion addresses the physiologic and clinical harms of anemia. A small prospective study of patients with aSAH with anemia found that RBC transfusion led to an 18% increase in CDO2, with global CBF remaining stable [47]. Similarly, in a study using oxygen-15 positron emission tomography, RBC transfusion in patients with aSAH who were at risk for DCI led to an increase in CBF and CDO2. Because the findings were similar to those of other interventions, including a fluid bolus (15 mL/kg) and induced hypertension (25% increase), the authors concluded that transfusion may be a meaningful alternative or adjunct to hemodynamic augmentation [48]. Another small cohort study of 15 patients with aSAH found that transfusion led to a significant increase in PbtO2 (every 1-g/dL increase in the Hb level was associated with a 1.39-mmHg increase in PbtO2 [p = 0.036]) [49]. Finally, a prospective cohort study evaluated 56 anemic (Hb level 7–13 g/dL) patients with aSAH who had their aneurysm secured and were deemed at risk for DCI [50]. Using oxygen-15 positron emission tomography scans to measure CDO2 pretransfusion and post transfusion of a single unit of RBCs, they found a 10% increase in global CDO2 and a 16% increase in CDO2 to vulnerable brain regions [50].
Transfusion and Clinical Outcomes: Observational Evidence
Despite this apparent physiologic rationale that transfusion improves CDO2, studies evaluating clinical outcomes have yielded conflicting results (Table 2). However, these results are affected by several limitations. First, the majority of these studies included data prior to the publication of the International Subarachnoid Aneurysm Trial and widespread acceptance of endovascular aneurysm treatment [51]. Patients requiring transfusion may have been more likely to have had open surgical treatment of their aneurysm. Second, given the observational nature of these studies, confounding may have played a significant role. Other factors that are associated with both transfusion and poor outcome (confounding by indication) could have biased the results. Third, there is significant variation in the transfusion thresholds used in these studies (from 7 to 11.5 g/dL), creating significant heterogeneity and limiting the comparison of results across studies. In actual practice, it appears that most clinicians are using transfusions thresholds in the 8–9-g/dL range. A North American survey from 2011 showed that for good-grade aSAH, the mean transfusion threshold was 7.85 g/dL, and for patients with DCI, it was 8.58 g/dL [52]. Fourth, the majority of studies on RBC transfusion in aSAH are retrospective in nature. These do not account for the timing of RBC transfusion (before, during, or after DCI), and as a result, DCI may be the cause of the unfavorable outcomes as opposed to the RBC transfusion [53]. Interestingly, a recent systematic review from 2021 describes a number of the same concerns but concludes that the overall benefits of transfusion likely outweigh the risks [54].
Transfusion and Clinical Outcomes: Randomized Controlled Trial Evidence
Clinical trials evaluating anemia and transfusion thresholds specific to aSAH are lacking. There is a single pilot trial evaluating the safety and feasibility of RBC transfusion in patients with aSAH at high risk of vasospasm [55]. The trial enrolled patients within 3 days of ictus who had a World Federation of Neurological Surgeons grade of 2–4 (or grade 1 with thick subarachnoid clot) and randomized them to an Hb transfusion trigger of 10 vs. 11.5 g/dL. The primary hypothesis was that an Hb level of 11.5 vs. 10 g/dL would not increase the number of days with a core temperature (> 100.4 °F) or decrease the number of ventilator-free days (during the first 13 days). The primary feasibility end point was that a higher Hb goal level would lead to a significantly higher Hb level during the study period. Radiographic (cerebral infarction) and clinical outcomes (impairment of functional status) were assessed. Cerebral infarction was evaluated at 14 days using magnetic resonance imaging, and clinical outcomes were measured at multiple time points over 3 months (using the National Institutes of Health Stroke Scale and/or the modified Rankin Scale). The results showed that the Hb level was different between the groups starting at day 4, but there was no difference in clinical outcomes. As a small pilot study, it was not powered to detect meaningful differences in outcomes, leading the study authors to conclude that a phase III trial is needed. Additionally, although the results confirmed both the safety and feasibility, the transfusion targets were considerably higher (10 vs. 11.5 g/dL) than those used by most clinicians, limiting the generalizability of the study protocol and, consequently, the external validity of the study results.
Guidelines
Not surprising given the existing physiologic and clinical data, the recommendations for the management of anemia are either inconsistent between guidelines or absent entirely (Table 3) [56,57,58,59,60]. The British Committee for Standards in Haematology guidelines recommend a target Hb level of 8–10 g/dL in patients with aSAH, while recognizing that the optimal Hb level has not be defined and that there is uncertainty surrounding the effects of transfusion on outcomes in this population [56]. The Neurocritical Care Society previously recommended maintaining a higher Hb level (8–10 g/dL) in patients with or at risk for DCI [57]. However, the new Neurocritical Care Society guidelines identify that there is insufficient evidence to recommend a transfusion threshold of greater than 7 g/dL in patients with aSAH [61]. The American Heart Association/American Stroke Association guidelines state that RBC transfusion “might be reasonable” for patients at risk of DCI, but the ideal Hb level is unclear [58]. The Chinese Stroke Association guidelines mention that a higher baseline Hb level is associated with better outcomes, but they recommend against RBC transfusion [62]. And finally, both the European Stroke Organization and the Japanese Society on Surgery for Cerebral Stroke guidelines do not address anemia or transfusion in aSAH [59, 60].
Upcoming Evidence/Future Considerations
Clinical trials evaluating the effect of RBC transfusion on meaningful clinical outcomes are needed. As afore reviewed, there is little existing evidence to guide clinicians on, specifically, when to transfuse, in which patients, and at what threshold. The effect of transfusion on the aSAH disease course, as well as patient-important clinical outcomes, needs to be better understood.
There are two relevant ongoing studies in the field:
-
1.
The SAHaRA (Aneurysmal SubArachnoid Hemorrhage - Red blood cell transfusion And outcome) trial (NCT03309579) addresses anemia and transfusion thresholds specifically in aSAH. It is a Canadian-led international prospective open-label, blinded end-point randomized controlled trial that plans to enroll 740 adults with aSAH and moderate anemia (Hb level ≤ 10 g/dL) [63]. The study compares a liberal versus a restrictive RBC transfusion trigger (≤ 10 vs. ≤ 8 g/dL) during the first 21 days. The primary outcome is functional neurologic status measured at 12 months using the modified Rankin Scale. The estimated study completion date is in the fall of 2024.
-
2.
The TRAIN (TRansfusion strategies in Acute brain INjured patients) trial (NCT02968654) is a European prospective randomized controlled trial that plans to enroll 1,000 patients within 10 days of acute brain injury, including aSAH (as well as other causes, such as traumatic brain injury) and randomize them to either a restrictive RBC transfusion trigger (7 g/dL) or a liberal RBC transfusion trigger (9 g/dL) for 28 days or until hospital discharge. The primary outcome is good neurological outcome at 180 days, defined as a Glasgow Outcome Score Extended score of 6–8 [64]. The estimated study completion date is the summer of 2023.
The results of these studies should help inform the timing and Hb trigger for transfusion in the acute phases of management of this particularly vulnerable patient population.
Suggested Practice Guidance
Evidence supporting a specific transfusion threshold in the general critical care population should not necessarily be extrapolated to aSAH because of the unique disease processes that can affect CDO2, including early brain injury and DCI. The majority of the current evidence, both physiologic and clinical, supports an association between anemia and unfavorable outcomes in patients with aSAH.
Evidence supporting a particular transfusion strategy in aSAH is extremely limited [65]. Physiologically, RBC transfusion in anemia leads to improvements in CDO2 and reduction in ischemic burden. However, studies evaluating clinical outcomes have yielded conflicting results. The studies reviewed in this article have transfusion triggers ranging from 7 to 11.5 g/dL. Most studies on anemia and transfusion seem to support a more liberal transfusion strategy, with a target Hb level ≥ 10 g/dL [54]. However, this must be balanced with the known risks of transfusion and the limitations of this evidence (Table 4).
In light of the limited evidence, a practical approach to anemia in aSAH includes avoidance of significant anemia with a transfusion threshold of 7–8 g/dL during the acute postrupture period (up to 21 days). Aiming for a higher Hb level in higher risk patients, such as those with clinical and/or physiologic risk factors, including poor clinical grade, presence of intracranial hypertension, active vasospasm or DCI, physiologic markers of hypoxic distress, or systemic factors that may negatively affect cerebral CDO2 (cardiac dysfunction, vasodilatory shock, etc.), is intuitively appealing but not supported by current evidence. With these points in mind and depending on local protocols, it is reasonable to transfuse patients with aSAH with the objective to aim for Hb levels above 7–10 g/dL. Randomized controlled trial evidence is needed to better inform RBC transfusion thresholds in anemic patients with aSAH, and such evidence is forthcoming.
Conclusions
In summary, evidence surrounding RBC transfusion in aSAH is bolstered by a strong physiologic rationale but limited to small largely single-center retrospective observational studies with conflicting results. The timing of transfusion in the aSAH disease course is poorly understood, as is its effect on outcome. Further trial evidence is needed.
References
Thomas J, Jensen L, Nahirniak S, Gibney RTN. Anemia and blood transfusion practices in the critically ill: a prospective cohort review. Heart Lung. 2010;39(3):217–25.
Hébert PC, Martin C, Yetisir E. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med. 1999;340:409–17.
Carson JL, Stanworth SJ, Dennis JA, et al. Transfusion thresholds for guiding red blood cell transfusion. Cochrane Database Syst Rev [internet]. 2021. https://doi.org/10.1002/14651858.CD002042.pub5.
Desjardins P, Turgeon AF, Tremblay M-H, et al. Hemoglobin levels and transfusions in neurocritically ill patients: a systematic review of comparative studies. Crit Care. 2012;16(2):R54.
Kumar M. Anemia and blood transfusion in subarachnoid hemorrhage. JHN J [Internet]. 2009;4(4):6.
Wang Z, Ying Z, Bosy-Westphal A, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. 2010;92(6):1369–77.
Tameem A, Krovvidi H. Cerebral physiology. Contin Educ Anaesth Crit Care Pain. 2013;13(4):113–8.
Williams LR, Leggett RW. Reference values for resting blood flow to organs of man. Clin Phys Physiol Meas. 1989;10(3):187–217.
Hoiland RL, Bain AR, Rieger MG, Bailey DM, Ainslie PN. Hypoxemia, oxygen content, and the regulation of cerebral blood flow. Am J Physiol Regul Integr Comp Physiol. 2016;310(5):R398-413.
Lassen NA. Normal average value of cerebral blood flow in younger adults is 50 ml/100 g/min. J Cereb Blood Flow Metab. 1985;5(3):347–9.
Cipolla M. The cerebral circulation. San Rafael: Morgan & Claypool Life Sciences; 2009.
Fan AP, An H, Moradi F, et al. Quantification of brain oxygen extraction and metabolism with [15O]-gas PET: a technical review in the era of PET/MRI. Neuroimage. 2020;220:117136.
Brown MM, Wade JPH, Marshall J. Fundamental importance of arterial oxygen content in the regulation of cerebral blood flow in man. Brain. 1985;108(1):81–93.
Hino A, Ueda S, Mizukawa N, Imahori Y, Tenjin H. Effect of hemodilution on cerebral hemodynamics and oxygen metabolism. Stroke. 1992;23(3):423–6.
Mühling J, Dehne MG, Sablotzki A, Hempelmann G. Cerebral blood flow velocity during isovolemic hemodilution and subsequent autologous blood retransfusion. Can J Anesth/J Can Anesth. 1999;46(6):550–7.
Carr JM, Ainslie PN, MacLeod DB, et al. Cerebral O2 and CO2 transport in isovolumic haemodilution: Compensation of cerebral delivery of O2 and maintenance of cerebrovascular reactivity to CO2. J Cereb Blood Flow Metab. 2023;43(1):99–114.
McLaren AT, David Mazer C, Zhang H, Liu E, Mok L, Hare GMT. A potential role for inducible nitric oxide synthase in the cerebral response to acute hemodilution. Can J Anesth/J Can Anesth. 2009;56(7):502–9.
Hudetz AG, Shen H, Kampine JP. Nitric oxide from neuronal NOS plays critical role in cerebral capillary flow response to hypoxia. Am J Physiol Heart Circ Physiol. 1998;274(3):H982–9.
Todd MM, Wu B, Maktabi M, Hindman BJ, Warner DS. Cerebral blood flow and oxygen delivery during hypoxemia and hemodilution: role of arterial oxygen content. Am J Physiol Heart Circ Physiol. 1994;267(5):H2025–31.
Tomiyama Y, Jansen K, Brian JE, Todd MM. Hemodilution, cerebral O2 delivery, and cerebral blood flow: a study using hyperbaric oxygenation. Am J Physiol Heart Circ Physiol. 1999;276(4):H1190–6.
Ekelund A, Reinstrup P, Ryding E, et al. Effects of iso- and hypervolemic hemodilution on regional cerebral blood flow and oxygen delivery for patients with vasospasm after aneurysmal subarachnoid hemorrhage. Acta Neurochir (Wien). 2002;144(7):703–13.
Tu Y-K, Liu H-M. Effects of isovolemic hemodilution on hemodynamics, cerebral perfusion, and cerebral vascular reactivity. Stroke. 1996;27(3):441–5.
Crystal GJ, Czinn EA, Salem MR. The mechanism of increased blood flow in the brain and spinal cord during hemodilution. Anesth Analg. 2014;118(3):637–43.
Willie CK, Tzeng Y-C, Fisher JA, Ainslie PN. Integrative regulation of human brain blood flow. J Physiol. 2014;592(5):841–59.
Fantini S, Sassaroli A, Tgavalekos KT, Kornbluth J. Cerebral blood flow and autoregulation: current measurement techniques and prospects for noninvasive optical methods. Neurophoton. 2016;3(3):031411.
Czosnyka M, Smielewski P, Piechnik S, Steiner LA, Pickard JD. Cerebral autoregulation following head injury. J Neurosurg. 2001;95(5):756–63.
Czosnyka M, Smielewski P, Kirkpatrick P, Menon DK, Pickard JD. Monitoring of cerebral autoregulation in head-injured patients. Stroke. 1996;27(10):1829–34.
Brown MM, Marshall J. Effect of plasma exchange on blood viscosity and cerebral blood flow. BMJ. 1982;284(6331):1733–6.
Tu Y-K, Kuo M-F, Liu H-M. Cerebral oxygen transport and metabolism during graded isovolemic hemodilution in experimental global ischemia. J Neurol Sci. 1997;150(2):115–22.
Hare GMT, Mazer CD, Hutchison JS, et al. Severe hemodilutional anemia increases cerebral tissue injury following acute neurotrauma. J Appl Physiol. 2007;103:9.
Dexter F, Hindman BJ. Effect of haemoglobin concentration on brain oxygenation in focal stroke: a mathematical modelling study. Br J Anaesth. 1997;79(3):346–51.
English SW, Chassé M, Turgeon AF, et al. Anemia prevalence and incidence and red blood cell transfusion practices in aneurysmal subarachnoid hemorrhage: results of a multicenter cohort study. Crit Care. 2018;22(1):169.
Sampson TR, Dhar R, Diringer MN. Factors associated with the development of anemia after subarachnoid hemorrhage. Neurocrit Care. 2010;12(1):4–9.
Hayden SJ, Albert TJ, Watkins TR, Swenson ER. Anemia in critical illness: insights into etiology, consequences, and management. Am J Respir Crit Care Med. 2012;185(10):1049–57.
Schmitt E, Meybohm P, Neef V, et al. Preoperative anaemia and red blood cell transfusion in patients with aneurysmal subarachnoid and intracerebral haemorrhage: a multicentre subanalysis of the German PBM network registry. Acta Neurochir (Wien). 2022;164(4):985–99.
Ayling OGS, Ibrahim GM, Alotaibi NM, Gooderham PA, Macdonald RL. Anemia after aneurysmal subarachnoid hemorrhage is associated with poor outcome and death. Stroke. 2018;49(8):1859–65.
Stein M, Brokmeier L, Herrmann J, et al. Mean hemoglobin concentration after acute subarachnoid hemorrhage and the relation to outcome, mortality, vasospasm, and brain infarction. J Clin Neurosci. 2015;22(3):530–4.
Kramer AH, Zygun DA, Bleck TP, Dumont AS, Kassell NF, Nathan B. Relationship between hemoglobin concentrations and outcomes across subgroups of patients with aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2009;10(2):157–65.
Kramer AH, Gurka MJ, Nathan B, Dumont AS, Kassell NF, Bleck TP. Complications associated with anemia and blood transfusion in patients with aneurysmal subarachnoid hemorrhage. Crit Care Med. 2008;36(7):2070–5.
Naidech AM, Jovanovic B, Wartenberg KE, et al. Higher hemoglobin is associated with improved outcome after subarachnoid hemorrhage*. Crit Care Med. 2007;35(10):2383–9.
Castella A, Attanasio L, Schuind S, et al. Association of anemia and transfusions with outcome after subarachnoid hemorrhage. Clin Neurol Neurosurg. 2021;206:106676.
Oddo M, Milby A, Chen I, et al. Hemoglobin concentration and cerebral metabolism in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2009;40(4):1275–81.
Kurtz P, Schmidt JM, Claassen J, et al. Anemia is associated with metabolic distress and brain tissue hypoxia after subarachnoid hemorrhage. Neurocrit Care. 2010;13(1):10–6.
Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KTS. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth. 2012;109(3):315–29.
Rass V, Helbok R. Early brain injury after poor-grade subarachnoid hemorrhage. Curr Neurol Neurosci Rep. 2019;19(10):78.
Li R, Lin F, Chen Y, et al. Elevated blood hemoglobin on admission as an independent predictor of unfavorable outcomes in patients with aneurysmal subarachnoid hemorrhage. Neurosurg Rev [Internet]. 2022. https://doi.org/10.1007/s10143-022-01780-w.
Dhar R, Zazulia AR, Videen TO, Zipfel GJ, Derdeyn CP, Diringer MN. Red blood cell transfusion increases cerebral oxygen delivery in anemic patients with subarachnoid hemorrhage. Stroke. 2009;40(9):3039–44.
Dhar R, Scalfani MT, Zazulia AR, Videen TO, Derdeyn CP, Diringer MN. Comparison of induced hypertension, fluid bolus, and blood transfusion to augment cerebral oxygen delivery after subarachnoid hemorrhage: clinical article. JNS. 2012;116(3):648–56.
Kurtz P, Helbok R, Claassen J, et al. The effect of packed red blood cell transfusion on cerebral oxygenation and metabolism after subarachnoid hemorrhage. Neurocrit Care. 2016;24(1):118–21.
Dhar R, Zazulia AR, Derdeyn CP, Diringer MN. RBC transfusion improves cerebral oxygen delivery in subarachnoid hemorrhage. Crit Care Med. 2017;45(4):653–9.
Molyneux A. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: a randomised trial. The Lancet. 2002;360(9342):1267–74.
Kramer AH, Diringer MN, Suarez JI, Naidech AM, Macdonald LR, Le Roux PD. Red blood cell transfusion in patients with subarachnoid hemorrhage: a multidisciplinary North American survey. Crit Care. 2011;15(1):R30.
LeRoux P. Haemoglobin management in acute brain injury. Curr Opin Crit Care. 2013;19(2):83–91.
Mofor P, Oduguwa E, Tao J, et al. Postoperative transfusion guidelines in aneurysmal cerebral subarachnoid hemorrhage: a systematic review and critical summary of available evidence. World Neurosurg. 2021;S1878–8750(21):01850–7.
Naidech AM, Shaibani A, Garg RK, et al. Prospective, randomized trial of higher goal hemoglobin after subarachnoid hemorrhage. Neurocrit Care. 2010;13(3):313–20.
Retter A, Wyncoll D, Pearse R, et al. Guidelines on the management of anaemia and red cell transfusion in adult critically ill patients. Br J Haematol. 2013;160(4):445–64.
Diringer MN, Bleck TP, Claude Hemphill J, et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s multidisciplinary consensus conference. Neurocrit Care. 2011;15(2):211.
Connolly ES, Rabinstein AA, Carhuapoma JR, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43(6):1711–37.
Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G. European stroke organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis. 2013;35(2):93–112.
Committee for Guidelines for Management of Aneurysmal Subarachnoid Hemorrhage, Japanese Society on Surgery for Cerebral Stroke. Evidence-based guidelines for the management of aneurysmal subarachnoid hemorrhage. Neurol Med Chir (Tokyo). 2012;52(6):355–429.
Treggiari MM, Rabinstein AA, Busl KM, et al. Guidelines for the neurocritical care management of aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2023. https://doi.org/10.1007/s12028-023-01713-5.
Dong Y, Guo Z-N, Li Q, et al. Chinese Stroke Association guidelines for clinical management of cerebrovascular disorders: executive summary and 2019 update of clinical management of spontaneous subarachnoid haemorrhage. Stroke Vasc Neurol. 2019;4(4):176–81.
English SW, McIntyre L. Is hemoglobin good for cerebral oxygenation and clinical outcome in acute brain injury? Curr Opin Crit Care. 2018;24(2):91–6.
Taccone F. Transfusion strategies in acute brain injured patients. A prospective multicenter randomized study. [Internet]. clinicaltrials.gov; 2021 [cited 2022 May 22]. https://clinicaltrials.gov/ct2/show/study/NCT02968654
Cable CA, Razavi SA, Roback JD, Murphy DJ. RBC transfusion strategies in the ICU: a concise review. Crit Care Med. 2019;47(11):1637–44.
Moman RN, Kor DJ, Chandran A, et al. Red blood cell transfusion in acute brain injury subtypes: an observational cohort study. J Crit Care. 2019;50:44–9.
Kumar MA, Levine J, Faerber J, et al. The effects of red blood cell transfusion on functional outcome after aneurysmal subarachnoid hemorrhage. World Neurosurg. 2017;108:807–16.
Pegoli M, Mandrekar J, Rabinstein AA, Lanzino G. Predictors of excellent functional outcome in aneurysmal subarachnoid hemorrhage. JNS. 2015;122(2):414–8.
Kumar MA, Boland TA, Baiou M, et al. Red blood cell transfusion increases the risk of thrombotic events in patients with subarachnoid hemorrhage. Neurocrit Care. 2014;20(1):84–90.
Festic E, Rabinstein AA, Freeman WD, et al. Blood transfusion is an important predictor of hospital mortality among patients with aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2013;18(2):209–15.
Broessner G, Lackner P, Hoefer C, et al. Influence of red blood cell transfusion on mortality and long-term functional outcome in 292 patients with spontaneous* subarachnoid hemorrhage. Crit Care Med. 2009;37(6):1886–92.
Springer MVMD, Schmidt JM, Wartenberg KEMD, Frontera JAMD, Badjatia NMD, Mayer SAMD. Predictors of global cognitive impairment 1 year after subarachnoid hemorrhage. Neurosurgery. 2009;65(6):1043–51.
Funding
This review did not receive any funding.
Author information
Authors and Affiliations
Contributions
LAT and SWE conceived and designed the manuscript. LAT performed the review of the literature and created the tables and figure. SWE, LM, and AFT reviewed each draft and provided critical feedback and content expertise. The final version was reviewed and approved by all authors.
Corresponding author
Ethics declarations
Conflict of interests
S. English is the recipient of a National New Investigator Award from the Heart and Stroke Foundation. A. Turgeon holds a Canada Research Chair in Critical Care Neurology and Trauma.
Ethical approval/informed consent
Because this is a review article, ethics and institutional review board approval was not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Terrett, L.A., McIntyre, L., Turgeon, A.F. et al. Anemia and Red Blood Cell Transfusion in Aneurysmal Subarachnoid Hemorrhage. Neurocrit Care 39, 91–103 (2023). https://doi.org/10.1007/s12028-023-01815-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-023-01815-0