
Abstract
Background
Transcranial Doppler (TCD) is widely used to detect and follow up cerebral vasospasm after subarachnoid hemorrhage (SAH). Therapeutic hypothermia might influence blood flow velocities assessed by TCD. The aim of the study was to evaluate the effect of hypothermia on Doppler blood flow velocity after SAH.
Methods
In 20 patients treated with hypothermia (33°) due to refractory intracranial hypertension or delayed cerebral ischemia (DCI), mean flow velocity of the middle cerebral artery (MFVMCA) was assessed by TCD. Thirteen patients were treated with combined hypothermia and barbiturate coma and seven with hypothermia alone. MFVMCA was obtained within 24 h before and after induction of hypothermia as well as before and after rewarming.
Results
Hypothermia was induced on average 5 days after SAH (range 1–12) and maintained for 144 h (range 29–270). After hypothermia induction, MFVMCA decreased from 113.7 ± 49.0 to 93.8 ± 44.7 cm/s (p = 0.001). The decrease was independent of SAH-related complications and barbiturate coma. MFVMCA further decreased by 28.2 cm/s between early and late hypothermia (p < 0.001). This second decrease was observed in patients with DCI (p < 0.001), but not in patients with intracranial hypertension (p = 0.715). Compared to late hypothermia, MFVMCA remained unchanged after rewarming (65.6 ± 32.1 vs 70.3 ± 36.8 cm/s; p = 0.219). However, patients treated with hypothermia alone showed an increase in MFVMCA after rewarming (p = 0.016).
Conclusion
Therapeutic hypothermia after SAH decreases Doppler blood flow velocity in both intracranial hypertension and DCI cases. The results can be the effect of hypothermia-related mechanisms or resolving cerebral vasospasm during prolonged hypothermia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Refractory high intracranial pressure (ICP) and delayed cerebral ischemia (DCI) are the most important causes of secondary brain injury after aneurysmal subarachnoid hemorrhage (SAH) [1, 2]. Recent animal studies have demonstrated that mild hypothermia (32–34 °C) has the potential to limit the extent of secondary brain injury by reversing acute cerebral perfusion pressure-independent hypoperfusion [3, 4], preventing brain edema formation [5, 6], and reducing cerebral vasospasm [7]. However, short-term hypothermia during aneurysm surgery failed to improve outcome in patients with good-grade SAH [8], suggesting that only a carefully selected group of patients with severe SAH suffering from intracranial hypertension and/or DCI might benefit from prolonged hypothermia treatment [9–11].
Although mild hypothermia likely interferes with a variety of processes leading to secondary brain injury, the most prevalent neuroprotective effect has been attributed to the suppression of cerebral metabolic oxygen and glucose demand [12, 13]. As a consequence, hypothermia has consistently been associated with reduced cerebral blood flow (CBF) under physiological conditions [14, 15]. However, CBF measurements during hypothermia in patients with acute brain injury are conflicting and the existing data are mainly limited to patients with traumatic brain injury [16–18]. The aim of this study was to evaluate changes in blood flow velocities via transcranial Doppler (TCD) sonography in patients with severe SAH who were treated with prolonged hypothermia due to refractory high ICP or DCI.
Materials and Methods
Patient Characteristics and Management
Between January 2007 and November 2010, 246 patients with SAH were admitted to the Neurointensive Care Unit, University Hospital Zurich. Of these, 46 patients with hypothermia treatment due to refractory high ICP or DCI were retrospectively identified from a prospective observational database approved by the local ethics committee. Thirty-eight of them underwent TCD examination within 24 h before induction of hypothermia. Patients with induction of hypothermia during already initiated barbiturate coma (n = 4) or immediately after endovascular treatment of cerebral vasospasm (n = 14) were excluded from the study.
Patients were managed according to a standardized protocol [10, 19]. Treatment of high ICP (>20 mmHg) consisted of cerebrospinal fluid drainage and osmotherapy (mannitol 20 %, hypertonic NaCl-hydroxyethyl-starch solution). Primary decompressive craniectomy was performed if signs of severe brain swelling were present during or immediately after aneurysm surgery and secondary decompressive craniectomy if elevated ICP was refractory to maximal medical treatment. The presence of DCI was clinically defined as secondary neurological deficit or radiological evidence of a delayed cerebral infarction. Cerebral vasospasm was confirmed or ruled out by regular catheter angiography. All patients were treated with oral nimodipine. Hypertensive therapy (systolic blood pressure >150 mmHg) was initiated if signs of DCI were apparent. If patients did not improve, balloon angioplasty and/or superselective papaverine infusion were performed. Hypothermia alone or in combination with barbiturate coma was applied as treatment of last resort if high ICP or DCI were refractory despite the above-mentioned treatment.
Hypothermia (target temperature 33.0–33.9 °C) was induced and maintained by endovascular cooling (Quattro™, Zoll Medical, Chelmsford, USA). In patients without contraindications for barbiturates, hypothermia was induced simultaneously with barbiturate coma. The thiopental loading dose was 10 mg/kg, followed by a continuous infusion adapted to an electroencephalographic (EEG) burst suppression pattern. Hypothermia and barbiturate coma were maintained until ICP normalized, DCI resolved or severe side effects occurred. Rewarming was performed ICP-controlled and no faster than 1 °C per 24 h.
Physiological Measurements
The following physiological parameters were obtained within 24 h before induction of hypothermia (Pre-HT, >35 °C), after reaching hypothermia (Early-HT, 33 °C), before rewarming (Late-HT, 33 °C), and after rewarming to normothermia (Post-HT, >35 °C): mean flow velocity in both middle cerebral arteries (MFVMCA), ICP, body core temperature (BCT), mean arterial pressure (MAP), arterial carbon dioxide tension (PaCO2), and hematocrit (HCT). Measurement values were obtained on average 7 h before hypothermia, 15 h after reaching hypothermia and 14 h before rewarming. Post-treatment values were available in 14 patients and obtained on average 9 h after reaching normothermia.
Bilateral MFVMCA was measured by transcranial color-coded duplex sonography with a 2-MHz probe (ACUSON 2000™, Siemens Healthcare, Germany). TCD examinations were performed daily by an independent neuroradiologist according to the method developed by Aaslid et al. [20]. The best signal obtained was insonated at a depth between 50 and 60 mm, and the mean flow velocity recorded from serial TCD. In healthy adults average MFVMCA is in the range of 73 ± 25 cm/s [21]. All patients had a ventricular catheter (Neurovent®, Raumedic AG, Germany) or intraparenchymatous probe (Neurovent-P®, Raumedic AG, Germany or Bactiseal™, Codman, Johnson & Johnson; USA) for continuous ICP monitoring. BCT and MAP were measured via a thermistor-tipped femoral arterial catheter. Blood gas analysis for determination of PaCO2 and HCT was performed via the same arterial line.
Statistical Analysis
Mean values of physiological parameters were compared by paired t test. Patients were classified according to specific SAH-related complications (ICP- vs DCI-group) and treatment protocol (combined hypothermia and barbiturate coma (cHTBC-group) versus hypothermia alone (HT-group)). Intergroup differences were compared by Mann–Whitney U test and Fisher’s exact test. The relation between mean differences in MFVMCA and MAP, PaCO2, and ICP were determined by linear regression analysis with calculation of the Pearson correlation coefficient. A p value <0.05 was considered statistically significant.
Results
A total of 20 patients were included in this study. Clinical and treatment characteristics are summarized in Table 1. The overall incidence of severe SAH according to the World Federation of Neurological Surgeon (WFNS) grades 4–5 and Fisher scores 3–4 was 50 and 95 %, respectively. Hypothermia was induced on average 5 days after SAH (range 1–12) and maintained for 144 h (range 29–270). The overall in-hospital mortality rate was 15 %. Hypothermia was induced earlier in the ICP-group compared to the DCI-group (p = 0.002). Otherwise, there were no significant intergroup differences with regards to patients’ age, admission scores, specific SAH-related complications, treatment protocol, duration of hypothermia, and outcome. The 12-month outcome according to the Glasgow Outcome Scale (GOS) showed favorable outcome (GOS 4–5) in 40 % (n = 8), severe disability (GOS 3) in 35 % (n = 7), and death (GOS 1) in 25 % (n = 5).
Effects of Hypothermia on Physiological Parameters
A total of 148 MFVMCA values were available from bilateral TCD examinations. MFVMCA was available in all patients before hypothermia (40 values), early after reaching hypothermia (40 values), and before rewarming during late hypothermia (40 values). After rewarming to normothermia MFVMCA was available in 14 patients (28 values). The mean values of physiological parameters are summarized in Table 2. Mean MFVMCA decreased significantly by 19.8 cm/sec after hypothermia induction (p = 0.001) and remained unchanged after rewarming (p = 0.219) (Fig. 1). Likewise, mean ICP decreased by 5.6 mmHg after hypothermia induction (p < 0.001) and remained unchanged after rewarming (p = 0.176). There were no significant changes in mean MAP, PaCO2, and HCT after hypothermia induction and after rewarming. There was no significant correlation between changes in MFVMCA and MAP (95 % CI −0.3 to 0.4, p = 0.749) and PaCO2 (95 % CI −0.1 to 0.5, p = 0.204). The correlation between changes in MFVMCA and ICP was statistically significant (95 % CI 0.0–0.6, p = 0.048).

The effect of hypothermia on mean flow velocity in the middle cerebral artery (MFVMCA) in all patients. Values are expressed as mean and 95 % CI. Dashed lines indicate lower and upper physiological limits of MFVMCA values according to Krejza et al. [21]. Asterisk indicates statistical significant difference between monitoring periods (paired t test, p value <0.05)
The following changes in physiological parameters were observed between early and late hypothermia: MFVMCA decreased by 28.2 cm/s (95 % CI −43.6 to −12.3, p = 0.0007) and PaCO2 increased by 0.6 kPa (95 % CI 0.4–0.9, p = 0.0002). There were no significant changes in ICP (95 % CI −9.8 to 13.5, p = 0.715), MAP (95 % CI −6.3 to 9.5, p = 0.680), and HCT (95 %CI −1.7 to 1.2, p = 0.723) between early and late hypothermia.
Effect of Hypothermia on MFVMCA According to Patient Subgroups
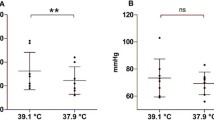
Mean MFVMCA for the patients’ subgroups according to specific SAH-related complications and treatment protocol are shown in Table 3. Hypothermia was induced due to refractory high ICP in 9 patients and DCI in 11 patients. MFVMCA before hypothermia and during early hypothermia was significantly higher in the DCI-group compared to the ICP-group (p = 0.002 and 0.003, respectively). After induction of hypothermia, MFVMCA significantly decreased in both the ICP-group (p = 0.016) and the DCI-group (p = 0.023, Fig. 2a). Between early and late hypothermia, MFVMCA significantly decreased in the DCI-group (95 % CI 25.6–72.8, p < 0.001), but remained unchanged in the ICP-group (95 % CI −14.6 to 10.4, p = 0.724). MFVMCA remained unchanged after rewarming to normothermia in both, the ICP-group (p = 0.564) and the DCI-group (p = 0.198).

The effect of hypothermia on mean flow velocity in the middle cerebral artery (MFVMCA) according to patients’ subgroups. a Changes in MFVMCA in patients with refractory high intracranial pressure versus delayed cerebral ischemia. b Changes in MFVMCA in patients treated with combined hypothermia and barbiturate coma versus hypothermia alone. Values are expressed as mean and 95 % CI. Dashed lines indicate lower and upper physiological limits of MFVMCA values according to Krejza et al. [21]. Asterisk indicates significant difference in the ICP- or cHTBC-group, hash indicates significant difference in the DCI- or HT-group
Thirteen patients were treated with combined hypothermia and barbiturate coma (cHTBC-group) and 7 patients with hypothermia alone (HT-group) due to contraindications for barbiturates. There were no significant differences in MFVMCA between both groups during the study (Table 3). After induction of hypothermia, MFVMCA significantly decreased in both the cHTBC-group (p = 0.020) and the HT-group (p = 0.013, Fig. 2b). MFVMCA further decreased between early and late hypothermia in the cHTBC-group (95 %CI 3.9–45.7, p = 0.022) and the HT-group (95 % CI 2.6–21.6, p = 0.017). After rewarming to normothermia, MFVMCA increased in the HT-group (p = 0.016), but remained unchanged in the cHTBC-group (p = 0.822). In 10 patients barbiturate administration was discontinued before rewarming (on average 48 h before, range 16–164) and in 3 patients after rewarming (on average 60 h post rewarming, range 30–87).
Discussion
The goal of this study was to evaluate the effect of hypothermia on blood flow velocity in patients with SAH via TCD. Mean differences in MFVMCA were used to estimate relative changes in CBF, as previously reported [22–24]. However, the expected linear relationship between MFVMCA and CBF only applies if the angle of isonation and blood vessel diameter remains constant (MFVMCA = CBF/angle of isonation * vessel diameter). The variations in the angle of isonation were minimized by using transcranial color-coded duplex sonography, a technique reported to improve consistency and accuracy of TCD [25]. In addition the diameter of the proximal segment of the middle cerebral artery has been shown to remain constant during moderate changes in MAP and PaCO2 [26, 27]. However, vasospasm of the middle cerebral artery is frequently observed after SAH and may increase MFVMCA, as confirmed previously [28]. If TCD is performed serially after SAH there is usually a steady rise in blood flow velocity with peak values around 1 week from SAH and recovery back to normal values over another week, reflecting the time course of pathological changes in arteries occurring during cerebral vasospasm [29]. Therefore, the results of this study need to be interpreted with particular care, as mean differences in MFVMCA might indicate (1) relative changes in CBF and/or (2) dynamic changes during cerebral vasospasm.
In the current study, the decrease in MFVMCA after induction of hypothermia was independent of the indication for hypothermia (high ICP or DCI), as well as from blood pressure, PaCO2, and hematocrit. This suggests that a lower body temperature (33 °C) itself is an important determinant of reduced blood flow velocities and possibly reflects lower CBF. Simultaneous induction of barbiturate coma may partly explain the decrease in MFVMCA in our patients. Barbiturates substantially reduce cerebral metabolism and, as a consequence, can lead to a decrease in CBF [30, 31]. Furthermore, hypothermia prolongs the duration of barbiturate-induced burst suppression pattern on EEG [32], possibly reflecting the unchanged MFVMCA after rewarming in patients with combined hypothermia and barbiturate coma. However, decreased MFVMCA after induction of hypothermia together with increased MFVMCA after rewarming in patients treated with hypothermia alone may suggest an independent temperature effect on CBF. Although the occurrence of cerebral vasospasm very likely influenced the absolute MFVMCA values, relative changes in MFVMCA after both induction of hypothermia (on average 5 days after SAH) and rewarming to normothermia (on average 13 days after SAH) were opposite to the time course of pathological changes in arteries after SAH. Finally, the study confirms previous reports on ICP reductions during hypothermia in patients with acute brain injury [16–18, 33]. In order to avoid rebound of ICP, rewarming should be performed ICP-controlled and no faster than 0.5–1 °C per 24 h. Furthermore, an abnormal increase in Doppler blood flow velocities during rewarming may serve as an index in the prediction of acute brain swelling, undermining the use of TCD as a rapid and non-invasive method to monitor cerebral hemodynamics during hypothermia [34].
Hypothermia is a therapeutic approach that likely interferes with a variety of processes leading to secondary brain injury, of these the suppression of cerebral metabolic rates of oxygen and glucose has been regarded as its most prevalent neuroprotective effect [12]. As a consequence, it is well known that hypothermia can lead to a reduction in CBF under physiological conditions [13, 14]. In juvenile pigs on cardiopulmonary bypass, Ehrlich et al. [15] demonstrated that flow-metabolism coupling was maintained and cerebrovascular resistance increased when body temperature was lowered from 37 to 28 °C. The findings of the current study support the physiological model of reduced CBF during hypothermia. Furthermore, the significant correlation between changes in MFVMCA and ICP might enhance the concept of increased cerebrovascular resistance during hypothermia [35]. Although the exact mechanisms of hypothermia-related ICP reductions remain to be elucidated, increased cerebrovascular resistance leading to reduced cerebral blood volume, and thus to reduced ICP, has been demonstrated previously [36]. Given the lack of controlled studies, no general recommendation for therapeutic hypothermia after SAH can currently be made. However, favorable outcome in 40 % of our patients may further support previous reports that patients with most severe SAH having refractory intracranial hypertension or DCI may benefit from prolonged hypothermia treatment [9, 10].
Hypothermia-related CBF reductions after acute brain injury are consistent with most [16, 17, 37, 38] but not all [18, 39] clinical studies, a possible reflection of the time point of hypothermia induction and different causes of primary brain injury. Shiozaki et al. [17] described a reduction in CBF by 15.4 ml/100 g/min during hypothermia in 5 patients developing refractory intracranial hypertension within 48 h after traumatic brain injury. On the other hand, Metz et al. [18] reported no change in CBF in 10 patients with traumatic brain injury after induction of hypothermia immediately after hospital admission. In our study, hypothermia was induced on average 5 days after SAH, when intracranial hypertension or DCI was refractory to conventional treatment. This might suggest that these results are independent of the impact of primary brain injury in the majority of patients, and thus might reflect true hypothermia-related changes on cerebral hemodynamics.
The effect of hypothermia on CBF after SAH is discussed controversially. Thome et al. [3] and Schubert et al. [4] demonstrated that hypothermia, induced before or immediately after cisternal blood injection in rats, improves CBF during cerebral perfusion pressure-independent hypoperfusion. In contrast, Török et al. [40] reported no change in CBF when hypothermia was induced up to 3 h after SAH using the endovascular perforation model in rats. The authors concluded that different severities of SAH in different experimental models might have contributed to these contradicting results. So far, two clinical reports have described CBF during hypothermia. Using positron emission tomography, Kawamura et al. [41] reported a decrease in CBF by 37–44 % during hypothermia in 5 patients with poor-grade SAH. However, the results are difficult to interpret as the CBF measurements were compared to normal values from normothermic volunteers. Furthermore, globally reduced CBF after SAH is also observed in normothermic patients [42, 43]. In the second study, Nagao et al. showed by means of single photon emission tomography that CBF decreased by 6.4 ml/100 g/min after induction of hypothermia and increased by 10.8 ml/100 g/min after rewarming in a total of 7 patients with cerebral vasospasm causing delayed ischemic neurological deficits. The authors noted that the change in CBF was detected in the region of cerebral vasospasm and CBF measurements were only performed if the patients’ condition allowed for technically difficult imaging studies. Therefore, the distinction between hypothermia- and vasospasm-related effects on CBF is difficult. For the current study routine TCD was used to evaluate relative changes in CBF during specific monitoring periods, i.e., before, during, and after hypothermia. Nevertheless, the dynamic changes during cerebral vasospasm are very likely to influence MFVMCA in particular during prolonged hypothermia.
Cerebral vasospasm is considered an important cause of DCI and can partly be explained by the mismatch between vasodilatatory and -constrictor substances as well as detrimental effects of SAH-related inflammatory reactions [44]. Animal studies have shown the potentially beneficial effects of hypothermia on cerebral vasospasm [3, 4, 7]. Furthermore, we previously demonstrated the attenuation of inflammatory reactions during combined hypothermia and barbiturate coma in patients with severe SAH [45]. The strong correlation between elevated blood flow velocities from TCD and radiological evidence of cerebral vasospasm has been reported previously [28, 46]. Accordingly, we observed higher MFVMCA before and during early hypothermia in patients with DCI (7 of 11 patients with MCA vasospasm) compared to patients with intracranial hypertension. As mentioned above, the rapid changes in MFVMCA after both hypothermia induction and rewarming are unlikely to reflect the pathological changes in arteries occurring during cerebral vasospasm. However, the decrease in MFVMCA during prolonged hypothermia (on average 144 h) might suggest resolved cerebral vasospasm in the subgroup of patients with DCI. These findings undermine the importance of carefully interpreting TCD findings in SAH patients with hypothermia, because changes in Doppler blood flow velocity might reflect hypothermia-related changes in CBF and/or the dynamic changes during cerebral vasospasm. Additional temperature effects on MFVMCA during prolonged hypothermia in the current study are unlikely, based on the tight temperature control (33.0–33.9 °C) using endovascular cooling [47].
The following limitations of this study need to be considered. First, the sample size of patients’ subgroups is small. Second, while hyperemia with high absolute CBF values has to be considered as differential diagnosis to cerebral vasospasm in patients with elevated flow velocities, the flow velocity in the extracranial internal carotid artery is not routinely measured at our institution, thus, the Lindegaard index could not be calculated. However, cerebral vasospasm was confirmed or excluded by catheter angiography in all patients. In addition, we investigated the mean differences in MFVMCA during specific monitoring periods, thus avoiding the difficulties of interpreting isolated TCD findings. Finally, we could not establish hypothermia-related changes in cerebral metabolic demands, although all patients were treated with bilateral extended cerebral monitoring (brain tissue oxygen tension and microdialysis) during prolonged hypothermia treatment. However, hypothermia was induced as early and fast as possible if SAH-specific complications were refractory to other therapies, a time at which extended cerebral monitoring was not installed in majority of patients.
Conclusion
Therapeutic hypothermia after SAH reduces MFVMCA in both intracranial hypertension and DCI cases. The results might reflect the physiological model of reduced CBF at lower body temperatures and undermine the use of TCD as a rapid and non-invasive method to monitor cerebral hemodynamics during hypothermia treatment. Further studies are needed to investigate flow-metabolism coupling during hypothermia in patients with SAH.
References
Heuer GG, Smith MJ, Elliott JP, Winn HR, LeRoux PD. Relationship between intracranial pressure and other clinical variables in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg. 2004;101:408–16.
Vergouwen MD, Ilodigwe D, Macdonald RL. Cerebral infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke. 2011;42:924–9.
Thome C, Schubert G, Piepgras A, Elste V, Schilling L, Schmiedek P. Hypothermia reduces acute vasospasm following SAH in rats. Acta Neurochir Suppl. 2001;77:255–8.
Schubert GA, Poli S, Mendelowitsch A, Schilling L, Thome C. Hypothermia reduces early hypoperfusion and metabolic alterations during the acute phase of massive subarachnoid hemorrhage: a laser-Doppler-flowmetry and microdialysis study in rats. J Neurotrauma. 2008;25:539–48.
Schubert GA, Poli S, Schilling L, Heiland S, Thome C. Hypothermia reduces cytotoxic edema and metabolic alterations during the acute phase of massive SAH: a diffusion-weighted imaging and spectroscopy study in rats. J Neurotrauma. 2008;25:841–52.
Piepgras A, Elste V, Frietsch T, Schmiedek P, Reith W, Schilling L. Effect of moderate hypothermia on experimental severe subarachnoid hemorrhage, as evaluated by apparent diffusion coefficient changes. Neurosurgery. 2001;48:1128–34 discussion 34–5.
Wang ZP, Chen HS, Wang FX. Influence of plasma and cerebrospinal fluid levels of endothelin-1 and no in reducing cerebral vasospasm after subarachnoid hemorrhage during treatment with mild hypothermia, in a dog model. Cell Biochem Biophys. 2011;61:137–43.
Todd MM, Hindman BJ, Clarke WR, Torner JC. Mild intraoperative hypothermia during surgery for intracranial aneurysm. N Engl J Med. 2005;352:135–45.
Gasser S, Khan N, Yonekawa Y, Imhof HG, Keller E. Long-term hypothermia in patients with severe brain edema after poor-grade subarachnoid hemorrhage: feasibility and intensive care complications. J Neurosurg Anesthesiol. 2003;15:240–8.
Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery. 2009;64:86–92 discussion 3.
Seule M, Keller E. Hypothermia after aneurysmal subarachnoid hemorrhage. Crit Care. 2012;16(Suppl 2):21–3.
Erecinska M, Thoresen M, Silver IA. Effects of hypothermia on energy metabolism in mammalian central nervous system. J Cereb Blood Flow Metab. 2003;23:513–30.
Yenari M, Wijman C, Stienberg G. Effects of hypothermia on cerebral metabolism, blood flow and autoregulation. New York: Marcel Dekker; 2004.
Rosomoff HL, Holaday DA. Cerebral blood flow and cerebral oxygen consumption during hypothermia. Am J Physiol. 1954;179:85–8.
Ehrlich MP, McCullough JN, Zhang N, et al. Effect of hypothermia on cerebral blood flow and metabolism in the pig. Ann Thorac Surg. 2002;73:191–7.
Marion DW, Obrist WD, Carlier PM, Penrod LE, Darby JM. The use of moderate therapeutic hypothermia for patients with severe head injuries: a preliminary report. J Neurosurg. 1993;79:354–62.
Shiozaki T, Sugimoto H, Taneda M, et al. Effect of mild hypothermia on uncontrollable intracranial hypertension after severe head injury. J Neurosurg. 1993;79:363–8.
Metz C, Holzschuh M, Bein T, et al. Moderate hypothermia in patients with severe head injury: cerebral and extracerebral effects. J Neurosurg. 1996;85:533–41.
Keller E, Krayenbuhl N, Bjeljac M, Yonekawa Y. Cerebral vasospasm: results of a structured multimodal treatment. Acta Neurochir Suppl. 2005;94:65–73.
Aaslid R, Markwalder TM, Nornes H. Noninvasive transcranial Doppler ultrasound recording of flow velocity in basal cerebral arteries. J Neurosurg. 1982;57:769–74.
Krejza J, Mariak Z, Walecki J, Szydlik P, Lewko J, Ustymowicz A. Transcranial color Doppler sonography of basal cerebral arteries in 182 healthy subjects: age and sex variability and normal reference values for blood flow parameters. AJR Am J Roentgenol. 1999;172:213–8.
Clyde BL, Resnick DK, Yonas H, Smith HA, Kaufmann AM. The relationship of blood velocity as measured by transcranial doppler ultrasonography to cerebral blood flow as determined by stable xenon computed tomographic studies after aneurysmal subarachnoid hemorrhage. Neurosurgery. 1996;38:896–904 discussion-5.
Bishop CC, Powell S, Rutt D, Browse NL. Transcranial Doppler measurement of middle cerebral artery blood flow velocity: a validation study. Stroke. 1986;17:913–5.
Lindegaard KF, Lundar T, Wiberg J, Sjoberg D, Aaslid R, Nornes H. Variations in middle cerebral artery blood flow investigated with noninvasive transcranial blood velocity measurements. Stroke. 1987;18:1025–30.
Baumgartner RW, Mathis J, Sturzenegger M, Mattle HP. A validation study on the intraobserver reproducibility of transcranial color-coded duplex sonography velocity measurements. Ultrasound Med Biol. 1994;20:233–7.
ter Minassian A, Melon E, Leguerinel C, Lodi CA, Bonnet F, Beydon L. Changes in cerebral blood flow during PaCO2 variations in patients with severe closed head injury: comparison between the Fick and transcranial Doppler methods. J Neurosurg. 1998;88:996–1001.
Giller CA, Bowman G, Dyer H, Mootz L, Krippner W. Cerebral arterial diameters during changes in blood pressure and carbon dioxide during craniotomy. Neurosurgery. 1993;32:737–41 discussion 41–2.
Fontanella M, Valfre W, Benech F, et al. Vasospasm after SAH due to aneurysm rupture of the anterior circle of Willis: value of TCD monitoring. Neurol Res. 2008;30:256–61.
Weir B, Macdonald RL, Stoodley M. Etiology of cerebral vasospasm. Acta Neurochir Suppl. 1999;72:27–46.
Nemoto EM, Klementavicius R, Melick JA, Yonas H. Suppression of cerebral metabolic rate for oxygen (CMRO2) by mild hypothermia compared with thiopental. J Neurosurg Anesthesiol. 1996;8:52–9.
Steen PA, Newberg L, Milde JH, Michenfelder JD. Hypothermia and barbiturates: individual and combined effects on canine cerebral oxygen consumption. Anesthesiology. 1983;58:527–32.
Kim JH, Kim SH, Yoo SK, Kim JY, Nam YT. The effects of mild hypothermia on thiopental-induced electroencephalogram burst suppression. J Neurosurg Anesthesiol. 1998;10:137–41.
Schwab S, Schwarz S, Aschoff A, Keller E, Hacke W. Moderate hypothermia and brain temperature in patients with severe middle cerebral artery infarction. Acta Neurochir Suppl. 1998;71:131–4.
Iida K, Kurisu K, Arita K, Ohtani M. Hyperemia prior to acute brain swelling during rewarming of patients who have been treated with moderate hypothermia for severe head injuries. J Neurosurg. 2003;98:793–9.
Giannotta SL, Raisis JE, McGillicuddy JE, Kindt GW. The effect of temperature on cerebrovascular resistance and cerebral metabolism in the primate. J Surg Res. 1978;25:105–10.
Van Bel F, Zeeuwe PE, Dorrepaal CA, Benders MJ, Van de Bor M, Hardjowijono R. Changes in cerebral hemodynamics and oxygenation during hypothermic cardiopulmonary bypass in neonates and infants. Biol Neonate. 1996;70:141–54.
Keller E, Steiner T, Fandino J, Schwab S, Hacke W. Changes in cerebral blood flow and oxygen metabolism during moderate hypothermia in patients with severe middle cerebral artery infarction. Neurosurg Focus. 2000;8:e4.
Nakamura T, Nagao S, Kawai N, Honma Y, Kuyama H. Significance of multimodal cerebral monitoring under moderate therapeutic hypothermia for severe head injury. Acta Neurochir Suppl. 1998;71:85–7.
Bisschops LL, van der Hoeven JG, Hoedemaekers CW. Effects of prolonged mild hypothermia on cerebral blood flow after cardiac arrest. Crit Care Med. 2012;40:2362–7.
Török E, Klopotowski M, Trabold R, Thal SC, Plesnila N, Scholler K. Mild hypothermia (33 degrees C) reduces intracranial hypertension and improves functional outcome after subarachnoid hemorrhage in rats. Neurosurgery. 2009;65:352–9 discussion 9.
Kawamura S, Suzuki A, Hadeishi H, Yasui N, Hatazawa J. Cerebral blood flow and oxygen metabolism during mild hypothermia in patients with subarachnoid haemorrhage. Acta Neurochir (Wien). 2000;142:1117–11121 discussion 21–2.
Voldby B, Enevoldsen EM, Jensen FT. Regional CBF, intraventricular pressure, and cerebral metabolism in patients with ruptured intracranial aneurysms. J Neurosurg. 1985;62:48–58.
Grubb RL Jr, Raichle ME, Eichling JO, Gado MH. Effects of subarachnoid hemorrhage on cerebral blood volume, blood flow, and oxygen utilization in humans. J Neurosurg. 1977;46:446–53.
Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3:256–63.
Muroi C, Frei K, El Beltagy M, Cesnulis E, Yonekawa Y, Keller E. Combined therapeutic hypothermia and barbiturate coma reduces interleukin-6 in the cerebrospinal fluid after aneurysmal subarachnoid hemorrhage. J Neurosurg Anesthesiol. 2008;20:193–8.
Jarus-Dziedzic K, Juniewicz H, Wronski J, et al. The relation between cerebral blood flow velocities as measured by TCD and the incidence of delayed ischemic deficits. A prospective study after subarachnoid hemorrhage. Neurol Res. 2002;24:582–92.
Keller E, Imhof HG, Gasser S, Terzic A, Yonekawa Y. Endovascular cooling with heat exchange catheters: a new method to induce and maintain hypothermia. Intensive Care Med. 2003;29:939–43.
Acknowledgments
M. S. was supported by a personal research Grant from the University Zurich, Switzerland.
Conflict of interest
The other authors have no financial or institutional conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Seule, M., Muroi, C., Sikorski, C. et al. Therapeutic Hypothermia Reduces Middle Cerebral Artery Flow Velocity in Patients with Severe Aneurysmal Subarachnoid Hemorrhage. Neurocrit Care 20, 255–262 (2014). https://doi.org/10.1007/s12028-013-9927-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-013-9927-x