
Abstract
Approximately one-tenth of the general population exhibit adrenal cortical nodules, and the incidence has increased. Afflicted patients display a multifaceted symptomatology—sometimes with rather spectacular features. Given the general infrequency as well as the specific clinical, histological, and molecular considerations characterizing these lesions, adrenal cortical tumors should be investigated by endocrine pathologists in high-volume tertiary centers. Even so, to distinguish specific forms of benign adrenal cortical lesions as well as to pinpoint malignant cases with the highest risk of poor outcome is often challenging using conventional histology alone, and molecular genetics and translational biomarkers are therefore gaining increased attention as a possible discriminator in this context. In general, our understanding of adrenal cortical tumorigenesis has increased tremendously the last decade, not least due to the development of next-generation sequencing techniques. Comprehensive analyses have helped establish the link between benign aldosterone-producing adrenal cortical proliferations and ion channel mutations, as well as mutations in the protein kinase A (PKA) signaling pathway coupled to cortisol-producing adrenal cortical lesions. Moreover, molecular classifications of adrenal cortical tumors have facilitated the distinction of benign from malignant forms, as well as the prognostication of the individual patients with verified adrenal cortical carcinoma, enabling high-resolution diagnostics that is not entirely possible by histology alone. Therefore, combinations of histology, immunohistochemistry, and next-generation multi-omic analyses are all needed in an integrated fashion to properly distinguish malignancy in some cases. Despite significant progress made in the field, current clinical and pathological challenges include the preoperative distinction of non-metastatic low-grade adrenal cortical carcinoma confined to the adrenal gland, adoption of individualized therapeutic algorithms aligned with molecular and histopathologic risk stratification tools, and histological confirmation of functional adrenal cortical disease in the context of multifocal adrenal cortical proliferations. We herein review the histological, genetic, and epigenetic landscapes of benign and malignant adrenal cortical neoplasia from a modern surgical endocrine pathology perspective and highlight key mechanisms of value for diagnostic and prognostic purposes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Approximately one-tenth of the general population exhibit adrenal cortical nodules, and the incidence has increased, most likely as a consequence of increased usage of abdominal imaging studies. These lesions might either be hormonally active or silent, and the former category is usually associated to specific hormone-specific symptoms depending on the secreted substance (mineralocorticoids, glucocorticoids, sex steroids) [1, 2]. The lesions can be multifocal, bilateral, or solitary, and recent advances have helped us understand that many of the synchronous manifestations of multiple cortical nodules are actually several, independent foci with mutation-driven neoplasia rather than true “hyperplastic” lesions. Of all adrenal cortical lesions, benign adrenal cortical adenoma is the most common tumor, occurring in all age groups and both sexes, while adrenal cortical carcinoma is exceedingly rare [2]. Current clinical and pathological challenges include the preoperative distinction of non-metastatic low-grade adrenal cortical carcinoma confined to the adrenal gland, adoption of individualized therapeutic algorithms aligned with molecular and histopathologic risk stratification tools, and histological confirmation of functional adrenal cortical disease in the context of multifocal adrenal cortical proliferations [2]. In this review, we aim to detail the current histological and molecular panorama of these lesions—with a specific connotation to the practicing endocrine pathologist.
Morphologic and Genomic Correlates of Primary Aldosteronism
Primary aldosteronism is the most common cause of secondary hypertension which can manifest with hypokalemia or normokalemia as well as non-specific symptoms including headache, fatigue, muscle weakness, nocturia, polyuria, and polydipsia [1, 2]. The diagnosis of primary aldosteronism requires laboratory confirmation of increased plasma aldosterone levels along with high plasma aldosterone-to-renin ratio subsequent to aldosterone suppression test [1, 2]. The autonomous or inappropriate aldosterone excess in primary aldosteronism can originate from unilateral or bilateral adrenal cortical disease. Bilateral aldosterone-excess is most commonly the result of aldosterone-producing adrenal cortical hyperplasia [3], which requires a lifelong anti-mineralocorticoid therapy, although bilateral adenomas are not that infrequent. Since incidental nonfunctional adrenal cortical nodules are frequent in the general population and not all radiologically identified adrenal lesions cause aldosterone excess in patients with primary aldosteronism [4,5,6], the appropriate lateralization of the aldosterone excess (versus potential bilateral disease) has been an important clinical task in the management of patients with primary aldosteronism. Several preoperative techniques (e.g., adrenal venous sampling with or without cosyntropin stimulation) are increasingly used to help distinguish the source of aldosterone-excess [7,8,9,10].
Since the first description of primary aldosteronism by Dr. Jerome W. Conn in 1955 [11, 12], the field has evolved tremendously to become an area of interest for multidisciplinary teams of endocrine oncology specialists including but not limited to endocrine pathologists and endocrinologists. The first well-documented patient by Dr. Conn was a 34-year-old female that underwent right adrenalectomy for a 4.0-cm clear cell (lipid-rich) adrenal cortical adenoma [13]. The traditional morphological correlates of primary aldosteronism include bilateral aldosterone-producing adrenal cortical hyperplasia (most common, accounting for up to 60–70% of primary aldosteronism [3]), unilateral adrenal cortical adenoma, and exceptional examples of adrenal cortical carcinoma) [1, 2, 14, 15]. Very rare examples of aldosterone-producing adenomas with oncocytic change have also been reported [16]. However, most conventional adenomas leading to Conn syndrome have a characteristic golden-yellow color due to enrichment of lipid-rich zona fasciculata (ZF)-type clear cells in the tumor; however, zona reticularis (ZR)- and zona glomerulosa (ZG)-like compact cells can be exclusively noted in some aldosterone-producing adenomas [17]. In addition, some tumors can feature mixed ZF- and ZG/ZR-like cells [17]. The overall morphological heterogeneity is now better reflected in molecular genotype-phenotype correlations of primary aldosteronism [17].
Advances in genomics of aldosterone-producing adrenal cortical proliferations, and the use of steroidogenic enzymes, especially the use of CYP11B2 (aldosterone synthase) and CYP11B1 (an enzyme in ZF that helps produce cortisol and corticosterone) immunohistochemistry in adrenalectomy specimens from patients with primary aldosteronism, demonstrated a level of morphological heterogeneity that one may fail to appreciate functional sites when relying only on H&E-stained conventional histological assessment [4, 6, 18,19,20,21,22]. The absence of CYP11B2 expression in paradoxical zona glomerulosa layer hyperplasia adjacent to an aldosterone-producing clear-cell-rich adenoma [19], the occurrence of CYP11B2-positive aldosterone-producing cell clusters (APCCs) or aldosterone-producing micronodules (APMs) without an adrenal mass, or multiple APCCs in association with a non-functional adenoma, and/or multifocal adrenal cortical nodular disease have challenged diagnosticians during the assessment of surgical specimens of patients with primary aldosteronism [2, 19, 21, 23,24,25,26].
Recently, a much-needed international histopathology nomenclature consensus study for reporting histological findings in patients with unilateral primary aldosteronism (HISTALDO) was introduced to address this important challenge (Fig. 1) [19]. The HISTALDO classification system combined histomorphology findings with CYP11B2 immunohistochemistry to refine a spectrum of clinically relevant six diagnostic entities as follows:
-
(i)
Aldosterone-Producing Carcinoma (APACC)
APACC is defined an aldosterone-producing adrenal cortical neoplasm that is diagnosed malignant using universal diagnostic criteria including multiparameter scoring schemes/algorithms.
-
(ii)
Aldosterone-Producing Adenoma (APA)
APA is defined as a solitary CY11B2-immunopositive benign adrenal cortical neoplasm that measures ≥ 1.0 cm and is composed either of zona fasciculata-like lipid-rich clear cells or zona reticularis- and/or zona glomerulosa-like compact cells, or variable amount of clear and compact cells.
-
(iii)
Aldosterone-Producing Nodule (APN)
APN is defined as a morphologically obviously visible sub-centimeter adrenal cortical nodular disease that shows positive reactivity for CYP11B2. Immunohistochemistry for CYP11B2 often shows a stronger staining intensity at the periphery of APN.
-
(iv)
Aldosterone-Producing Micronodule (APM; formerly known as APCCs)
APM is a CYP11B2-immunopositive adrenal cortical proliferation composed of zona glomerulosa cells. APMs are cytologically indistinguishable from cells of the adjacent zona glomerulosa layer, and often feature the same gradient CYP11B2 staining pattern as APN.
-
(v)
Multiple Aldosterone-Producing Nodules (MAPN) or multiple aldosterone-producing micronodules (MAPM)
CYP11B2-immunopositive MAPN and MAPM are seen at the periphery of the adrenal cortex with regions of normal zona glomerulosa layer. Primary aldosteronism can synchronously feature both MAPN and MAPM.
-
(vi)
Aldosterone-Producing Diffuse Hyperplasia (APDH) This diagnostic category is applied to CYP11B2-positive broad or continuous zona glomerulosa hyperplasia accounting for more than 50% of zona glomerulosa layer.

Graphic depiction of the HISTALDO classification model. Aldosterone producing adrenal cortical carcinoma (APACC) and adenoma (APA) are solitary lesions clearly visible by both routine hematoxylin-eosin (H&E) and immunohistochemical (IHC) staining for CYP11B2 (aldosterone synthase). Smaller solitary lesions (sub-centimeter) visible by H&E and IHC are denoted aldosterone producing nodules (APNs), while the counterpart that may be hard to distinguish using H&E but always visualized on IHC are entitled aldosterone producing micronodules (APMs) (formerly known as aldosterone producing cell clusters). When multifocal, these entities are termed “multiple APN” (MAPN) and “multiple APM” (MAPM), respectively—corresponding to the older term “micronodular hyperplasia.” Finally, aldosterone producing diffuse hyperplasia is characterized by a continuous CYP11B2 staining along the zona glomerulosa. Image created with www.BioRender.com
Unilateral primary aldosteronism with solitary APA or APN were referred to as exhibiting classical histology, whereas those with APDH, MAPN, and/or MAPM were termed as non-classical histology by the HISTALDO classification (Fig. 2) [19]. The classical and non-classical histology had distinct molecular findings (see genotype-phenotype correlations), but more importantly, this approach correlated with a reproducible reporting of histological findings that correlated with lack of postoperative biochemical benefit in 4.5% of patients with classical histology compared with almost 42% of patients with non-classical histology [19].

Histological and immunohistochemical attributes of aldosterone producing adrenal cortical lesions. Top row depicts a 40-mm large adrenal cortical adenoma with diffuse CYP11B2 immunoreactivity at × 200 magnification. Middle row illustrates a 9-mm aldosterone producing adrenal cortical microadenoma (or aldosterone producing adrenal cortical nodule measuring less than 1.0 cm; APN) visible by both routine H&E (hematoxylin-eosin) and CYP11B2 immunohistochemistry at × 40 magnification. There were no other culprit lesions in this adrenal. Bottom row portrays a 3-mm aldosterone producing adrenal cortical micronodule (APM) only clearly demonstrable using immunohistochemistry
Molecular Pathogenesis of Primary Aldosteronism: A Disease of Ion Channels
Understanding cellular mechanisms involved in normal physiology shed light into the molecular pathogenies of primary aldosteronism (Fig. 3). In normal physiology, renin-angiotensin system and extra-cellular potassium ion (K+) levels regulate aldosterone synthesis [1]. The resting state of zona glomerulosa cells and the constitutively active K+ channels mediate the extra-cellular transport of K+, thereby keeping the cell in a hyperpolarized state. The latter in turn keeps voltage-dependent calcium (Ca2+) channels closed. When AT2 binds its receptor, a conformational change results in the closure of potassium channels and leads to the accumulation of intracytoplasmic K+ and depolarization of the zona glomerulosa cell membrane potential. This in turn will open voltage-dependent Ca2+ channels. The influx of Ca2+ in turn activates the cell cycle and CYP11B2 gene transcription that encodes CYP11B2 (aldosterone synthase) [1].

Schematic representation of mutated ion channels in aldosterone-producing adrenal cortical adenoma. The left aspect depicts the resting state of an adrenal zona glomerulosa cell in the absence of angiotensin 2 (AT2) stimulus. Potassium channels are constitutively active, mediating the extracellular transportation of potassium ions (K+), thereby keeping the cell in a hyperpolarized state. This in turn keeps voltage-gated calcium (Ca2+) channels closed. Middle aspect illustrates the physiological response to AT2. When binding the angiotensin 2 receptor (AT2-R), a conformational change will stimulate the K+ channels to close, leading to the intracellular accumulation of K+. This in turn will lead to depolarization of the membrane potential, and the opening of voltage-gated Ca2+ channels. The influx of Ca2+ in turn will mediate transduction-mediated activation of the cell cycle as well as transcription of target genes, of which CYP11B2 (aldosterone synthase) is one. The right aspect demonstrates how various gene mutations simulate the physiological activation of the AT2-R pathway; for example, mutations of the K+ transporter KCNJ5 leading to an aberrant intracellular accumulation of Ca2+, as well as mutations of the sodium/potassium ATPase ATP1A1 resulting in the inhibited extracellular transportation of sodium (Na+). Similarly, mutations in the Ca2+ ATPase ATP2B3 lead to decreased extracellular conveyance of Ca2+, and CACNA1 subunit mutations will lead to an increased Ca2+ influx. Image created with www.BioRender.com
Autonomous or inappropriate aldosterone secretion is due to increased transcription of CYP11B2 (aldosterone synthase) and is also correlated with CYP11B2 hypomethylation in APAs [27, 28]. Increased intracellular Ca2+ (as discussed in normal physiology) and the activation of the calcium-calmodulin dependent protein kinase pathway are the genomic hallmark of primary aldosteronism [1, 2, 29]. For this reason, the pathogenesis of benign aldosterone-producing adrenal cortical disease (e.g., APA, APM/APCC, APN) is typically linked to somatic mutations in several ion channels (Fig. 3) including the potassium channel mutation-KCNJ5 (encodes G-protein activated inward rectifier potassium channel 4; GIRK4) [30, 31], sodium/potassium ATPase mutation-ATP1A1 (encodes alpha-1 subunit of the sodium/potassium ATPase) [31,32,33], calcium ATPase mutation-ATP2B3 (encodes the plasma cell membrane calcium ATPase isoform; PMCA3) [31, 32], voltage-dependent calcium channel subunit mutations including the high-voltage activated L-type subunit-CACNA1D (encodes Cav1.3) [33,34,35] and the low-voltage activated T-type subunit-CACNA1H (encodes Cav3.2) [35, 36], and the recently described voltage-gated chloride channel mutation-CLCN2 (encodes CIC-2) [37].
In general, somatic ion channel-related mutations were reported to account for around 50–60% of sporadic APAs [38, 39]; however, subsequent precise approaches using CYP11B2-positivity as a rigid inclusion criterion for the confirmation of the source of aldosterone excess identified ion channels-related aldosterone-driver mutations in 90% of APAs in Americans [40, 41] and 96% of APAs in Japanese [35].
Unlike most APAs, the lack of ion channels-related mutations in aldosterone-producing ACCs (APACCs) suggests alternative molecular mechanisms that regulate CYP11B2 transcription in the setting of malignancy [42].
Additional noteworthy somatic alterations that have been identified in APAs include CTNNB1 mutations which occur in up to 5% of sporadic APAs [43, 44]. While the Wnt/beta-catenin pathway is required during the development of adrenal glands, aberrant activation of this pathway due to somatic CTNNB1 mutations is a well-characterized event in a subset of ACCs [45, 46] as well as in benign adrenal cortical neoplasms, including adrenal cortical adenomas with mild autonomous cortisol secretion (subclinical Cushing syndrome) and non-functional adrenal cortical adenomas [27, 47].
CTNNB1 and previously discussed ion channel-related mutations are mutually exclusive in APAs. Activation of the Wnt/beta-catenin pathway, however, has been noted in up to 70% of APAs [48]. While some researchers have restricted the role of CTNNB1 to cortical tumorigenesis (rather than aldosterone synthesis) in general, high CYP11B2 (mRNA and protein) expression in CTNNB1-mutant APAs [44] would support a cross-talk between the Wnt/beta-catenin and calcium calmodulin–dependent kinase pathways. Berthon et al. was the first to link aldosterone secretion in the context of aberrant beta-catenin expression to overexpression of angiotensin II type 1 receptor (AT1R), CYP21, and CYP11B2 [48]. In mice, the activation of Wnt/beta-catenin pathway resulted in disrupted adrenal cortical zonation with altered aldosterone synthesis [49, 50]. Furthermore, significantly high levels of gonadal receptors including gonadotropin-releasing hormone receptor and luteinizing hormone-chorionic gonadotropin receptor expression in -CTNNB1mutant APAs in 3 females (2 pregnant and 1 post-menopausal) were also recorded [51]. Nevertheless, the underlying complex molecular mechanisms regulating the cross-talk between the CYP11B2 transcription and Wnt/beta-catenin pathways activation requires further work.
Germline Variants in Primary Aldosteronism
Although most patients with primary aldosteronism are associated with sporadic disease due to somatic genomic alterations, a very small fraction of patients with primary aldosteronism can manifest with germline pathogenic variants (autosomal dominant) including CYP11B1/CYP11B2 chimeric fusions (Familial Hyperaldosteronism type I; also known as glucocorticoid-remediable primary aldosteronism) [52], CLCN2 (Familial Hyperaldosteronism type II) [53], KCNJ5 (Familial Hyperaldosteronism type III) [54, 55], CACNA1H (Familial hyperaldosteronism type IV; results in early onset familial or de novo hypertension) [56, 57], and CACNA1D (early onset primary aldosteronism with seizure and neurological abnormalities; also known as PASNA syndrome) [34, 58].
The discovery of germline Armadillo Repeat Containing 5 (ARMC5) variants in association with KCNJ5-driven sporadic APAs [59, 60], rare occurrence of germline phosphodiesterase (PDE2A and PDE3B) variants [61], and germline ATP2B4 variants in some patients [62] has expanded the spectrum of germline variants of primary aldosteronism. These findings also underscore germline susceptibility may be well underestimated for patients with primary aldosteronism.
Genotype-Phenotype Correlations in Sporadic Forms of Primary Aldosteronism
Molecular studies of primary aldosteronism have helped develop genotype-phenotype correlations with respect to tumor characteristics (e.g., cytomorphology, size, and focality), degree of autonomous aldosterone secretion, patient demographics (age, gender, and ethnic background), higher risk of recurrence of postoperative hypertension, and expression of steroidogenic enzymes (ZG-related biomarkers: CYP11B2, ZF-related biomarkers: CYP11B1 and CYP17) in various aldosterone-producing adrenal cortical lesions (e.g., APAs, APM/APCCs, and APN) [17, 24, 33, 63,64,65].
Among all ion channel-related mutations, KCNJ5 was the most frequent mutation with an overall prevalence of 43% (ranging from 35% in Europe/Australia/USA to 63% in East Asia) in a meta-analysis of 1636 patients [39]. In addition, somatic KCNJ5 mutations were reported in 71.2% of APAs in a Korean series [66]. Interestingly, CACNA1D mutations (with the rate of 42%) surpassed the frequency of KCNJ5 mutations (with the rate 34%) in blacks with CYP11B2-expressing APAs [40].
In general, KCNJ5-related primary aldosteronism is common in females with pronounced and earlier age of disease-onset and is likely to manifest with a solitary or dominant APA composed predominantly of ZF-like clear cells [33, 39, 64, 65]. KCNJ5-related phenotype is on a par with the original description of APA (4.0-cm clear-cell-rich APA in a young female) by Dr. J. Conn [11, 12]. KCNJ5-wild type disease is more frequent in males (often later age of disease-onset) and is likely to manifest with a smaller tumor size (often less than 1.0 cm) and/or multifocal disease including MAPM/APCCs and/or MAPNs, and frequent compact cell cytomorphology [17, 33]. These observations are also in line with the fact that the classical histology designation of the HISTALDO classification is enriched in KCJN5-mutant aldosterone-producing lesions when compared with the non-classical histology group [19].
Molecular heterogeneity, which is characterized by the occurrence of various ion channels-related somatic drivers (both KCNJ5 and KNCJ5-wild type somatic alterations) in multinodular asynchronous aldosterone-producing proliferations, can occur within the same adrenal gland [24, 67]. In addition, somatic CACNA1H mutations were restricted to the CYP11B2-expressing cellular component within the adenoma substance, whereas the remaining CYP11B2-negative cellular component of the adenoma lacked CACNA1H mutations [36]. The latter not only supports intra-tumoral clonal heterogeneity with respect to aldosterone synthesis but also supports the hypothesis that the pathophysiologic functionality of aldosterone excess may be independent of tumorigenesis. In fact, this observation is of significance given the high rate of Wnt/beta-catenin pathway activation (despite the low frequency of CTNNB1 mutations) in primary aldosteronism.
From a steroidogenic enzyme expression perspective, all aldosterone-producing lesions show variable expression for CYP11B2 [37, 63, 68, 69]. ACAs harboring ATP1A1, ATP2B3, and CACNA1D mutations often feature higher levels of mRNA transcripts for CYP11B2 when compared with KCNJ5-dependent primary aldosteronism [17, 33, 37, 63, 64, 69]. KCNJ5-driven APAs show abundant CYP17A1 and CYP11B1 mRNA transcripts among all APAs [37]. These findings help explain variations in cytomorphology and immunohistochemical staining patterns for steroidogenic enzymes [63, 68, 69]. In addition, KCNJ5 mutations have been implicated in the pathogenesis of synchronous aldosterone- and cortisol-secreting adrenal cortical neoplasms [70,71,72]. The latter may also be explained with ZF-like steroidogenic enzyme profiling in these tumors. CLCN2-mutant APAs are composed of compact cells that show high CYP17A1 and CY11B1 expression as seen in KCNJ5-related APAs [37].
Despite its rarity, CTNNB1-related primary aldosteronism seems to be more frequent in females that manifest with a later age of disease-onset, heterogenous cytomorphology, and nuclear beta-catenin expression, as well as a higher risk of postoperative disease recurrence [43, 44].
Aldosterone-driver mutation status also showed correlation with the adrenal vein sampling (AVS) lateralization index after cosyntropin stimulation in patients with primary aldosteronism [8]. For instance, individuals harboring KCNJ5-mutant adrenal cortical lesion(s) were reported to have descending lateralization index, whereas those harboring ATP1A1 and ATP2B3 were associated with an increased AVS index value [8], although this has to be confirmed by another study.
Morphologic and Genomic Correlates of Adrenal Cushing Syndrome
Endogenous Cushing syndrome is caused by autonomous and elevated cortisol secretion. Not all patients with endogenous cortisol excess manifest with florid clinical and biochemical findings; thus, a subset of patients may show evidence of mild autonomous cortisol secretion which can result in subclinical Cushing syndrome. Static and dynamic endocrine function tests and imaging findings are required in the confirmation of hypercortisolism and the source of cortisol excess [1, 17, 73]. The major cause of endogenous cortisol excess is due to a pituitary ACTH-dependent bilateral diffuse adrenal cortical hyperplasia [73]. Together, ACTH-dependent (pituitary and ectopic ACTH excess) and ACTH-independent (primary adrenal cortisol excess) Cushing syndrome encompasses a wide-spectrum of morphological entities ranging from primary bilateral nodular adrenal cortical disease to adrenal cortical neoplasms [1, 17, 73].
Cortisol-secreting adrenal cortical neoplasms are most often unilateral. From a morphological perspective, cortisol-producing adrenal cortical adenomas are distinguished from carcinomas using the universally accepted multi-parameter scoring systems along with immunohistochemical and molecular biomarkers [2, 68, 74]. Cortisol-producing adenomas are often enriched in ZF-like clear cells but variable amount of compact cells and/or pigment deposition can also occur [1, 17, 73]. The presence of cortical atrophy in the non-tumorous adrenal cortex is also a pathognomonic feature of autonomous cortisol secretion in the absence of exogenous cortisol administration [75].
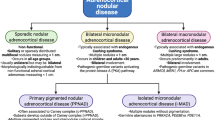
Primary bilateral nodular adrenal cortical disease is often divided into two distinct morphologic entities including (i) Primary bilateral micronodular adrenal cortical disease (nodules measure less than 1.0 cm, often 0.1–0.4 cm) and (ii) Primary bilateral macronodular adrenal cortical disease (nodules often exceed 1.0 cm) (Fig. 4) [1, 17, 73]. In modern endocrine pathology practices, the historical term of bilateral nodular adrenal cortical hyperplasia is no longer appreciated since both micronodular and macronodular types of this disorder often consist of genetically modified adrenal cortical cells [2]. While the appropriate terminology has been widely adopted for the micronodular form of this disorder (e.g., primary pigmented micronodular adrenal cortical disease), the term “bilateral macronodular adrenal cortical disease” is now being increasingly used by endocrine pathologists instead of a frequently used misnomer of primary bilateral macronodular adrenal cortical hyperplasia (PBMAH) [2].

Schematic overview of cortisol-secreting adrenal cortical lesions. While unilateral cortisol-producing lesions consist of adrenal cortical carcinoma and adenoma, bilateral disease is characterized by either diffuse hyperplasia due to ACTH-secretion (most often Cushing’s disease), or micronodular (< 1 cm) or macronodular (> 1 cm) adrenal cortical disease. The former is subdivided into primary pigmented micronodular adrenal cortical disease (PPNAD) and micronodular adrenal cortical disease (MAD). PPNAD is frequently associated with Carney complex, but can also be seen in non-syndromic forms. MAD lacks the classic pigmentation seen in PPNAD. Macronodular adrenal cortical disease is also entitled primary bilateral macronodular adrenal hyperplasia (PBMAH), but the latter terminology is a misnomer as the nodules of this entity is neoplastic rather than true hyperplastic. Image created with www.BioRender.com
Primary bilateral micronodular adrenal cortical disease encompasses primary pigmented nodular adrenal cortical disease (PPNAD) and micronodular adrenal cortical disease (MAD) (Fig. 4). The MAD lacks obvious pigmentation in micronodules. Micronodules are composed of compact adrenal cortical cells, and they are frequently located in the intersection of deep ZF and ZR [1, 17, 73]. Unlike MADs, internodular adrenal cortex is often atrophic in the setting of PPNADs [1, 17, 73]. The prototypical macronodular form of macronodular adrenal cortical hyperplasia, so-called “PBMAH,” is associated with bilateral adrenal gland enlargement due to multiple benign macronodular adrenal cortical nodules that are enriched in ZF-like cells [73].
Studies on molecular pathogenesis of cortisol-producing adrenal cortical nodules have significantly improved our understanding of adrenal cortical tumorigenesis and also shed light to mechanisms responsible for autonomous cortisol secretion. The next section will provide a summary for the molecular pathogenesis of endogenous adrenal Cushing syndrome.
Molecular Pathogenesis of Adrenal Cushing Syndrome
Understanding adrenal cortical cellular mechanisms involved in normal physiological response to ACTH shed light into the molecular pathogenies of Cushing syndrome (Fig. 5). In the ZF layer, cortisol is secreted in response to ACTH stimulus. In normal physiology, upon ACTH binding to the 7-transmembrane G-protein coupled melanocortin 2 receptor (MC2R), the G-protein stimulatory alpha subunit activates membrane-bound adenylyl cyclase (AC), which in turn catalyzes the conversion of cAMP from ATP, a turnover process that is also down-regulated by phosphodiesterase (PDE). When intracellular cAMP levels rise, the regulatory subunits of PKA (PKA-R) are inhibited, thereby lifting repression of the catalytic subunits of PKA (PKA-C). The free PKA-C will phosphorylate the PKA substrates in various cellular compartiments. The free PKA-C enters the nucleus and activates the transcription factor cAMP response element-binding protein (CREB), leading to transcription of target genes promoting proliferation and cortisol-production [1, 17].

Aberrant protein kinase A (PKA) signaling in cortisol-producing adrenal cortical adenoma. The left aspect demonstrates the physiological response to ACTH stimulus in a zona fasciculata cell of the adrenal cortex. Upon binding the G-protein coupled melanocortin 2 receptor (MC2R), the G stimulatory alpha subunit will activate membrane-bound adenylyl cyclase (AC), which in turn catalyzes the conversion of cAMP from ATP, a turnover process also regulated by phosphodiesterase (PDE). When levels of cAMP rise, the regulatory subunits of PKA (PKA-R) are inhibited, thereby lifting repression of the catalytic subunits of PKA (entitled PKA-C). PKA-C will enter the nucleus activate the cAMP response element-binding protein, leading to transcription of target genes promoting proliferation and cortisol-production. Right aspect of the image represents aberrant molecular PKA signaling in cortisol-producing adrenal cortical adenoma. For instance, aberrant expression of G protein coupled receptors (GPCRs) might activate the PKA pathway, which can also be achieved by activating mutations in the MC2R, GNAS, and PRKACA genes; encoding MC2R; the G stimulatory alpha subunit; and PKA-C, respectively. Moreover, inactivating mutations in superfamily members PDE8B and PDE11A encoding PDE, as well as PRKAR1A, encoding the inhibitory PKA-R subunit, will have similar effects. Image created with www.BioRender.com
Autonomous or inappropriate cortisol secretion in most adrenal cortical neoplasms and bilateral nodular adrenal cortical disease (e.g., PPNAD, MAD, a subset of PBMAH) is related to constitutive activation of the cAMP/PKA signaling pathway [1, 17, 76]. Common mechanisms involved in aberrant cAMP/PKA signaling pathway activation include (i) aberrant expression of G-protein coupled receptors (GPCRs); (ii) activating mutations in the MC2R, GNAS, and PRKACA genes, encoding MC2R, the G-protein stimulatory alpha subunit, and PKA-C, respectively; (iii) inactivating mutations in PRKAR1A, encoding the type I alpha regulatory subunit of PKA (PKA-R); and (iv) inactivating mutations in PDE superfamily members PDE8B and PDE11A encoding PDE (Fig. 5) [1, 17, 76].
Bilateral micronodular adrenal cortical disease is often associated with germline pathogenic variants in the PKA signaling pathway [2, 17, 77,78,79,80]. Around 80% of PPNADs harbor pathogenic PRKAR1A variants [78]. PRKAR1A wild-type PPNADs are linked to genomic alterations of the CNC2 gene locus [81]. PPNAD is frequently seen in association with Carney Complex (referred to c-PPNAD) [1]. In the absence of family history or Carney Complex-related manifestations, the term “isolated PPNAD” (i-PPNAD) is applied by some experts [1]. Germline PDE variants (PDE11A and PDE8B) [80, 82, 83] or germline PRKACA duplication [80, 84] were identified in PRKAR1A-wild type i-PPNADs and MADs. Moreover, a recent study showed a missense ARMC5 variant in PPNAD [27]. Furthermore, a current study expanded our knowledge on germline correlates of PKA signaling pathway by demonstrating germline PRKACB variants that may cause PPNAD-like manifestations [85]. Some adrenal cortical adenomas arising in the background of PPNAD show somatic CTNNB1 mutations [86]; moreover, altered microRNA regulation, especially in the context of PRKAR1A-related PPNAD is shown to affect the Wnt/beta-catenin signaling pathway [87].
Since its initial description by Kirschner et al. in 1964 [88], primary bilateral macronodular adrenal cortical disease has been an area of interest given its complex pathogenesis that has been explained by several mechanisms including (i) germline pathogenic ARMC5 variants (around 25–55% of PBMAH in various series) [89,90,91]; (ii) germline susceptibility due to MEN1, FH, APC, PDE11A, and PDE8B variants [17, 92,93,94,95,96,97,98]; (iii) postzygotic somatic GNAS mosaicism in the context of McCune Albright syndrome [2, 99]; (iv) aberrant G-protein coupled receptor expression [100, 101]; (v) ectopic hormone receptor expression or dysregulation of membrane receptors [101,102,103]; (vi) paracrine regulatory effect of aberrant intra-adrenal ACTH synthesis and secretion by adrenal cortical cells [104, 105]; and (vii) increased PRKAR2B using real-time PCR and protein expression levels [106]. A recent study identified a specific group of ARMC5-wild type disease with miRNA142 expression [27]; this finding was in support of one of suggested pathogenetic mechanisms via ectopic GIP receptors in adrenals with PBMAH [107].
Somatic PRKACA mutations are the most common alterations identified in around 40% of adrenal cortical adenomas with overt Cushing syndrome [70, 108,109,110,111] (Fig. 5). Other relatively infrequent somatic alterations include allelic loses or inactivating somatic mutations in PRKAR1A [112], activating mutations in GNAS [70] and PRKACB [113].
The Wnt/beta-catenin pathway activation due to CTNNB1 mutations occurs in around 25% of cortisol-producing adrenal cortical adenomas [27, 70, 114]. While virtually all mutations in the PKA signaling pathways are mutually exclusive, rare examples of synchronous GNAS- and CTNNB1-harboring cortisol-producing adrenal cortical adenomas were also identified [70]. In addition, rare examples of aldosterone- and cortisol co-secreting adrenal cortical neoplasms due to somatic KCNJ5 mutations [70,71,72] expand our knowledge even further regarding pathogenic alterations of adrenal Cushing syndrome.
The heterogeneous spectrum of benign cortisol-producing clonal adrenal cortical proliferations was reflected in 3 distinct integrated genomic groups in a recent pan-genomic study (Fig. 6) [27]. These groups are summarized as follows:
-
(i)
PPNADs and adrenal cortical adenomas with overt Cushing syndrome are linked to an active cAMP/PKA signaling pathway:
In this group, tumors exhibited higher expression of the DLK1-MEG3 miRNA cluster. Moreover, adenomas were distinguished from PPNADs given their differential segregation into a specific miRNA signature.
-
(ii)
Adrenal cortical adenomas with mild autonomous cortisol secretion are linked to an active Wnt/beta-catenin signaling pathway:
Among somatic copy number alterations, chromosome 17q losses (including the PRKAR1A locus) were more frequent in the group of lesions with overt Cushing syndrome, compared to tumors with active Wnt/beta-catenin signaling pathway due to CTNNB1 and ZNRF3 mutations. These lesions were enriched for gain of chromosome 9q (including the Nuclear Receptor Subfamily 5 Group A Member 1 NR5A1 locus encoding SF1). These tumors also featured lower methylation of CpG islands. Adenomas with mild autonomous cortisol secretion were also segregated together with non-functional adrenal cortical adenomas in this genomic group.
-
(iii)
ARMC5-driven primary bilateral macronodular adrenal cortical disease is linked to an ovarian gene expression signature

Integrated genomics of benign adrenocortical lesions. Transcriptome analyses reveal distinct mRNA profiles for aldosterone-producing adrenocortical adenoma, cortisol-producing adrenocortical lesions (mix of adrenal cortical adenoma, ACTH-dependent hyperplasia in Cushing’s disease, and primary pigmented nodular adrenal cortical disease (PPNAD)), primary macronodular adrenal cortical disease, and non-producing/subclinical cortisol-producing adenomas. While aldosterone-producing adenomas exhibited strong CYP11B2 expression (encoding aldosterone synthase), the joint group of cortisol-producing lesions displayed a steroidogenic mRNA profile reminiscent of the C1A adrenal cortical carcinoma expressional profile [45, 190]. Primary macronodular adrenal cortical disease (primary macronodular adrenal hyperplasia) expressed genes in accordance with an ovarian phenotype (here exemplified by FOXL2), whereas non-producing/subclinical cortisol-producing adenomas displayed an mRNA profile similar to the C1B adrenal cortical carcinoma cluster. Cortisol-producing lesions were further collectively characterized by protein kinase A (PKA) activation, a coupling to the DLK1-MEG3 miRNA cluster, and when additionally profiled for miRNA signatures—divided into a group of adrenal cortical adenomas (mi5 signature) and multifocal lesions (ACTH-dependent hyperplastic lesions and PPNAD; mi4 signature). Primary macronodular adrenal cortical disease was characterized by inactivating ARMC5 mutations, a MIR100 miRNA cluster profile, and global promoter hypermethylation, while non-producing or subclinical adenomas displayed Wnt pathway aberrances (CTNNB1 mutations and ZNRF3 mutations/deletions), frequent gain of chromosome 9q, and a global hypomethylation status. Image created with www.BioRender.com
An ovarian gene expression signature (FOXL2, CYP19A1, PTHLH), increased expression of the MIR100 cluster and global hypermethylation of CpG islands were distinct features of this homogenous group of cortisol-producing adrenal cortical lesions.
Genotype-Phenotype Correlations in Benign Lesions of Adrenal Cushing Syndrome
Cortisol-producing adenomas with PRKACA and PRKAR1A alterations were often associated with a smaller tumor size [17, 112, 115], and PRKACA-related tumors were more frequently reported in younger patients with overt Cushing syndrome [17, 108]. Several lines of evidence showed that CTNNB1-driven adenomas were preferably large, non-functional tumors as well as lesions with mild autonomous cortisol secretion [47]. These observations were also validated in a recent study that compared steroidogenic biomarker expression profile in PRKACA-, GNAS- and CTNNB1-mutant cortisol-producing adrenal cortical adenomas [116]. In terms of steroidogenic biomarker expression profile, PRKACA-mutant adrenal cortical adenomas displayed increased immunohistochemical reactivity for CYP11B1, CYP17A1, and 3βHSD compared with PRKACA-wild type (GNAS and CTNNB1 mutant) adenomas [116]. The same study also reported for the first time that cytomorphological heterogeneity was reflected in steroidogenic biomarker expression profile as the compact cell component of cortisol-producing adenomas had the highest expression. In addition, PRKACA- and GNAS-related adenomas were found to be more senescent and hormonally active compared with wild-type cortisol-producing adrenal cortical adenomas.
Initially considered a diagnostic feature of c-PPNAD [117], paradoxical dexamethasone-induced increase in urinary cortisol levels also occurs in rare PRKAR1A-related adrenal cortical adenomas [112]; this phenomenon is largely explained via a glucocorticoid receptor-related effect on PKA catalytic subunits [117]. The CNC2-locus-related c-PPNADs disease was more frequently diagnosed later in life compared with those with germline PRKAR1A variants [118]. A recent study showed that ARMC5 polymorphic variants may interfere with the occurrence and severity of adrenal Cushing syndrome in patients with PRKAR1A-driven PPNADs [119]. Moreover, while most patients with c-PPNAD are associated with benign adrenal cortical nodular proliferations, rare examples of well-documented adrenal cortical carcinomas were reported in association with this disorder [120, 121].
Germline ARMC5 alterations are responsible for the most common form familial primary bilateral macronodular adrenal cortical disease (PBMAH) [89]. In addition to its extensive genetic variance [122] and the occurrence of distinct secondary mutations in individual nodules of PBMAH [89], ARMC5-induced molecular mechanisms that regulate steroidogenesis, apoptosis and cellular proliferation have been an area of interest [123]. The general statement that ARMC5-driven disease affects proliferation as well as functionality may also reflect the genetic landscape of this particular association. Espiard et al. showed that patients with ARMC5-mutant PBMAH were more frequently associated with larger and more nodular adrenals as well as more pronounced Cushing syndrome (characterized by higher levels of midnight cortisol, urinary free cortisol and cortisol after dexamethasone suppression test) than those with ARMC5-wild type disease [90]. For instance, despite being classified as a benign process, paradoxically increased genomic instability of ARMC5-related macronodular adrenal cortical disease can be attributable to global hypermethylation of CpG islands identified in the most recent pan-genomic study [27].
Morphologic and Genomic Correlates of Adrenal Cortical Carcinoma
Although much rarer than its benign counterpart tumors of the adrenal cortex, adrenal cortical carcinoma (ACC) is the most lethal malignancy derived from this organ and responsible for the majority of deaths in patients with primary adrenal neoplasia. The incidence is estimated to 0.7–2 cases per million population annually in the USA, and the age-span is wide, ranging from early childhood (syndromic ACC; 5–10% of cases) to older ages (mostly sporadic ACC) [124, 125]. Female patients are overrepresented, and the majority of patients are detected through symptoms related to hypersecretion of various adrenal cortical hormones, most usually cortisol and androgens, rarely aldosterone [126]. Among the other half of patients, the ACC is hormonally silent, and clinical signs of disease might be delayed until further tumor growth causes localized symptoms—alternatively, the tumor is found en passant during radiological investigations for unrelated reasons. The prognosis is poor if the tumor is locally advanced or spread to distant sites upon diagnosis, and the overall 5-year survival rate is 30% [126]. However, in lower stage ACCs localized to the adrenal, the vast majority of patients live five year or longer following adrenalectomy, although recurrence rates are high [127]. In this aspect, complete surgical resection with negative margins is an important prognostic factor [126]. If locally advanced or spread to distant sites, administration of the adrenolytic agent mitotane is usually recommended [128,129,130].
Histologically, ACCs are circumscribed by a fibrous capsule, and usually growing in large nests intermingled with solid and trabecular areas [124]. Cells are usually eosinophilic or vacuolated (lipid-rich), but subsets of cases will present with oncocytic, myxoid or sarcomatoid features [131,132,133,134,135]. The usual hallmarks of malignant tumors are present, such as nuclear pleomorphism, increased mitotic counts, tumor necrosis, and invasive behavior (capsular or vascular invasion, extension into the periadrenal fat) (Fig. 7) [124, 136]. From an immunohistochemical standpoint, ACCs were early-on known to be vimentin positive while most often negative for cytokeratins and epithelial membrane antigen (EMA), allowing an expression-based separation from renal cell carcinomas—which is not always possible using morphological assessment alone [137]. Building on this, the current differential diagnostics is usually focused on excluding a non-adrenal cortical origin (most often pheochromocytoma and metastatic lesions), and for this reason a panel consisting of SF1, melan A (clone A103), calretinin, inhibin alpha, and chromogranin A could be considered (Fig. 8). Among these, SF1 is regarded as the best universal diagnostic biomarker of adrenal cortical differentiation [68]. While the combination of synaptophysin immunoreactivity and pan-cytokeratin negativity alone would argue in favor of ACC as opposed to many metastatic carcinomas, this expressional pattern is also true for pheochromocytoma [138, 139]. Chromogranin A expression strongly supports a diagnosis of pheochromocytoma over a cortical neoplasm [68].

Histological attributes of adrenal cortical carcinoma (ACC). All photomicrographs are routine hematoxylin-eosin staining magnified × 200 if not otherwise stated. a Overall architecture of an ACC. The tumor cells are lipid-rich or eosinophilic and arranged in large nests and solid areas. Magnification × 100. b Same tumor, with confluent tumor necrosis. c ACC displaying venous angioinvasion characterized by intravascular tumor cells admixed with thrombus. d Capsular invasion and extra-adrenal extension into the surrounding adipose tissue are sometimes noted. Magnification × 100. e Aggravated nuclear pleomorphism with occasional multinucleated, bizarre cells in an oncocytic ACC. Note the presence of an intranuclear inclusion body. Magnification × 400. f Myxoid features can be present in some cases

Typical immunohistochemical expression profile in adrenal cortical carcinoma (ACC). All photomicrographs are magnified × 200. ACCs are regularly uniformly positive for synaptophysin and SF1 and usually exhibit some grade of calretinin and melan A expression. P53 immunostainings are frequently diffusely positive, and the Ki-67 proliferation index is often higher than 5%

IGF2 immunohistochemistry in adrenal cortical carcinoma (ACC). Paranuclear dot-like IGF2 immunoreactivity is a diagnostic feature of ACC and correlates with IGF2 overexpression. Scale bar is 70 µm
Histological Evaluation of Malignant Behavior
The main diagnostic predicament for endocrine pathologists in this context remains the distinction between adrenal cortical adenoma and ACC [140]. To stratify the risk of malignancy in adrenal cortical neoplasms, the current WHO classification of endocrine tumors [124] endorses the application of the multi-parameter schemes including the Weiss criteria for adult, non-oncocytic adrenal cortical tumors [141], the Lin-Weiss-Bisceglia criteria for oncocytic tumors [142], and the Wieneke classification for pediatric cases [143]. The Weiss criteria focus on identifying ACCs by an accumulated score provided by the identification of nuclear pleomorphism, elevated mitotic count, atypical mitoses, reduced number of clear cells, diffuse architecture, presence of necrosis, venous or sinusoidal invasion, and capsular invasion [141]. Three or more of these criteria in a given adrenal cortical tumor would signify a significant risk of malignant behavior, and the algorithm has been reproduced and also modified in large, independent series [144, 145]. As always in endocrine pathology whenever malignancy is wholly or partially dictated by the tumoral relationship to blood vessels and capsule, extensive gross sampling of the tumor is required—which is sometimes burdensome given the impressive sizes of malignant adrenal cortical neoplasms. In fact, the identification of venous angioinvasion characterized by tumor cells invading through a vessel wall and admixed with thrombus is not only regarded a diagnostic feature of ACC, but it is also an important prognostic factor in patients with ACC [74].
As oncocytic tumors per definition are eosinophilic and often display confluent growth and prominent nuclear atypia, this “head-start” of three points by the Weiss criteria would make any oncocytic adrenal cortical tumor potentially malignant—which of course is not the case. To counter this, the Lin-Weiss-Bisceglia criteria were put forward as an alternate algorithm tailor-made for pinpointing oncocytic ACC [142]. In this aspect, one “major” criterion is needed (increased mitotic rate, atypical mitoses, or venous invasion), while the presence of one or more of minor criteria would classify as an adrenal cortical tumor with uncertain malignant potential (aggravated size/weight, necrosis, and sinusoidal or capsular invasion).
Pediatric ACCs have proven difficult to tackle from a histological viewpoint, not least the risk of histologically overstating the risk of malignancy when applying conventional criteria normally used for adult patients [143]. As of this, a separate scoring system proposed by Wieneke et al. combine nine macroscropic and microscopic features of pediatric adrenal cortical neoplasms associated to clinically malignant cases. This model has since been reproduced by other groups and could potentially also benefit from the inclusion of P53, IGF2, and Ki-67 immunostainings [146,147,148].
More recent proposed multiparameter schemes include the reticulin algorithm and the Helsinki scoring systems, which have also been promising in the distinction of ACC [136, 149,150,151]. While the reticulin algorithm builds on the inclusion of reticulin staining to identify loss of nested architecture as a parameter together with additional histological features (necrosis, mitotic rate and vascular invasion), the Helsinki scoring system relies of Ki-67 immunohistochemistry along with the identification of necrosis and mitotic activity [149,150,151].
Immunohistochemistry to Aid in Clinical Routine Practice
While the histological criteria above are useful to diagnose ACC in most patients, there is a consensus that not all malignant adrenal cortical neoplasms are correctly identified through routine histology alone [136]. This is not least exemplified by borderline cases with uncertain malignant potential, which is a term reserved for adrenal cortical tumors fulfilling some criteria for ACC, although not yet reaching the proposed cut-offs [124]. For this reason, endocrine pathologists have turned their attention to immunohistochemistry as a cheap and reproducible method to highlight cases at risk of future dissemination. In adult patients, histochemical and immunohistochemical markers that have proven useful in the distinction between ACCs and benign adrenal cortical nodules include the reticulin stain [149], the Ki-67 proliferation labeling index [151, 152], P53 [153,154,155,156], and IGF2 [74]. In general, intense P53 nuclear immunostaining helps identify a subset of ACCs that are often associated with more obvious histological features of malignancy; therefore, an altered reticulin network in association with an increased Ki-67 count (often above 5%) and paranuclear IGF2 expression is often regarded more useful in the distinction of ACCs from adrenal cortical adenomas in adults [74] (Figs. 8 and 9).
Given the fact that most predictive algorithms are based on establishing a mitotic index in order to highlight malignant cases, it may come as little surprise that researches have been trying to benefit from analyzing the proliferative potential of ACCs. Besides Ki-67, phospohistone (PHH) immunostainings may help to highlight mitoses, and BUB1B, HURP, and NEK2 are additional markers of the mitotic machinery that may help identifying ACCs [74, 157]. Partly mirroring the development of risk assessment of pheochromocytoma and paraganglioma [158], the combination of immunohistochemistry and histology has proved particularly valuable to develop histological predictive models also for ACCs. As discussed earlier, the so-called Helsinki algorithm combine histological parameters and a Ki-67 labeling index as a model to pinpoint ACCs with metastatic potential—thereby providing a small but necessary step for cohesive, multimodal risk assessment of adrenal cortical tumors [150, 151]. Evaluation of the Ki-67 (MIB1) labeling index should be performed on the tumor region with the highest mitotic density preferably using an automated image analysis nuclear algorithm or manual counting; however, eyeballing is no longer recommended [136, 159, 160]. Even so, combinations of histology, immunohistochemistry, and next-generation multi-omics analyses are all probably needed in an integrated fashion to properly distinguish malignancy (Fig. 10).

Molecular attributes of adrenal cortical carcinoma (ACC) and adenoma (ACA). Mechanisms underlying the development of ACCs (depicted to the left, in red color) include over-expression of IGF2 and mutations in the Wnt and P53 signaling pathways. Cell cycle–related genes are often up-regulated, and chromosomal aberrations (usually deletions) are plentiful. ACCs are furthermore characterized by global hypmethylation, but increased promoter-specific hypermethylation, as opposed to ACAs (right, in blue color). ACAs in turn often lack Wnt and P53 pathway mutations, but instead exhibit mutations in genes encoding various ion channels (KCNJ5, ATP1A1, ATP2B3, CACNA1D, and CACNA1H) and in genes regulating protein kinase A (PKA)-related pathways (PRKACA and PRKAR1A). The transcriptome is overrepresented in genes associated to steroidogenesis, and chromosomal aberrations are few. Image created with www.BioRender.com
The Genomic Landscape of Adrenal Cortical Carcinoma
Early Clues from Associated Tumor Syndromes
Prior to the advent of next-generation sequencing (NGS) techniques, recurrent somatic genetic events in ACC were mostly found due to targeted analyses of genes also responsible for syndromic forms of the disease (reviewed below). For example, germline mutations of the tumor suppressor gene TP53 predispose for the Li-Fraumeni syndrome, in which patients are at increased risk of developing ACCs, sarcomas, breast cancer, and various tumors of the central nervous system [161, 162]. TP53 encodes P53, a well-characterized tumor suppressor gene and a master regulator of cellular response to environmental stress and DNA damage (Fig. 11). Following the linkage between germline TP53 mutations and Li-Fraumeni syndrome, somatic TP53 gene mutations were early on reported in subsets of sporadic ACC, while exceedingly rarely observed in ACAs [163]. TP53 was listed as the top mutated gene in ACC (18% of cases) according to the Catalogue of Somatic Mutations in Cancer (COSMIC) database (accessed December 2020) (Table 1); however, if other genetic mechanisms besides mutations are considered, ZNRF3 is the most commonly altered gene in ACCs with an average 20% of cases displaying bilallelic inactivation [45, 164].

Schematic representation of the Wnt and P53 pathways in resting states and in adrenal cortical carcinoma (ACC). In its idle state, the Wnt pathway main effector beta-catenin is sent for ubiquitin-mediated proteolysis by a tri-molecular complex (APC, GSK3-beta, and Axin), and the P53 protein is regulated negatively in a similar manner by MDM2 (left). ACCs recurrently demonstrate activating CTNNB1 mutations (illustrated by a green star), which allows beta-catenin to escape degradation and instead transport to the nucleus, where it will initiate transcription of Wnt target genes, in turn promoting proliferation. This can also be achieved by inactivating mutations or deletions of the negative Wnt regulators ZNRF3 and APC (illustrated by pink stars). Upon cellular stress (for example, extensive DNA damage), MDM2 mediated inhibition of P53 is lifted, leading to the activation of P53 which will promote DNA repair and apoptosis. However, in ACCs with inactivating TP53 gene mutations (pink star), this process is inhibited—leading to inhibited proliferation and accumulation of additional DNA damage. Image created with www.BioRender.com
Moreover, in several other syndromic diseases in which ACC is an established feature (Lynch syndrome [165, 166], familial adenomatous polyposis (FAP) [167], multiple endocrine neoplasia type 1 (MEN1) [168,169,170], and neurofibromatosis type 1 (NF1)) [171, 172], the corresponding genetic aberrancy was also found as somatic mutations in sporadic ACCs (MSH2, APC, MEN1, and NF1, respectively) [164, 173]. In all, among the top 20 mutated genes on the somatic level in ACC, five (25%) are ACC syndromic genes (TP53, MSH2, APC, MEN1, and NF1)—thereby truly manifesting their importance as contributors in the development of ACC (Table 1).
Additionally, loss of the maternal 11p15 allele (with synchronous duplication of the paternal allele) is a frequent finding in ACCs [174,175,176,177]. This locus contains the imprinted H19 and IGF2 genes, which are exclusive expressed from maternal and paternal alleles, respectively [177]. The selective loss of the maternal allele and duplication of the paternal allele is therefore reflected in ACCs as evident down-regulation of H19 and overexpression of IGF2. The H19 gene encodes a long non-coding RNA with tumor suppressive properties, while IGF2 bears oncogenic features—and this change in gene dosage therefore is thought to propel the development of ACC [175, 178]. Interestingly, aberrant methylation patterns in imprinting regions at chromosome 11p15 as well as uniparental disomy (in this case, two copies of the paternal allele is inherited) are the two main mechanisms underlying the Beckwith-Wiedemann syndrome (BWS) [179, 180]. BWS is a developmental disorder in which the afflicted also carries an increased risk of tumor formation, including ACCs.
Chromosomal Aberrations
Besides clues from hereditary syndromes, the gross genetic landscape of ACCs was early-on dissected using loss of heterozygosity (LOH) and comparative genomic hybridization (CGH) techniques. ACCs regularly display high-grade nuclear atypia, atypical mitoses, and aneuploidy histologically, features mirrored on the molecular level by the identification of complex karyotype changes with widespread losses and gain of genetic material. Recurrent regions of gain include 9q34, encompassing NR5A1 encoding the nuclear transcription factor steroidogenic factor 1 (SF1), and 5p15 (including the telomerase reverse transcriptase (TERT) gene encoding the catalytic subunit of telomerase [46, 164, 181]. Gain of these loci lead to increased gene output, in turn instigating adrenal cortical cell proliferation (NR5A1) as well as immortalization due to elongation of telomeric repeats (TERT)—two mechanisms of importance for the development of ACC [182, 183]. Loss of genetic material on the other hand was regularly noted at loci encoding important tumor suppressors, such as MEN1, TP53, and RB1 [184,185,186].
Global Transcriptome Profiling Studies
In addition to the identification of recurrently mutated genes discussed above, researchers turned their attention to global expressional analyses in order to pinpoint important molecular mechanisms driving the ACC development. These types of sub-classifications often facilitate the potential separation of tumors into different prognostic clusters to highlight cases exhibiting the highest risk of disseminated disease. In one of the earliest efforts, Giordano and colleagues interrogated the expressional status of approximately 10,000 genes in a small cohort of ACCs and adrenal cortical adenomas using microarray technique, and verified 91 genes as differentially expressed between these groups [187]. Among the significantly up-regulated genes, IGF2, TOP2A, and Ki-67 emerged. Following this study, de Fraipont et al. corroborated the importance of IGF2 when analyzing the expression of 230 candidate genes in a somewhat larger tumor cohort [188]. Unsupervised hierarchical analyses revealed two distinct expressional clusters, the IGF2 cluster and the steroidogenesis cluster. Interestingly, ACCs tended to express high and low amounts respectively of genes associated to the IGF2 and steroidogenesis clusters, while adenomas on the opposite expressed low and high quantities, respectively, for the same clusters (Fig. 10). In this aspect, the previously suggested association of increased IGF2 expression in ACCs was validated, and the authors furthermore presented a 22-gene panel that allowed for an accurate separation of ACCs from adenomas, on par with results obtained from the Weiss algorithm [188]. Other microarray studies have since reinforced the notion that ACCs have a distinct expressional profile which sets them apart from their benign counterparts, also verifying IGF2 and/or TOP2A as significantly upregulated in malignant adrenal cortical tumors [157, 189,190,191]. Moreover, in one of the most comprehensive expressional profiling studies to date, ACCs with exceedingly poor prognosis was found to cluster together using an unsupervised hierarchical analysis [157]. Interestingly, this cluster (denoted “cluster 1”) was enriched for genes associated to chromosomal integrity and proliferation, suggesting a more chaotic genomic profile than cluster 2 ACCs [157]. This study thus exemplifies how expressional profiling might help to distinguish ACCs from adenomas, as well as to stratify the risk for adverse outcomes within the ACC group itself. The idea that expressional sub-clustering of ACCs bears clinical relevance was even more reinforced by a separate study from France, revealing two main expressional clusters (C1 and C2) [190]. The C1 group contained almost all adrenal cortical tumors that exhibited high Weiss scores, relapses, and distant metastases (i.e., bona fide ACCs), whereas the C2 group was considered clinically benign [190]. Interestingly, the C1 group was enriched for genes responsible for the mitotic machinery and DNA replication, while the C2 group (adenomas) was overrepresented in terms of genes associated to immune response mechanisms. The malignant C1 group was further sub-divided into C1A and C1B, as C1A-related ACCs were strongly associated with poor prognostic patient outcome than those clustered in C1B—thereby again verifying the notion that ACCs could be stratified into two prognostic groups based on expressional profiling [190]. Interestingly, C1A type ACCs have since been shown to harbor mutually exclusive TP53 or CTNNB1 gene mutations, thereby allowing for an important association between mutational profiles and expressional clustering (Fig. 11) [192].
To conclude this section, ACCs regularly overexpress IGF2, cell cycle-related genes as well as genes coupled to chromosomal integrity, while adenomas overexpress genes associated to steroidogenesis (Fig. 10). These findings also help endocrine pathologists validate the usefulness of IGF2 and cell cycle–related immunohistochemical biomarkers in the diagnosis of ACC [74]. Moreover, ACCs can be sub-classified into high and low risk cases based on their expressional output. The usefulness of P53 and beta-catenin immunohistochemistry in adrenalectomy specimens is also acknowledged to help stratify C1A-related disease [68, 74]. Nowadays, based on subsequent pan-genomic characterization using multi-omics platforms, we know that the clustering provided by the early transcriptome analyses reviewed above still stands as valid observations and constitute cornerstone studies in our efforts to characterize and profile ACCs.
Early Studies of Epigenetic Aberrancies
In various human tumors, other mechanisms besides mutations have been linked to expressional and translational regulation of gene output, not least epigenetic modifications as well as expression of specific micro-RNAs (miRs). Epigenetic aberrancies especially have gained ground as contributors of tumor development, and the most common underlying mechanism revolves around altered levels of methylation at specific cytosine residues (often entitled CpG sites) in promoter regions of genes commonly associated to cancer. As changes in methylation patterns of CpG sites may confer either increased or reduced gene output, promoter hypermethylationand hypomethylation are nowadays considered driver events in tumor formation. The first comprehensive description of the ACC methylome was published in 2012 by Fonseca et al., in which normal adrenal tissues, adenomas, and ACCs were assessed for global methylation levels across > 27,000 CpG sites [193]. The authors found a significant over-representation of CpG island hypermethylation in ACCs regarding genes coupled to transcription factor regulation, the cell cycle machinery, and apoptosis, including CDKN2A, GATA4, and DLEC1. These genes showed a significant downregulation of mRNA, while treatment of the H295R ACC cell line using a demethylation agent restored the expression. Expanding on this fact that epigenetic silencing of cancer-related genes might play a role in ACCs, a subsequent study from the National Institute of Health employed a global methylome array covering more than 485,000 CpG sites [194]. The authors observed that normal adrenal tissues compared with benign tissue samples had the least number of differences in methylation, and were predominantly hypermethylated, while primary and metastatic ACCs displayed the largest difference compared with normal adrenal samples, and were in general hypomethylated (Fig. 10). Moreover, gene-specific aberrant methylation in ACCs was found in genes involved in the IGF2 signaling pathway as well as in pathways associated to lipid metabolism, which was also noted on the transcriptional level as altered mRNA levels of the corresponding gene products [194]. The potential for analyses of the global methylome as a diagnostic instrument for ACCs was even more substantiated when a French study identified specific methylation signatures (denoted “CpG island methylator phenotypes”; CIMPs) in ACCs [195]. When performing an unsupervised clustering, ACCs aggregated as according to the level of global CpG methylation, and were classified either as non-CIMP, CIMP-low, or CIMP-high ACCs. Non-CIMP ACCs displayed similar levels of global methylation as adenomas, while CIMP-low and CIMP-high cases exhibited higher levels than benign tumors. Intriguingly, patients with CIMP ACCs displayed worse survival than patients with non-CIMP ACCs, and CIMP-high ACCs were even more pronounced in this aspect than CIMP-low cases [195]. In all, global methylome analyses seem to be able to pinpoint ACC cases with dismal prognosis, similar to what has been shown by global transcriptome analyses discussed earlier.
Dysregulation of micro-RNAs
Micro-RNAs (miRNAs) are a class of non-coding, short, single-stranded RNA molecules with the potential to regulate gene output through the translational inhibition of mRNAs. The role of miRNAs in cancer is expanding, and dysregulation of miRNAs currently constitutes a common phenomenon in cancer development, including ACC [196]. For example, specific miRNAs found to be aberrantly expressed include the observed downregulation of miR-195 and upregulation of miR-483-5p—a feature that was also coupled to poorer disease-specific survival [197]. Moreover, circulating levels of these two miRNAs were linked to an increased risk of recurrence in ACC patients, furthermore validating the important roles of miR-195 and miR-483-5p.
Pan-Genomic Characterization of Adrenal Cortical Carcinoma
The development of the NGS technique has vastly improved the ability to interrogate genomic sequences in tumor samples on a large scale. By modern methodology, researchers have been able to characterize various underlying genetic aberrancies in ACC, and the field is rapidly evolving. Recently, the mutational landscape of ACCs was deciphered by whole-exome sequencing (WES) analyses, and additional analyses of fusion genes, gene copy number, global methylation, and miRNA profiling have laid the foundation for a comprehensive molecular coverage with clear-cut associations to tumor characteristics and patient outcome.
In 2014, Assié and co-workers published the first whole-exome sequencing analysis of ACC and integrated the mutational landscape with data from analyses covering gene expression, copy number alterations, methylation, and miRNA profiling [164]. The most commonly observed somatic genetic event were mutations and/or deletions of the Wnt pathway members ZNRF3 and CTNNB1, followed by mutations in p53 pathway members TP53, CDK2NA, and RB1. Interestingly, these events were mutually exclusive to a large degree. Additional mutations in chromatin remodeling genes were also seen (DAXX, MEN1, ATRX), and recurrent gain of the TERT gene locus was also noted. Using miRNA profiling, ACCs were sub-divided into three groups (Mi1, Mi2, and Mi3). This clustering revealed differential expression of several miRNAs located at the DLK1-MEG3 locus on chromosome 14q as a main contributor of this unsupervised clustering. The authors then combined these above-mentioned analyses with transcriptome and methylome analyses (in which ACCs were divided into the C1A and C1B expressional clusters and non-CIMP, CIMP-low, and CIMP-high groups, respectively), thereby presenting the first integrated, multi-omic characterization of ACC [164]. In this model, two main molecular groups were discerned based on their transcriptomic output, namely, C1A and C1B. C1A tumors displayed significant correlation to a high number of chromosomal alterations, associated to CIMP ACCs as opposed to non-CIMP ACCs, and clustered with the Mi3 miRNA profiling group characterized by up-regulation of DLK-MEG3 locus related miRNAs. The C1A group was furthermore associated to increased mutational burden, as well as to mutations and deletions in Wnt and P53 signaling pathway members (ZNRF3, CTNNB1, TP53), while the C1B group was generally devoid of these genetic events. As expected, C1A patients exhibited worse clinical outcome than C1B patients. Even within the C1A group, outcomes were coupled to the CIMP profiles, in which CIMP-high ACC cases in the C1A group exhibited the worst survival. This study thus solidifies some 20 years of ACC research in which analyses of mutations, transcriptome, methylome, and miRNA patterns have helped us recognize that ACCs correspond to at least two distinct molecular entities with significantly different patient outcomes [164]. Shortly after this study was published, a collaborative effort from Yale and Karolinska verified the abundance of ZNRF3 gene deletions in ACC—thereby adding yet another Wnt pathway member to the list of recurrently altered genes in ACC [46]. From a molecular biology standpoint, ZNRF3 is a membrane-bound ubiquitin-protein ligase that mediates ubiquitination of the Wnt pathway activating receptor Frizzled, which will lead to a proteosomal degradation and hence negative regulation of the Wnt cascade [198]. The observed loss-of-function mutations and gene deletions demonstrated in ACCs thus suggest that ZNRF3 is a tumor suppressor (Fig. 11), and further strengthen the coupling between aberrant Wnt signaling and ACCs. As dysregulation of the Wnt network in turn seem to associate strongly to a specific molecular signature with poor clinical outcome, immunohistochemical analysis of the Wnt effector protein beta-catenin has emerged as a prognostic marker in ACC (Fig. 12) [199].

Beta-catenin immunohistochemistry in adrenal cortical carcinoma (ACC). An active Wnt/beta-catenin pathway in ACCs can be highlighted using beta-catenin immunohistochemistry. Upon Wnt pathway activation, beta-catenin is translocated into the nucleus to initiate the activation of transcriptional programs leading to augmented proliferation. As Wnt activated ACCs usually exhibit worse clinical outcomes, nuclear beta-catenin immunoreactivity could therefore serve as a marker of poor prognosis. Scale bar is 80 µm
Following the groundbreaking work of Assié and colleagues, the Cancer Genome Atlas (TCGA) network published a genome-wide project on ACC in 2016 [45]. A global cohort consisting of 91 ACCs were interrogated for DNA mutations, copy number alterations, methylation, and expression levels of mRNA and miRNAs. The authors identified several novel mutated genes of potential impact for ACC development, including PRKAR1A, RPL22, TERF2, and CCNE1. Genome-wide copy number analyses pinpointed deletions as much more common events than gains/amplifications, and furthermore identified three main CNA clusters; “quiet,” “noisy,” and “chromosomal,” occurring in 9%, 30%, and 61% of ACCs, respectively. “Quiet” ACCs were euploid with few arm-level CNAs, the “noisy” subset was characterized by many focal CNAs, and the “chromosomal” group consisted of ACCs with abundant whole-chromosomal deletions. The “noisy” group exhibited worse clinical outcome, suggesting that many focal, arm-level losses of genetic material are associated with more aggressive disease. Interestingly, within the “noisy” and “chromosomal groups,” ACCs could be sub-divided into two classes; those with and without whole-genome doubling (WGD)—a hyper-diploid karyotype. WGD in turn associated to worse patient outcome, expressional cluster C1A, the CIMP-high profile, TERT alterations, mutations in CTNNB1 and TP53, as well as specific expression of TERT and other telomere-regulating genes. In all, the authors proposed a three-cluster tier, entitled Cluster of Cluster (CoC) I–III, in addition to scattered cases in a fourth, unassigned group (Fig. 13) [45]. CoCI tumors exhibited the best prognosis, were overrepresented in ACCs with a C1B mRNA profile, a CIMP-low phenotype and adhering to the “chromosomal” profile of CNAs. CoCII cases were intermediate in terms of prognosis and were predominantly associated to expression cluster C1A, a CIMP-intermediate profile, and seemed to have more cases with WGD than the CoCI group. Lastly, the CoCIII group were characterized by ACCs with poor survival, and associated to the C1A expressional cluster, a CIMP-high profile, “noisy” CNA profile with associated WGD, and mutations in TP53 or CTNNB1 (Fig. 13).

Generalized multi-OMICs profiles of adrenal cortical carcinoma (ACC). Via pan-genomic characterization of the ACC mutational, chromosomal, expressional, and epigenetic landscapes, three main clusters have emerged, entitled Cluster of Clusters I–III (CoCI–III). CoCI ACCs exhibit the best prognosis of the three and are regularly associated to lower TNM stages. These tumors are characterized by a low frequency of mutations in Wnt and P53 signaling pathway genes and associate to the mRNA expressional cluster C1B enriched for genes regulating immune response mechanisms. CoCI ACCs furthermore exhibit low levels of gene-specific methylation, thereby characterized as “CpG island methylator phenotype-low” (CIMP-low). Amplifications and whole-chromosome deletions are commonly seen, but whole-genome doubling (WGD) is rare. While CoCII ACCs are considered an intermediate group of tumors in terms of prognosis and genetic aberrancies, CoCIII ACCs exhibit the worst prognosis and the most advanced genetic imbalances, characterized by frequent mutations in Wnt and P53 pathway gene members, expression of genes primarily associated to mitotic regulation (cluster C1A), and a CIMP-high profile. Chromosomal aberrations are frequent and scattered across the genome (“noisy”), and WGD is evident in the majority of CoCIII ACCs. Image created with www.BioRender.com
In all, these comprehensive studies have laid the foundation for a molecular approach to stratify the prognosis of ACCs, in which histology alone is insufficient—and might pinpoint novel aspects as to how to triage cases for adjuvant therapies, as surgery and mitotane alone are insufficient to cure most patients of this often-lethal disease.
Molecular Tools for Prognostication of Adrenal Cortical Carcinomas
As molecular genetics is emerging as a powerful diagnostic and prognostic tool in surgical pathology, this will also have consequences for how we assess adrenal cortical neoplasms in the near future. Histological assessment and immunohistochemistry are still gold-standard techniques in diagnosing ACC; the need for molecular analyses to guide the clinicians will probably increase given the rapid development of individualized treatment options for cancer patients in general. For example, metastatic ACCs that are unresponsive to mitotane could potentially benefit from NGS screening of underlying genetic aberrancies of possible therapeutic value. Moreover, this screening could in theory also aid in the identification of hereditary syndromes. For example, the detection of a TP53 mutation in ACC tissue should mandate additional genetic analyses also in germline DNA. The chance of detecting germline TP53 alterations in ACC patients decreases with age, with approximately 50% of children diagnosed with ACC harboring germline TP53 mutations, compared with less than 10% of adults diagnosed with ACC [124].
Even though the integrated OMIC analyses discussed above have provided us with an increased understanding of the molecular profiles distinguishing ACCs from adenomas (Fig. 10) as well as to pinpoint ACCs with exceptionally poor clinical outcomes (Fig. 13), surgical pathology laboratories will still look for simplified molecular schemes to aid in these matters. In fact, the TCGA consortium suggested a focused methylation signature consisting of 68 probes that correctly sorted ACCs into the CoCI-III prognostic groups discussed above [45]. Although this methylation panel in theory could be employed for clinical FFPE material, interpretation of methylation levels of numerous genes might be burdensome for clinical purposes. In terms of single-gene markers, G0/G1Switch2 (G0S2) gene hypermethylation has been reported as an indicator of the CIMP-high group of ACCs [200]. The G0S2 methylation status correlated strongly with fatal ACCs and could therefore constitute a beneficial marker to single out ACCs with exceptional poor prognosis. Moreover, the identification of aberrant signaling in telomerase-related pathways in ACCs with poor prognosis (especially TERT gene expression, gene copy gains, promoter mutations, and aberrant methylation) might be a way forward to pinpoint cases at risk of worse clinical outcomes [46, 183, 201, 202]. Interestingly, telomere length itself, which in turn is coupled to the activity of TERT, might serve as a prognostic marker for ACCs [45]. However, as mutational compositions may vary due to spatial heterogeneity in ACCs, the benefits of using single-gene markers must be weighed against the risk of false negative results due to a heterogeneous tumor exhibiting sub-clonal mutations [203].
Germline Susceptibility for Adrenal Cortical Neoplasia
As mentioned in earlier sections, many clues regarding aberrant genetic mechanisms propelling the development of adrenal cortical neoplasia come from careful investigations of genes associated to various tumor syndromes in which adrenal lesions constitute an established feature. These hereditary conditions in which adrenal cortical tumors are overrepresented compared with the general population are detailed in Table 2 and include the Li-Fraumeni syndrome, the Beckwith-Wiedemann syndrome, the MEN1 syndrome, the NF1 syndrome, the Lynch syndrome, the FAP syndrome, and Carney complex as well as familial forms of primary hyperaldosteronism.
Approximately 10% of ACC patients will harbor an underlying germline mutation in an ACC susceptibility gene [204]. For patients with ACCs, the most common syndromic manifestation is Li-Fraumeni syndrome and might be evident in 50–80% of children with ACC, as well as in 3–5% of adults [124]. Moreover, rare ACCs might be associated to patients with Lynch syndrome, an autosomal-dominant hereditary cancer predisposition syndrome caused by inactivating constitutional mutations in mismatch repair genes MSH2, MSH6, MLH1, and PSM2. In ACCs arising in the Lynch syndrome setting, absent MMR protein immunoreactivity might be noted, and immunohistochemistry targeting MSH2, MSH6, MLH1, and PMS2 could therefore be a rather cost-effective way to triage cases for genetic counseling (Fig. 14) [204, 205]. Small subsets of ACC patients have also been shown to be MEN1 syndrome carriers, in which the afflicted often display primary hyperparathyroidism as well as neuroendocrine neoplasia of the pituitary and pancreas [124].

Mismatch repair (MMR) protein immunohistochemistry. Routine MMR immunohistochemistry is strongly encouraged not only to screen for the possibility of a germline susceptibility (extra-colonic manifestations of Lynch syndrome) but may also shape therapeutic decisions as microsatellite instable tumors might confer specific opportunities for adjuvant treatments. Depicted here is an adrenal cortical carcinoma displaying retained nuclear MLH1 and PMS2 immunoreactivity, while displaying absent staining for MSH2 and MSH6. Note the internal positive controls for the two latter markers. Scale bar is 300 µm
Interestingly, an association between ACC and germline mutations in the Succinate Dehydrogenase Complex Subunit A (SDHA) and Succinate Dehydrogenase Complex Subunit C (SDHC) genes has also been reported. As these genes are inactivated on the constitutional level in two different familial paraganglioma-pheochromocytoma syndromes (paraganglioma syndrome type 5 and 3, respectively), the authors speculate that ACCs may be rare manifestations of these entities [124, 206].
As a rather significant subset of adult patients with ACC might carry germline alterations of any of these genes above, genetic counseling and mutational screening for syndromic forms could be recommended [204]. Moreover, as comprehensive genetic screening of somatic DNA acquired from ACCs will be a likely cornerstone of future diagnostic work-up in pathology laboratories, there will probably be an increased volume of ACC patients with MMR-related pathogenesis due MSH2, MSH6, PMS2 and MLH1, as well as TP53 and MEN1 gene mutations that need to be excluded or confirmed on the germline level as well.
Conclusion
Modern molecular analyses of adrenal cortical neoplasia have skyrocketed our understanding of these tumors. The notion that aldosterone- and cortisol-producing adrenal cortical adenomas are driven by ion channel mutations and aberrant PKA signaling, respectively, are pivotal advances that helped the scientific community understand how downstream alterations in the AT2 and ACTH pathways of the zona glomerulosa and fasciculat, respectively, lead to tumor formation. Moreover, this knowledge might also open up for hypothetical, non-surgical therapeutic strategies for this patient category in instances when surgery is deemed too risky. Furthermore, the in-depth genomic characterization of ACC has identified high-risk subsets with exceedingly poor prognosis, suggesting that molecular triaging might be of clinical value where conventional histology is insufficient. In the near future, comprehensive molecular diagnostics will most likely be considered as a natural part of the funded pathologist work-up of adrenal cortical neoplasia, in which clinical, histological, and genetic analyses are merged into a comprehensive final endocrine pathologist’s report—giving the treating clinician the proper prognostic information needed to individualize the risk assessment, adjuvant treatment options and patient follow-up.
References
Duan K, Mete O (2015) Clinicopathologic Correlates of Primary Aldosteronism. Arch Pathol Lab Med 139:948–954. https://doi.org/10.5858/arpa.2014-0156-RS
Hodgson A, Pakbaz S, Mete O (2019) A Diagnostic Approach to Adrenocortical Tumors. Surg Pathol Clin 12:967–995. https://doi.org/10.1016/j.path.2019.08.005
Zennaro M-C, Fernandes-Rosa F, Boulkroun S, Jeunemaitre X (2015) Bilateral Idiopathic Adrenal Hyperplasia: Genetics and Beyond. Horm Metab Res 47:947–952. https://doi.org/10.1055/s-0035-1565198
Nanba AT, Nanba K, Byrd JB, Shields JJ, Giordano TJ, Miller BS, Rainey WE, Auchus RJ, Turcu AF (2017) Discordance between imaging and immunohistochemistry in unilateral primary aldosteronism. Clin Endocrinol (Oxf) 87:665–672. https://doi.org/10.1111/cen.13442
Yamazaki Y, Nakamura Y, Omata K, Ise K, Tezuka Y, Ono Y, Morimoto R, Nozawa Y, Gomez-Sanchez CE, Tomlins SA, Rainey WE, Ito S, Satoh F, Sasano H (2017) Histopathological Classification of Cross-Sectional Image-Negative Hyperaldosteronism. J Clin Endocrinol Metab 102:1182–1192. https://doi.org/10.1210/jc.2016-2986
Kempers MJE, Lenders JWM, van Outheusden L, van der Wilt GJ, Schultze Kool LJ, Hermus ARMM, Deinum J (2009) Systematic review: diagnostic procedures to differentiate unilateral from bilateral adrenal abnormality in primary aldosteronism. Ann Intern Med 151:329–337. https://doi.org/10.7326/0003-4819-151-5-200909010-00007
Sung T-Y, Alobuia WM, Tyagi MV, Ghosh C, Kebebew E (2020) Adrenal Vein Sampling to Distinguish Between Unilateral and Bilateral Primary Hyperaldosteronism: To ACTH Stimulate or Not? J Clin Med 9. https://doi.org/10.3390/jcm9051447
Wannachalee T, Zhao L, Nanba K, Nanba AT, Shields JJ, Rainey WE, Auchus RJ, Turcu AF (2019) Three Discrete Patterns of Primary Aldosteronism Lateralization in Response to Cosyntropin During Adrenal Vein Sampling. J Clin Endocrinol Metab 104:5867–5876. https://doi.org/10.1210/jc.2019-01182
Williams TA, Burrello J, Sechi LA, Fardella CE, Matrozova J, Adolf C, Baudrand R, Bernardi S, Beuschlein F, Catena C, Doumas M, Fallo F, Giacchetti G, Heinrich DA, Saint-Hilary G, Jansen PM, Januszewicz A, Kocjan T, Nishikawa T, Quinkler M, Satoh F, Umakoshi H, Widimský J, Hahner S, Douma S, Stowasser M, Mulatero P, Reincke M (2018) Computed Tomography and Adrenal Venous Sampling in the Diagnosis of Unilateral Primary Aldosteronism. Hypertension 72:641–649. https://doi.org/10.1161/HYPERTENSIONAHA.118.11382
Campbell RA, Young DS, Shaver CN, Snyder SK, Milan SA, Lairmore TC, McDonald DK (2019) Influence of Adrenal Venous Sampling on Management in Patients with Primary Aldosteronism Independent of Lateralization on Cross-Sectional Imaging. J Am Coll Surg 229:116–124. https://doi.org/10.1016/j.jamcollsurg.2019.03.012
Conn JW (1955) Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med 45:3–17
Conn JW (1955) Primary aldosteronism. J Lab Clin Med 45:661–664
Chalmers TM (1960) Conn’s syndrome. Postgrad Med J 36:198–200. https://doi.org/10.1136/pgmj.36.413.198
Duan K, Giordano TJ, Mete O (2016) Adrenal cortical proliferations. In: In: Mete O, Asa SL, eds. Endocrine Pathology. Cambridge University Press, pp 602–627
Griffin AC, Kelz R, LiVolsi VA (2014) Aldosterone-secreting adrenal cortical carcinoma. A case report and review of the literature. Endocr Pathol 25:344–349. https://doi.org/10.1007/s12022-014-9307-x
Mete O, Asa SL (2009) Aldosterone-producing adrenal cortical adenoma with oncocytic change and cytoplasmic eosinophilic globular inclusions. Endocr Pathol 20:182–185. https://doi.org/10.1007/s12022-009-9082-2
Mete O, Duan K (2018) The Many Faces of Primary Aldosteronism and Cushing Syndrome: A Reflection of Adrenocortical Tumor Heterogeneity. Front Med (Lausanne) 5:54. https://doi.org/10.3389/fmed.2018.00054
Nishimoto K, Koga M, Seki T, Oki K, Gomez-Sanchez EP, Gomez-Sanchez CE, Naruse M, Sakaguchi T, Morita S, Kosaka T, Oya M, Ogishima T, Yasuda M, Suematsu M, Kabe Y, Omura M, Nishikawa T, Mukai K (2017) Immunohistochemistry of aldosterone synthase leads the way to the pathogenesis of primary aldosteronism. Mol Cell Endocrinol 441:124–133. https://doi.org/10.1016/j.mce.2016.10.014
Williams TA, Gomez-Sanchez CE, Rainey WE, Giordano TJ, Lam AK, Marker A, Mete O, Yamazaki Y, Zerbini MCN, Beuschlein F, Satoh F, Burrello J, Schneider H, Lenders JWM, Mulatero P, Castellano I, Knösel T, Papotti M, Saeger W, Sasano H, Reincke M (2021) International Histopathology Consensus for Unilateral Primary Aldosteronism. J Clin Endocrinol Metab 106:42–54. https://doi.org/10.1210/clinem/dgaa484
Volpe C, Hamberger B, Zedenius J, Juhlin CC (2020) Impact of immunohistochemistry on the diagnosis and management of primary aldosteronism: An important tool for improved patient follow-up. Scand J Surg 109:133–142. https://doi.org/10.1177/1457496918822622
Ito A, Yamazaki Y, Sasano H, Matsubara D, Fukushima N, Tamba M, Tabata K, Ashizawa K, Takei A, Koizumi M, Sakuma Y, Sata N, Oshiro H (2017) A case of primary aldosteronism caused by unilateral multiple adrenocortical micronodules presenting as muscle cramps at rest: The importance of functional histopathology for identifying a culprit lesion. Pathol Int 67:214–221. https://doi.org/10.1111/pin.12521
Stenman A, Shabo I, Ramström A, Zedenius J, Juhlin CC (2019) Synchronous aldosterone- and cortisol-producing adrenocortical adenomas diagnosed using CYP11B immunohistochemistry. SAGE Open Med Case Rep 7:2050313X19883770. https://doi.org/10.1177/2050313X19883770
Oba K, Chiba Y, Matsuda Y, Kumakawa T, Aoyama R, Akahoshi M, Hashimoto S, Tachibana A, Toyoshima K, Kodera R, Toyoshima K, Tamura Y, Nagata T, Yamazaki Y, Sasano H, Araki A (2020) Primary Aldosteronism Associated with Multiple Adrenocortical Micronodules in a Patient with Renal Cell Carcinoma. Case Rep Endocrinol 2020:2808101. https://doi.org/10.1155/2020/2808101
Nanba K, Chen AX, Omata K, Vinco M, Giordano TJ, Else T, Hammer GD, Tomlins SA, Rainey WE (2016) Molecular Heterogeneity in Aldosterone-Producing Adenomas. J Clin Endocrinol Metab 101:999–1007. https://doi.org/10.1210/jc.2015-3239
Gomez-Sanchez CE, Gomez-Sanchez EP, Nishimoto K (2020) Immunohistochemistry of the Human Adrenal CYP11B2 in Normal Individuals and in Patients with Primary Aldosteronism. Horm Metab Res 52:421–426. https://doi.org/10.1055/a-1139-2079
Nanba K, Omata K, Tomlins SA, Giordano TJ, Hammer GD, Rainey WE, Else T (2016) Double adrenocortical adenomas harboring independent KCNJ5 and PRKACA somatic mutations. Eur J Endocrinol 175:K1-6. https://doi.org/10.1530/EJE-16-0262
Faillot S, Foulonneau T, Néou M, Espiard S, Garinet S, Vaczlavik A, Jouinot A, Rondof W, Septier A, Drougat L, Hécale-Perlemoine K, Ragazzon B, Rizk-Rabin M, Sibony M, Bonnet-Serrano F, Guibourdenche J, Libé R, Groussin L, Dousset B, de Reyniès A, Bertherat J, Assié G (2021) Genomic classification of benign adrenocortical lesions. Endocr Relat Cancer 28:79–95. https://doi.org/10.1530/ERC-20-0128
Howard B, Wang Y, Xekouki P, Faucz FR, Jain M, Zhang L, Meltzer PG, Stratakis CA, Kebebew E (2014) Integrated analysis of genome-wide methylation and gene expression shows epigenetic regulation of CYP11B2 in aldosteronomas. J Clin Endocrinol Metab 99:E536-543. https://doi.org/10.1210/jc.2013-3495
Nanba K, Chen A, Nishimoto K, Rainey WE (2015) Role of Ca(2+)/calmodulin-dependent protein kinase kinase in adrenal aldosterone production. Endocrinology 156:1750–1756. https://doi.org/10.1210/en.2014-1782
Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Åkerström G, Wang W, Carling T, Lifton RP (2011) K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331:768–772. https://doi.org/10.1126/science.1198785
Williams TA, Monticone S, Schack VR, Stindl J, Burrello J, Buffolo F, Annaratone L, Castellano I, Beuschlein F, Reincke M, Lucatello B, Ronconi V, Fallo F, Bernini G, Maccario M, Giacchetti G, Veglio F, Warth R, Vilsen B, Mulatero P (2014) Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 63:188–195. https://doi.org/10.1161/HYPERTENSIONAHA.113.01733
Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, Walther A, Tauber P, Schwarzmayr T, Diener S, Graf E, Allolio B, Samson-Couterie B, Benecke A, Quinkler M, Fallo F, Plouin P-F, Mantero F, Meitinger T, Mulatero P, Jeunemaitre X, Warth R, Vilsen B, Zennaro M-C, Strom TM, Reincke M (2013) Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 45:440–444, 444e1–2. https://doi.org/10.1038/ng.2550
Azizan EAB, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W, Shaikh LH, Brighton CA, Teo AED, Davenport AP, Dekkers T, Tops B, Küsters B, Ceral J, Yeo GSH, Neogi SG, McFarlane I, Rosenfeld N, Marass F, Hadfield J, Margas W, Chaggar K, Solar M, Deinum J, Dolphin AC, Farooqi IS, Striessnig J, Nissen P, Brown MJ (2013) Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 45:1055–1060. https://doi.org/10.1038/ng.2716
Scholl UI, Goh G, Stölting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson-Williams C, Libutti SK, Mane S, Hellman P, Westin G, Åkerström G, Björklund P, Carling T, Fahlke C, Hidalgo P, Lifton RP (2013) Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 45:1050–1054. https://doi.org/10.1038/ng.2695
Nanba K, Yamazaki Y, Bick N, Onodera K, Tezuka Y, Omata K, Ono Y, Blinder AR, Tomlins SA, Rainey WE, Satoh F, Sasano H (2020) Prevalence of Somatic Mutations in Aldosterone-Producing Adenomas in Japanese Patients. J Clin Endocrinol Metab 105. https://doi.org/10.1210/clinem/dgaa595
Nanba K, Blinder AR, Rege J, Hattangady NG, Else T, Liu C-J, Tomlins SA, Vats P, Kumar-Sinha C, Giordano TJ, Rainey WE (2020) Somatic CACNA1H Mutation As a Cause of Aldosterone-Producing Adenoma. Hypertension 75:645–649. https://doi.org/10.1161/HYPERTENSIONAHA.119.14349
Rege J, Nanba K, Blinder AR, Plaska S, Udager AM, Vats P, Kumar-Sinha C, Giordano TJ, Rainey WE, Else T (2020) Identification of Somatic Mutations in CLCN2 in Aldosterone-Producing Adenomas. J Endocr Soc 4:bvaa123. https://doi.org/10.1210/jendso/bvaa123
Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, Strom TM, Monticone S, Amar L, Meatchi T, Mantero F, Cicala M-V, Quinkler M, Fallo F, Allolio B, Bernini G, Maccario M, Giacchetti G, Jeunemaitre X, Mulatero P, Reincke M, Zennaro M-C (2014) Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 64:354–361. https://doi.org/10.1161/HYPERTENSIONAHA.114.03419
Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP (2015) A Meta-Analysis of Somatic KCNJ5 K(+) Channel Mutations In 1636 Patients With an Aldosterone-Producing Adenoma. J Clin Endocrinol Metab 100:E1089-1095. https://doi.org/10.1210/jc.2015-2149
Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, Thompson LDR, Cohen DL, Luther JM, Gellert L, Vaidya A, Barletta JA, Else T, Giordano TJ, Tomlins SA, Rainey WE (2019) Genetic Characteristics of Aldosterone-Producing Adenomas in Blacks. Hypertension 73:885–892. https://doi.org/10.1161/HYPERTENSIONAHA.118.12070
Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, Miller BS, Giordano TJ, Tomlins SA, Rainey WE (2018) Targeted Molecular Characterization of Aldosterone-Producing Adenomas in White Americans. J Clin Endocrinol Metab 103:3869–3876. https://doi.org/10.1210/jc.2018-01004
Mouat IC, Omata K, McDaniel AS, Hattangady NG, Talapatra D, Cani AK, Hovelson DH, Tomlins SA, Rainey WE, Hammer GD, Giordano TJ, Else T (2019) Somatic mutations in adrenocortical carcinoma with primary aldosteronism or hyperreninemic hyperaldosteronism. Endocr Relat Cancer 26:217–225. https://doi.org/10.1530/ERC-18-0385
Wu V-C, Wang S-M, Chueh S-CJ, Yang S-Y, Huang K-H, Lin Y-H, Wang J-J, Connolly R, Hu Y-H, Gomez-Sanchez CE, Peng K-Y, Wu K-D (2017) The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci Rep 7:39121. https://doi.org/10.1038/srep39121
Åkerström T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, Stålberg P, Robinson B, Alexander Iwen K, Dralle H, Walz MK, Lehnert H, Sidhu S, Gomez-Sanchez C, Hellman P, Björklund P (2016) Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep 6:19546. https://doi.org/10.1038/srep19546
Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, Kim S, Assie G, Morozova O, Akbani R, Shih J, Hoadley KA, Choueiri TK, Waldmann J, Mete O, Robertson AG, Wu H-T, Raphael BJ, Shao L, Meyerson M, Demeure MJ, Beuschlein F, Gill AJ, Sidhu SB, Almeida MQ, Fragoso MCBV, Cope LM, Kebebew E, Habra MA, Whitsett TG, Bussey KJ, Rainey WE, Asa SL, Bertherat J, Fassnacht M, Wheeler DA, Cancer Genome Atlas Research Network, Hammer GD, Giordano TJ, Verhaak RGW (2016) Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 29:723–736. https://doi.org/10.1016/j.ccell.2016.04.002
Juhlin CC, Goh G, Healy JM, Fonseca AL, Scholl UI, Stenman A, Kunstman JW, Brown TC, Overton JD, Mane SM, Nelson-Williams C, Bäckdahl M, Suttorp A-C, Haase M, Choi M, Schlessinger J, Rimm DL, Höög A, Prasad ML, Korah R, Larsson C, Lifton RP, Carling T (2015) Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J Clin Endocrinol Metab 100:E493-502. https://doi.org/10.1210/jc.2014-3282
Bonnet S, Gaujoux S, Launay P, Baudry C, Chokri I, Ragazzon B, Libé R, René-Corail F, Audebourg A, Vacher-Lavenu M-C, Groussin L, Bertagna X, Dousset B, Bertherat J, Tissier F (2011) Wnt/β-catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: a study in cortisol-secreting and -nonsecreting tumors. J Clin Endocrinol Metab 96:E419-426. https://doi.org/10.1210/jc.2010-1885
Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, Samson-Couterie B, Zennaro M-C, Plouin P-F, Skah S, Plateroti M, Lefèbvre H, Sahut-Barnola I, Batisse-Lignier M, Assié G, Lefrançois-Martinez A-M, Bertherat J, Martinez A, Val P (2014) WNT/β-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet 23:889–905. https://doi.org/10.1093/hmg/ddt484
Bhandaru M, Kempe DS, Rotte A, Rexhepaj R, Kuhl D, Lang F (2009) Hyperaldosteronism, hypervolemia, and increased blood pressure in mice expressing defective APC. Am J Physiol Regul Integr Comp Physiol 297:R571-575. https://doi.org/10.1152/ajpregu.00070.2009
Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, Taketo MM, Tissier F, Bertherat J, Lefrançois-Martinez A-M, Martinez A, Val P (2010) Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet 19:1561–1576. https://doi.org/10.1093/hmg/ddq029
Teo AED, Garg S, Shaikh LH, Zhou J, Karet Frankl FE, Gurnell M, Happerfield L, Marker A, Bienz M, Azizan EAB, Brown MJ (2015) Pregnancy, Primary Aldosteronism, and Adrenal CTNNB1 Mutations. N Engl J Med 373:1429–1436. https://doi.org/10.1056/NEJMoa1504869
Ise T, Shimoda A, Takakuwa H, Kato T, Izumiya Y, Shimizu K, Suzuki T, Sasano H, Yokoyama H, Kobayashi K (2001) A chimeric CYP11B1/CYP11B2 gene in glucocorticoid-insuppressible familial hyperaldosteronism. Clin Endocrinol (Oxf) 55:131–134. https://doi.org/10.1046/j.1365-2265.2001.01192.x
Scholl UI, Stölting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, Vichot AA, Jin SC, Loring E, Untiet V, Yoo T, Choi J, Xu S, Wu A, Kirchner M, Mertins P, Rump LC, Onder AM, Gamble C, McKenney D, Lash RW, Jones DP, Chune G, Gagliardi P, Choi M, Gordon R, Stowasser M, Fahlke C, Lifton RP (2018) CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 50:349–354. https://doi.org/10.1038/s41588-018-0048-5
Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, Couch R, Hammer LK, Harley FL, Farhi A, Wang W-H, Lifton RP (2012) Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A 109:2533–2538. https://doi.org/10.1073/pnas.1121407109
Mulatero P, Tauber P, Zennaro M-C, Monticone S, Lang K, Beuschlein F, Fischer E, Tizzani D, Pallauf A, Viola A, Amar L, Williams TA, Strom TM, Graf E, Bandulik S, Penton D, Plouin P-F, Warth R, Allolio B, Jeunemaitre X, Veglio F, Reincke M (2012) KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 59:235–240. https://doi.org/10.1161/HYPERTENSIONAHA.111.183996
Daniil G, Fernandes-Rosa FL, Chemin J, Blesneac I, Beltrand J, Polak M, Jeunemaitre X, Boulkroun S, Amar L, Strom TM, Lory P, Zennaro M-C (2016) CACNA1H Mutations Are Associated With Different Forms of Primary Aldosteronism. EBioMedicine 13:225–236. https://doi.org/10.1016/j.ebiom.2016.10.002
Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, Prasad ML, Goh G, Carling T, Juhlin CC, Quack I, Rump LC, Thiel A, Lande M, Frazier BG, Rasoulpour M, Bowlin DL, Sethna CB, Trachtman H, Fahlke C, Lifton RP (2015) Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 4:e06315. https://doi.org/10.7554/eLife.06315
Semenova NA, Ryzhkova OR, Strokova TV, Taran NN (2018) [The third case report a patient with primary aldosteronism, seizures, and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene]. Zh Nevrol Psikhiatr Im S S Korsakova 118:49–52. https://doi.org/10.17116/jnevro201811812149
Zilbermint M, Xekouki P, Faucz FR, Berthon A, Gkourogianni A, Schernthaner-Reiter MH, Batsis M, Sinaii N, Quezado MM, Merino M, Hodes A, Abraham SB, Libé R, Assié G, Espiard S, Drougat L, Ragazzon B, Davis A, Gebreab SY, Neff R, Kebebew E, Bertherat J, Lodish MB, Stratakis CA (2015) Primary Aldosteronism and ARMC5 Variants. J Clin Endocrinol Metab 100:E900-909. https://doi.org/10.1210/jc.2014-4167
Mulatero P, Schiavi F, Williams TA, Monticone S, Barbon G, Opocher G, Fallo F (2016) ARMC5 mutation analysis in patients with primary aldosteronism and bilateral adrenal lesions. J Hum Hypertens 30:374–378. https://doi.org/10.1038/jhh.2015.98
Rassi-Cruz M, Maria AG, Faucz FR, London E, Vilela LAP, Santana LS, Benedetti AFF, Goldbaum TS, Tanno FY, Srougi V, Chambo JL, Pereira MAA, Cavalcante ACBS, Carnevale FC, Pilan B, Bortolotto LA, F Drager L, Lerario AM, Latronico AC, Fragoso MCBV, Mendonca BB, Zerbini MCN, Stratakis CA, Almeida MQ (2021) Phosphodiesterase 2A and 3B variants are associated with primary aldosteronism. Endocr Relat Cancer 28:1–13. https://doi.org/10.1530/ERC-20-0384
Hattangady NG, Foster J, Lerario AM, Ponce-Balbuena D, Rege J, Monticone S, Rainey WE, Mulatero P, Else T (2020) Molecular and Electrophysiological Analyses of ATP2B4 Gene Variants in Bilateral Adrenal Hyperaldosteronism. Horm Cancer 11:52–62. https://doi.org/10.1007/s12672-019-00375-0
Tan GC, Negro G, Pinggera A, Tizen Laim NMS, Mohamed Rose I, Ceral J, Ryska A, Chin LK, Kamaruddin NA, Mohd Mokhtar N, A Jamal AR, Sukor N, Solar M, Striessnig J, Brown MJ, Azizan EA (2017) Aldosterone-Producing Adenomas: Histopathology-Genotype Correlation and Identification of a Novel CACNA1D Mutation. Hypertension 70:129–136. https://doi.org/10.1161/HYPERTENSIONAHA.117.09057
Azizan EAB, Lam BYH, Newhouse SJ, Zhou J, Kuc RE, Clarke J, Happerfield L, Marker A, Hoffman GJ, Brown MJ (2012) Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J Clin Endocrinol Metab 97:E819-829. https://doi.org/10.1210/jc.2011-2965
Åkerström T, Crona J, Delgado Verdugo A, Starker LF, Cupisti K, Willenberg HS, Knoefel WT, Saeger W, Feller A, Ip J, Soon P, Anlauf M, Alesina PF, Schmid KW, Decaussin M, Levillain P, Wängberg B, Peix J-L, Robinson B, Zedenius J, Bäckdahl M, Caramuta S, Iwen KA, Botling J, Stålberg P, Kraimps J-L, Dralle H, Hellman P, Sidhu S, Westin G, Lehnert H, Walz MK, Åkerström G, Carling T, Choi M, Lifton RP, Björklund P (2012) Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS One 7:e41926. https://doi.org/10.1371/journal.pone.0041926
Hong AR, Kim JH, Song YS, Lee KE, Seo SH, Seong M-W, Shin CS, Kim SW, Kim SY (2016) Genetics of Aldosterone-Producing Adenoma in Korean Patients. PLoS One 11:e0147590. https://doi.org/10.1371/journal.pone.0147590
Fernandes-Rosa FL, Giscos-Douriez I, Amar L, Gomez-Sanchez CE, Meatchi T, Boulkroun S, Zennaro M-C (2015) Different Somatic Mutations in Multinodular Adrenals With Aldosterone-Producing Adenoma. Hypertension 66:1014–1022. https://doi.org/10.1161/HYPERTENSIONAHA.115.05993
Mete O, Asa SL, Giordano TJ, Papotti M, Sasano H, Volante M (2018) Immunohistochemical Biomarkers of Adrenal Cortical Neoplasms. Endocr Pathol 29:137–149. https://doi.org/10.1007/s12022-018-9525-8
Monticone S, Castellano I, Versace K, Lucatello B, Veglio F, Gomez-Sanchez CE, Williams TA, Mulatero P (2015) Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol Cell Endocrinol 411:146–154. https://doi.org/10.1016/j.mce.2015.04.022
Thiel A, Reis A-C, Haase M, Goh G, Schott M, Willenberg HS, Scholl UI (2015) PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases. Eur J Endocrinol 172:677–685. https://doi.org/10.1530/EJE-14-1113
Yamada M, Nakajima Y, Taguchi R, Okamura T, Ishii S, Tomaru T, Ozawa A, Shibusawa N, Yoshino S, Toki A, Ishida E, Hashimoto K, Satoh T, Mori M (2012) KCNJ5 mutations in aldosterone- and cortisol-co-secreting adrenal adenomas. Endocr J 59:735–741. https://doi.org/10.1507/endocrj.ej12-0247
Tang L, Li X, Wang B, Ma X, Li H, Gao Y, Gu L, Nie W, Zhang X (2018) Clinical Characteristics of Aldosterone- and Cortisol-Coproducing Adrenal Adenoma in Primary Aldosteronism. Int J Endocrinol 2018:4920841. https://doi.org/10.1155/2018/4920841
Vaidya A, Nehs M, Kilbridge K (2019) Treatment of Adrenocortical Carcinoma. Surg Pathol Clin 12:997–1006. https://doi.org/10.1016/j.path.2019.08.010
Mete O, Gucer H, Kefeli M, Asa SL (2018) Diagnostic and Prognostic Biomarkers of Adrenal Cortical Carcinoma. Am J Surg Pathol 42:201–213. https://doi.org/10.1097/PAS.0000000000000943
Mete O, Asa SL (2012) Morphological distinction of cortisol-producing and aldosterone-producing adrenal cortical adenomas: not only possible but a critical clinical responsibility. Histopathology 60:1015–1016; author reply 1016–1017. https://doi.org/10.1111/j.1365-2559.2011.04141.x
Berthon A, Bertherat J (2020) Update of Genetic and Molecular Causes of Adrenocortical Hyperplasias Causing Cushing Syndrome. Horm Metab Res 52:598–606. https://doi.org/10.1055/a-1061-7349
Groussin L, Jullian E, Perlemoine K, Louvel A, Leheup B, Luton JP, Bertagna X, Bertherat J (2002) Mutations of the PRKAR1A gene in Cushing’s syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab 87:4324–4329. https://doi.org/10.1210/jc.2002-020592
Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer B, Zacharieva S, Pignatelli D, Carney JA, Luton JP, Bertagna X, Stratakis CA, Bertherat J (2002) Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet 71:1433–1442. https://doi.org/10.1086/344579
Groussin L, Horvath A, Jullian E, Boikos S, Rene-Corail F, Lefebvre H, Cephise-Velayoudom F-L, Vantyghem M-C, Chanson P, Conte-Devolx B, Lucas M, Gentil A, Malchoff CD, Tissier F, Carney JA, Bertagna X, Stratakis CA, Bertherat J (2006) A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab 91:1943–1949. https://doi.org/10.1210/jc.2005-2708
Kamilaris CDC, Hannah-Shmouni F, Stratakis CA (2020) Adrenocortical tumorigenesis: Lessons from genetics. Best Pract Res Clin Endocrinol Metab 34:101428. https://doi.org/10.1016/j.beem.2020.101428
Matyakhina L, Pack S, Kirschner LS, Pak E, Mannan P, Jaikumar J, Taymans SE, Sandrini F, Carney JA, Stratakis CA (2003) Chromosome 2 (2p16) abnormalities in Carney complex tumours. J Med Genet 40:268–277. https://doi.org/10.1136/jmg.40.4.268
Horvath A, Giatzakis C, Tsang K, Greene E, Osorio P, Boikos S, Libè R, Patronas Y, Robinson-White A, Remmers E, Bertherat J, Nesterova M, Stratakis CA (2008) A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. Eur J Hum Genet 16:1245–1253. https://doi.org/10.1038/ejhg.2008.85
Boikos SA, Horvath A, Heyerdahl S, Stein E, Robinson-White A, Bossis I, Bertherat J, Carney JA, Stratakis CA (2008) Phosphodiesterase 11A expression in the adrenal cortex, primary pigmented nodular adrenocortical disease, and other corticotropin-independent lesions. Horm Metab Res 40:347–353. https://doi.org/10.1055/s-2008-1076694
Carney JA, Lyssikatos C, Lodish MB, Stratakis CA (2015) Germline PRKACA amplification leads to Cushing syndrome caused by 3 adrenocortical pathologic phenotypes. Hum Pathol 46:40–49. https://doi.org/10.1016/j.humpath.2014.09.005
Espiard S, Drougat L, Settas N, Haydar S, Bathon K, London E, Levy I, Faucz FR, Calebiro D, Bertherat J, Li D, Levine MA, Stratakis CA (2020) PRKACB variants in skeletal disease or adrenocortical hyperplasia: effects on protein kinase A. Endocr Relat Cancer 27:647–656. https://doi.org/10.1530/ERC-20-0309
Tadjine M, Lampron A, Ouadi L, Horvath A, Stratakis CA, Bourdeau I (2008) Detection of somatic beta-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD). Clin Endocrinol (Oxf) 69:367–373. https://doi.org/10.1111/j.1365-2265.2008.03273.x
Iliopoulos D, Bimpaki EI, Nesterova M, Stratakis CA (2009) MicroRNA signature of primary pigmented nodular adrenocortical disease: clinical correlations and regulation of Wnt signaling. Cancer Res 69:3278–3282. https://doi.org/10.1158/0008-5472.CAN-09-0155
Kirschner MA, Powell RD, Lipsett MB (1964) CUSHING’S SYNDROME: NODULAR CORTICAL HYPERPLASIA OF ADRENAL GLANDS WITH CLINICAL AND PATHOLOGICAL FEATURES SUGGESTING ADRENOCORTICAL TUMOR. J Clin Endocrinol Metab 24:947–955. https://doi.org/10.1210/jcem-24-10-947
Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, Barreau O, Lefèvre L, Sibony M, Guignat L, Rodriguez S, Perlemoine K, René-Corail F, Letourneur F, Trabulsi B, Poussier A, Chabbert-Buffet N, Borson-Chazot F, Groussin L, Bertagna X, Stratakis CA, Ragazzon B, Bertherat J (2013) ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med 369:2105–2114. https://doi.org/10.1056/NEJMoa1304603
Espiard S, Drougat L, Libé R, Assié G, Perlemoine K, Guignat L, Barrande G, Brucker-Davis F, Doullay F, Lopez S, Sonnet E, Torremocha F, Pinsard D, Chabbert-Buffet N, Raffin-Sanson M-L, Groussin L, Borson-Chazot F, Coste J, Bertagna X, Stratakis CA, Beuschlein F, Ragazzon B, Bertherat J (2015) ARMC5 Mutations in a Large Cohort of Primary Macronodular Adrenal Hyperplasia: Clinical and Functional Consequences. J Clin Endocrinol Metab 100:E926-935. https://doi.org/10.1210/jc.2014-4204
Faucz FR, Zilbermint M, Lodish MB, Szarek E, Trivellin G, Sinaii N, Berthon A, Libé R, Assié G, Espiard S, Drougat L, Ragazzon B, Bertherat J, Stratakis CA (2014) Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab 99:E1113-1119. https://doi.org/10.1210/jc.2013-4280
Burgess JR, Harle RA, Tucker P, Parameswaran V, Davies P, Greenaway TM, Shepherd JJ (1996) Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg 131:699–702. https://doi.org/10.1001/archsurg.1996.01430190021006
Yoshida M, Hiroi M, Imai T, Kikumori T, Himeno T, Nakamura Y, Sasano H, Yamada M, Murakami Y, Nakamura S, Oiso Y (2011) A case of ACTH-independent macronodular adrenal hyperplasia associated with multiple endocrine neoplasia type 1. Endocr J 58:269–277. https://doi.org/10.1507/endocrj.k10e-218
Libé R, Fratticci A, Coste J, Tissier F, Horvath A, Ragazzon B, Rene-Corail F, Groussin L, Bertagna X, Raffin-Sanson ML, Stratakis CA, Bertherat J (2008) Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res 14:4016–4024. https://doi.org/10.1158/1078-0432.CCR-08-0106
Hsiao H-P, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, Robinson-White AJ, Nesterova M, Lacroix A, Stratakis CA (2009) Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab 94:2930–2937. https://doi.org/10.1210/jc.2009-0516
Gaujoux S, Pinson S, Gimenez-Roqueplo A-P, Amar L, Ragazzon B, Launay P, Meatchi T, Libé R, Bertagna X, Audebourg A, Zucman-Rossi J, Tissier F, Bertherat J (2010) Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin Cancer Res 16:5133–5141. https://doi.org/10.1158/1078-0432.CCR-10-1497
Hannah-Shmouni F, Faucz FR, Stratakis CA (2016) Alterations of Phosphodiesterases in Adrenocortical Tumors. Front Endocrinol (Lausanne) 7:111. https://doi.org/10.3389/fendo.2016.00111
Matyakhina L, Freedman RJ, Bourdeau I, Wei M-H, Stergiopoulos SG, Chidakel A, Walther M, Abu-Asab M, Tsokos M, Keil M, Toro J, Linehan WM, Stratakis CA (2005) Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab 90:3773–3779. https://doi.org/10.1210/jc.2004-2377
Hamajima T, Maruwaka K, Homma K, Matsuo K, Fujieda K, Hasegawa T (2010) Unilateral adrenalectomy can be an alternative therapy for infantile onset Cushing’ s syndrome caused by ACTH-independent macronodular adrenal hyperplasia with McCune-Albright syndrome. Endocr J 57:819–824. https://doi.org/10.1507/endocrj.k10e-003
Lacroix A, Baldacchino V, Bourdeau I, Hamet P, Tremblay J (2004) Cushing’s syndrome variants secondary to aberrant hormone receptors. Trends Endocrinol Metab 15:375–382. https://doi.org/10.1016/j.tem.2004.08.007
Lacroix A, Bourdeau I, Lampron A, Mazzuco TL, Tremblay J, Hamet P (2010) Aberrant G-protein coupled receptor expression in relation to adrenocortical overfunction. Clin Endocrinol (Oxf) 73:1–15. https://doi.org/10.1111/j.1365-2265.2009.03689.x
Lacroix A, N’Diaye N, Mircescu H, Hamet P, Tremblay J (1998) Abnormal expression and function of hormone receptors in adrenal Cushing’s syndrome. Endocr Res 24:835–843. https://doi.org/10.3109/07435809809032694
Mazzuco TL, Thomas M, Martinie M, Cherradi N, Sturm N, Feige J-J, Chabre O (2007) Cellular and molecular abnormalities of a macronodular adrenal hyperplasia causing beta-blocker-sensitive Cushing’s syndrome. Arq Bras Endocrinol Metabol 51:1452–1462. https://doi.org/10.1590/s0004-27302007000900007
Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, Libé R, Bram Z, Groussin L, Caron P, Tabarin A, Grunenberger F, Christin-Maitre S, Bertagna X, Kuhn J-M, Anouar Y, Bertherat J, Lefebvre H (2013) Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med 369:2115–2125. https://doi.org/10.1056/NEJMoa1215245
Lefebvre H, Duparc C, Chartrel N, Jegou S, Pellerin A, Laquerriere A, Ivell R, Vaudry H, Kuhn J-M (2003) Intraadrenal adrenocorticotropin production in a case of bilateral macronodular adrenal hyperplasia causing Cushing’s syndrome. J Clin Endocrinol Metab 88:3035–3042. https://doi.org/10.1210/jc.2002-030014
Bourdeau I, Matyakhina L, Stergiopoulos SG, Sandrini F, Boikos S, Stratakis CA (2006) 17q22-24 chromosomal losses and alterations of protein kinase a subunit expression and activity in adrenocorticotropin-independent macronodular adrenal hyperplasia. J Clin Endocrinol Metab 91:3626–3632. https://doi.org/10.1210/jc.2005-2608
Lacroix A, Bolté E, Tremblay J, Dupré J, Poitras P, Fournier H, Garon J, Garrel D, Bayard F, Taillefer R (1992) Gastric inhibitory polypeptide-dependent cortisol hypersecretion--a new cause of Cushing’s syndrome. N Engl J Med 327:974–980. https://doi.org/10.1056/NEJM199210013271402
Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, Schaak K, Schmittfull A, Schwarzmayr T, Barreau O, Vezzosi D, Rizk-Rabin M, Zabel U, Szarek E, Salpea P, Forlino A, Vetro A, Zuffardi O, Kisker C, Diener S, Meitinger T, Lohse MJ, Reincke M, Bertherat J, Strom TM, Allolio B (2014) Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med 370:1019–1028. https://doi.org/10.1056/NEJMoa1310359
Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kunstman JW, Kuntsman JW, Korah R, Suttorp A-C, Dietrich D, Haase M, Willenberg HS, Stålberg P, Hellman P, Akerström G, Björklund P, Carling T, Lifton RP (2014) Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet 46:613–617. https://doi.org/10.1038/ng.2956
Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, Quinkler M, Rayes N, Tabarin A, Laure Jullié M, Mantero F, Rubin B, Waldmann J, Bartsch DK, Pasquali R, Lohse M, Allolio B, Fassnacht M, Beuschlein F, Reincke M (2014) Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab 99:E2093-2100. https://doi.org/10.1210/jc.2014-2152
Nakajima Y, Okamura T, Gohko T, Satoh T, Hashimoto K, Shibusawa N, Ozawa A, Ishii S, Tomaru T, Horiguchi K, Okada S, Takata D, Rokutanda N, Horiguchi J, Tsushima Y, Oyama T, Takeyoshi I, Yamada M (2014) Somatic mutations of the catalytic subunit of cyclic AMP-dependent protein kinase (PRKACA) gene in Japanese patients with several adrenal adenomas secreting cortisol [Rapid Communication]. Endocr J 61:825–832. https://doi.org/10.1507/endocrj.ej14-0282
Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, Papageorgiou T, Bourdeau I, Kirschner LS, Vincent-Dejean C, Perlemoine K, Gicquel C, Bertagna X, Stratakis CA (2003) Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res 63:5308–5319
Espiard S, Knape MJ, Bathon K, Assié G, Rizk-Rabin M, Faillot S, Luscap-Rondof W, Abid D, Guignat L, Calebiro D, Herberg FW, Stratakis CA, Bertherat J (2018) Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight 3. https://doi.org/10.1172/jci.insight.98296
Ronchi CL, Di Dalmazi G, Faillot S, Sbiera S, Assié G, Weigand I, Calebiro D, Schwarzmayr T, Appenzeller S, Rubin B, Waldmann J, Scaroni C, Bartsch DK, Mantero F, Mannelli M, Kastelan D, Chiodini I, Bertherat J, Reincke M, Strom TM, Fassnacht M, Beuschlein F, European Network for the Study of Adrenocortical Tumors (ENSAT) (2016) Genetic Landscape of Sporadic Unilateral Adrenocortical Adenomas Without PRKACA p.Leu206Arg Mutation. J Clin Endocrinol Metab 101:3526–3538. https://doi.org/10.1210/jc.2016-1586
Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, Yoshida K, Nagata Y, Sato-Otsubo A, Yoshizato T, Suzuki H, Shiozawa Y, Kataoka K, Kon A, Aoki K, Chiba K, Tanaka H, Kume H, Miyano S, Fukayama M, Nureki O, Homma Y, Ogawa S (2014) Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 344:917–920. https://doi.org/10.1126/science.1252328
Gao X, Yamazaki Y, Tezuka Y, Pieroni J, Ishii K, Atsumi N, Ono Y, Omata K, Morimoto R, Nakamura Y, Satoh F, Sasano H (2020) Intratumoral heterogeneity of the tumor cells based on in situ cortisol excess in cortisol-producing adenomas; ∼An association among morphometry, genotype and cellular senescence∼. J Steroid Biochem Mol Biol 204:105764. https://doi.org/10.1016/j.jsbmb.2020.105764
Louiset E, Stratakis CA, Perraudin V, Griffin KJ, Libé R, Cabrol S, Fève B, Young J, Groussin L, Bertherat J, Lefebvre H (2009) The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. J Clin Endocrinol Metab 94:2406–2413. https://doi.org/10.1210/jc.2009-0031
Correa R, Salpea P, Stratakis CA (2015) Carney complex: an update. Eur J Endocrinol 173:M85-97. https://doi.org/10.1530/EJE-15-0209
Maria AG, Tatsi C, Berthon A, Drougat L, Settas N, Hannah-Shmouni F, Bertherat J, Faucz FR, Stratakis CA (2020) ARMC5 variants in PRKAR1A-mutated patients modify cortisol levels and Cushing’s syndrome. Endocr Relat Cancer 27:509–517. https://doi.org/10.1530/ERC-20-0273
Morin E, Mete O, Wasserman JD, Joshua AM, Asa SL, Ezzat S (2012) Carney complex with adrenal cortical carcinoma. J Clin Endocrinol Metab 97:E202-206. https://doi.org/10.1210/jc.2011-2321
Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M, Lyssikatos C, Horvath A, Carney JA, Stratakis CA (2012) A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab 97:351–359. https://doi.org/10.1210/jc.2011-2244
Correa R, Zilbermint M, Berthon A, Espiard S, Batsis M, Papadakis GZ, Xekouki P, Lodish MB, Bertherat J, Faucz FR, Stratakis CA (2015) The ARMC5 gene shows extensive genetic variance in primary macronodular adrenocortical hyperplasia. Eur J Endocrinol 173:435–440. https://doi.org/10.1530/EJE-15-0205
Stratakis CA, Berthon A (2019) Molecular mechanisms of ARMC5 mutations in adrenal pathophysiology. Curr Opin Endocr Metab Res 8:104–111. https://doi.org/10.1016/j.coemr.2019.07.010
Lloyd RV, Osamura RY, Klöppel G, Rosai J, International Agency for Research on Cancer (2017) WHO classification of tumours of endocrine organs, 4th edition. International Agency for Research on Cancer, Lyon
Libé R (2019) Clinical and molecular prognostic factors in adrenocortical carcinoma. Minerva Endocrinol 44:58–69. https://doi.org/10.23736/S0391-1977.18.02900-0
Mihai R (2015) Diagnosis, treatment and outcome of adrenocortical cancer. Br J Surg 102:291–306. https://doi.org/10.1002/bjs.9743
Ayala-Ramirez M, Jasim S, Feng L, Ejaz S, Deniz F, Busaidy N, Waguespack SG, Naing A, Sircar K, Wood CG, Pagliaro L, Jimenez C, Vassilopoulou-Sellin R, Habra MA (2013) Adrenocortical carcinoma: clinical outcomes and prognosis of 330 patients at a tertiary care center. Eur J Endocrinol 169:891–899. https://doi.org/10.1530/EJE-13-0519
Corso CR, Acco A, Bach C, Bonatto SJR, de Figueiredo BC, de Souza LM (2020) Pharmacological profile and effects of mitotane in adrenocortical carcinoma. Br J Clin Pharmacol. https://doi.org/10.1111/bcp.14721
Puglisi S, Calabrese A, Basile V, Pia A, Reimondo G, Perotti P, Terzolo M (2020) New perspectives for mitotane treatment of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab 34:101415. https://doi.org/10.1016/j.beem.2020.101415
Megerle F, Kroiss M, Hahner S, Fassnacht M (2019) Advanced Adrenocortical Carcinoma - What to do when First-Line Therapy Fails? Exp Clin Endocrinol Diabetes 127:109–116. https://doi.org/10.1055/a-0715-1946
Mills JK, Khalil M, Pasieka J, Kong S, Xu Y, Harvey A (2019) Oncocytic subtypes of adrenal cortical carcinoma: Aggressive in appearance yet more indolent in behavior? Surgery 166:524–533. https://doi.org/10.1016/j.surg.2019.05.049
Erickson LA (2018) Challenges in surgical pathology of adrenocortical tumours. Histopathology 72:82–96. https://doi.org/10.1111/his.13255
Sung TY, Choi YM, Kim WG, Lee YM, Kim TY, Shong YK, Kim WB, Song DE (2017) Myxoid and Sarcomatoid Variants of Adrenocortical Carcinoma: Analysis of Rare Variants in Single Tertiary Care Center. J Korean Med Sci 32:764–771. https://doi.org/10.3346/jkms.2017.32.5.764
Weissferdt A, Phan A, Suster S, Moran CA (2013) Myxoid adrenocortical carcinoma: a clinicopathologic and immunohistochemical study of 7 cases, including 1 case with lipomatous metaplasia. Am J Clin Pathol 139:780–786. https://doi.org/10.1309/AJCPCDZLC13RSXRZ
Papathomas TG, Duregon E, Korpershoek E, Restuccia DF, van Marion R, Cappellesso R, Sturm N, Rossi G, Coli A, Zucchini N, Stoop H, Oosterhuis W, Ventura L, Volante M, Fassina A, Dinjens WNM, Papotti M, de Krijger RR (2016) Sarcomatoid adrenocortical carcinoma: a comprehensive pathological, immunohistochemical, and targeted next-generation sequencing analysis. Hum Pathol 58:113–122. https://doi.org/10.1016/j.humpath.2016.08.006
Giordano TJ, Berney D, de Krijger RR, Erickson L, Fassnacht M, Mete O, Papathomas T, Papotti M, Sasano H, Thompson LDR, Volante M, Gill AJ (2020) Data set for reporting of carcinoma of the adrenal cortex: explanations and recommendations of the guidelines from the International Collaboration on Cancer Reporting. Hum Pathol. https://doi.org/10.1016/j.humpath.2020.10.001
Wick MR, Cherwitz DL, McGlennen RC, Dehner LP (1986) Adrenocortical carcinoma. An immunohistochemical comparison with renal cell carcinoma. Am J Pathol 122:343–352
Saeger W, Fassnacht M, Chita R, Prager G, Nies C, Lorenz K, Bärlehner E, Simon D, Niederle B, Beuschlein F, Allolio B, Reincke M (2003) High diagnostic accuracy of adrenal core biopsy: results of the German and Austrian adrenal network multicenter trial in 220 consecutive patients. Hum Pathol 34:180–186. https://doi.org/10.1053/hupa.2003.24
Chetty R, Pillay P, Jaichand V (1998) Cytokeratin expression in adrenal phaeochromocytomas and extra-adrenal paragangliomas. J Clin Pathol 51:477–478. https://doi.org/10.1136/jcp.51.6.477
Papotti M, Duregon E, Volante M, McNicol AM (2014) Pathology of the adrenal cortex: a reappraisal of the past 25 years focusing on adrenal cortical tumors. Endocr Pathol 25:35–48. https://doi.org/10.1007/s12022-013-9291-6
Weiss LM, Medeiros LJ, Vickery AL (1989) Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol 13:202–206. https://doi.org/10.1097/00000478-198903000-00004
Bisceglia M, Ludovico O, Di Mattia A, Ben-Dor D, Sandbank J, Pasquinelli G, Lau SK, Weiss LM (2004) Adrenocortical oncocytic tumors: report of 10 cases and review of the literature. Int J Surg Pathol 12:231–243. https://doi.org/10.1177/106689690401200304
Wieneke JA, Thompson LDR, Heffess CS (2003) Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol 27:867–881. https://doi.org/10.1097/00000478-200307000-00001
Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C, Wemeau JL, Lecomte-Houcke M, Leteurtre E (2002) Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol 26:1612–1619. https://doi.org/10.1097/00000478-200212000-00009
Wang C, Sun Y, Wu H, Zhao D, Chen J (2014) Distinguishing adrenal cortical carcinomas and adenomas: a study of clinicopathological features and biomarkers. Histopathology 64:567–576. https://doi.org/10.1111/his.12283
Das S, Sengupta M, Islam N, Roy P, Datta C, Mishra PK, Banerjee S, Chaudhuri MK, Chatterjee U (2016) Weineke criteria, Ki-67 index and p53 status to study pediatric adrenocortical tumors: Is there a correlation? J Pediatr Surg 51:1795–1800. https://doi.org/10.1016/j.jpedsurg.2016.07.014
Guntiboina VA, Sengupta M, Islam N, Barman S, Biswas SK, Chatterjee U, Mishra PK, Roy P, Mallick MG, Datta C (2019) Diagnostic and prognostic utility of SF1, IGF2 and p57 immunoexpression in pediatric adrenal cortical tumors. J Pediatr Surg 54:1906–1912. https://doi.org/10.1016/j.jpedsurg.2018.12.002
Magro G, Esposito G, Cecchetto G, Dall’Igna P, Marcato R, Gambini C, Boldrini R, Collini P, D’Onofrio V, Salfi N, d’Amore E, Ferrari A, Bisogno G, Alaggio R (2012) Pediatric adrenocortical tumors: morphological diagnostic criteria and immunohistochemical expression of matrix metalloproteinase type 2 and human leucocyte-associated antigen (HLA) class II antigens. Results from the Italian Pediatric Rare Tumor (TREP) Study project. Hum Pathol 43:31–39. https://doi.org/10.1016/j.humpath.2011.04.016
Duregon E, Fassina A, Volante M, Nesi G, Santi R, Gatti G, Cappellesso R, Dalino Ciaramella P, Ventura L, Gambacorta M, Dei Tos AP, Loli P, Mannelli M, Mantero F, Berruti A, Terzolo M, Papotti M (2013) The reticulin algorithm for adrenocortical tumor diagnosis: a multicentric validation study on 245 unpublished cases. Am J Surg Pathol 37:1433–1440. https://doi.org/10.1097/PAS.0b013e31828d387b
Duregon E, Cappellesso R, Maffeis V, Zaggia B, Ventura L, Berruti A, Terzolo M, Fassina A, Volante M, Papotti M (2017) Validation of the prognostic role of the “Helsinki Score” in 225 cases of adrenocortical carcinoma. Hum Pathol 62:1–7. https://doi.org/10.1016/j.humpath.2016.09.035
Pennanen M, Heiskanen I, Sane T, Remes S, Mustonen H, Haglund C, Arola J (2015) Helsinki score-a novel model for prediction of metastases in adrenocortical carcinomas. Hum Pathol 46:404–410. https://doi.org/10.1016/j.humpath.2014.11.015
Renaudin K, Smati S, Wargny M, Al Ghuzlan A, Aubert S, Leteurtre E, Patey M, Sibony M, Sturm N, Tissier F, Amar L, Bertherat J, Berthozat C, Chabre O, Do Cao C, Haissaguerre M, Pierre P, Briet C, Vezzosi D, Lifante JC, Pattou F, Mirallie E, Baudin E, Cariou B, Libe R, Drui D, for Comete-Cancer Network (2018) Clinicopathological description of 43 oncocytic adrenocortical tumors: importance of Ki-67 in histoprognostic evaluation. Mod Pathol 31:1708–1716. https://doi.org/10.1038/s41379-018-0077-8
McNicol AM, Nolan CE, Struthers AJ, Farquharson MA, Hermans J, Haak HR (1997) Expression of p53 in adrenocortical tumours: clinicopathological correlations. J Pathol 181:146–152. https://doi.org/10.1002/(SICI)1096-9896(199702)181:2146::AID-PATH7443.0.CO;2-7
Reincke M, Karl M, Travis WH, Mastorakos G, Allolio B, Linehan HM, Chrousos GP (1994) p53 mutations in human adrenocortical neoplasms: immunohistochemical and molecular studies. J Clin Endocrinol Metab 78:790–794. https://doi.org/10.1210/jcem.78.3.8126158
Stojadinovic A, Brennan MF, Hoos A, Omeroglu A, Leung DHY, Dudas ME, Nissan A, Cordon-Cardo C, Ghossein RA (2003) Adrenocortical adenoma and carcinoma: histopathological and molecular comparative analysis. Mod Pathol 16:742–751. https://doi.org/10.1097/01.MP.0000081730.72305.81
Babinska A, Sworczak K, Wisniewski P, Nałecz A, Jaskiewicz K (2008) The role of immunohistochemistry in histopathological diagnostics of clinically “silent” incidentally detected adrenal masses. Exp Clin Endocrinol Diabetes 116:246–251. https://doi.org/10.1055/s-2007-993164
Giordano TJ, Kuick R, Else T, Gauger PG, Vinco M, Bauersfeld J, Sanders D, Thomas DG, Doherty G, Hammer G (2009) Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res 15:668–676. https://doi.org/10.1158/1078-0432.CCR-08-1067
Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N, Rakugi H, Ikeda Y, Tanabe A, Nigawara T, Ito S, Kimura I, Naruse M, Phaeochromocytoma Study Group in Japan (2014) Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer 21:405–414. https://doi.org/10.1530/ERC-13-0494
Papathomas TG, Pucci E, Giordano TJ, Lu H, Duregon E, Volante M, Papotti M, Lloyd RV, Tischler AS, van Nederveen FH, Nose V, Erickson L, Mete O, Asa SL, Turchini J, Gill AJ, Matias-Guiu X, Skordilis K, Stephenson TJ, Tissier F, Feelders RA, Smid M, Nigg A, Korpershoek E, van der Spek PJ, Dinjens WNM, Stubbs AP, de Krijger RR (2016) An International Ki67 Reproducibility Study in Adrenal Cortical Carcinoma. Am J Surg Pathol 40:569–576. https://doi.org/10.1097/PAS.0000000000000574
Mete O, Davis J, Rudzinski E, Asa S, Baker T, Berman M, Erickson L, Giordano T, Thompson L, Tischler A, Weiss L (2020) College of American Pathologists (CAP) Protocol for the Examination of Specimens From Patients With Carcinoma of the Adrenal Gland. Version: 4.1.0.0.
Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233–1238. https://doi.org/10.1126/science.1978757
Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH (1990) Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 348:747–749. https://doi.org/10.1038/348747a0
Ohgaki H, Kleihues P, Heitz PU (1993) p53 mutations in sporadic adrenocortical tumors. Int J Cancer 54:408–410. https://doi.org/10.1002/ijc.2910540310
Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, René-Corail F, Elarouci N, Sbiera S, Kroiss M, Allolio B, Waldmann J, Quinkler M, Mannelli M, Mantero F, Papathomas T, De Krijger R, Tabarin A, Kerlan V, Baudin E, Tissier F, Dousset B, Groussin L, Amar L, Clauser E, Bertagna X, Ragazzon B, Beuschlein F, Libé R, de Reyniès A, Bertherat J (2014) Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46:607–612. https://doi.org/10.1038/ng.2953
Medina-Arana V, Delgado L, González L, Bravo A, Díaz H, Salido E, Riverol D, González-Aguilera JJ, Fernández-Peralta AM (2011) Adrenocortical carcinoma, an unusual extracolonic tumor associated with Lynch II syndrome. Fam Cancer 10:265–271. https://doi.org/10.1007/s10689-010-9416-8
Berends MJ, Cats A, Hollema H, Karrenbeld A, Beentjes JA, Sijmons RH, Mensink RG, Hofstra RM, Verschueren RC, Kleibeuker JH (2000) Adrenocortical adenocarcinoma in an MSH2 carrier: coincidence or causal relation? Hum Pathol 31:1522–1527. https://doi.org/10.1053/hupa.2000.20409
Smith TG, Clark SK, Katz DE, Reznek RH, Phillips RK (2000) Adrenal masses are associated with familial adenomatous polyposis. Dis Colon Rectum 43:1739–1742. https://doi.org/10.1007/BF02236860
Skogseid B, Larsson C, Lindgren PG, Kvanta E, Rastad J, Theodorsson E, Wide L, Wilander E, Oberg K (1992) Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 75:76–81. https://doi.org/10.1210/jcem.75.1.1352309
Haase M, Anlauf M, Schott M, Schinner S, Kaminsky E, Scherbaum WA, Willenberg HS (2011) A new mutation in the menin gene causes the multiple endocrine neoplasia type 1 syndrome with adrenocortical carcinoma. Endocrine 39:153–159. https://doi.org/10.1007/s12020-010-9424-3
Griniatsos JE, Dimitriou N, Zilos A, Sakellariou S, Evangelou K, Kamakari S, Korkolopoulou P, Kaltsas G (2011) Bilateral adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1 (MEN1) and a novel mutation in the MEN1 gene. World J Surg Oncol 9:6. https://doi.org/10.1186/1477-7819-9-6
Wagner AS, Fleitz JM, Kleinschmidt-Demasters BK (2005) Pediatric adrenal cortical carcinoma: brain metastases and relationship to NF-1, case reports and review of the literature. J Neurooncol 75:127–133. https://doi.org/10.1007/s11060-005-0376-z
Menon RK, Ferrau F, Kurzawinski TR, Rumsby G, Freeman A, Amin Z, Korbonits M, Chung T-TLL (2014) Adrenal cancer in neurofibromatosis type 1: case report and DNA analysis. Endocrinol Diabetes Metab Case Rep 2014:140074. https://doi.org/10.1530/EDM-14-0074
Kieler M, Müllauer L, Koperek O, Bianconi D, Unseld M, Raderer M, Prager GW (2018) Analysis of 10 Adrenocortical Carcinoma Patients in the Cohort of the Precision Medicine Platform MONDTI. Oncology 94:306–310. https://doi.org/10.1159/000486678
Henry I, Jeanpierre M, Couillin P, Barichard F, Serre JL, Journel H, Lamouroux A, Turleau C, de Grouchy J, Junien C (1989) Molecular definition of the 11p15.5 region involved in Beckwith-Wiedemann syndrome and probably in predisposition to adrenocortical carcinoma. Hum Genet 81:273–277. https://doi.org/10.1007/BF00279003
Gicquel C, Bertagna X, Schneid H, Francillard-Leblond M, Luton JP, Girard F, Le Bouc Y (1994) Rearrangements at the 11p15 locus and overexpression of insulin-like growth factor-II gene in sporadic adrenocortical tumors. J Clin Endocrinol Metab 78:1444–1453. https://doi.org/10.1210/jcem.78.6.7911125
Nielsen HM, How-Kit A, Guerin C, Castinetti F, Vollan HKM, De Micco C, Daunay A, Taieb D, Van Loo P, Besse C, Kristensen VN, Hansen LL, Barlier A, Sebag F, Tost J (2015) Copy number variations alter methylation and parallel IGF2 overexpression in adrenal tumors. Endocr Relat Cancer 22:953–967. https://doi.org/10.1530/ERC-15-0086
Sasaki H, Ishihara K, Kato R (2000) Mechanisms of Igf2/H19 imprinting: DNA methylation, chromatin and long-distance gene regulation. J Biochem 127:711–715. https://doi.org/10.1093/oxfordjournals.jbchem.a022661
Yoshimizu T, Miroglio A, Ripoche M-A, Gabory A, Vernucci M, Riccio A, Colnot S, Godard C, Terris B, Jammes H, Dandolo L (2008) The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci U S A 105:12417–12422. https://doi.org/10.1073/pnas.0801540105
Ohlsson R, Nyström A, Pfeifer-Ohlsson S, Töhönen V, Hedborg F, Schofield P, Flam F, Ekström TJ (1993) IGF2 is parentally imprinted during human embryogenesis and in the Beckwith-Wiedemann syndrome. Nat Genet 4:94–97. https://doi.org/10.1038/ng0593-94
Henry I, Bonaiti-Pellié C, Chehensse V, Beldjord C, Schwartz C, Utermann G, Junien C (1991) Uniparental paternal disomy in a genetic cancer-predisposing syndrome. Nature 351:665–667. https://doi.org/10.1038/351665a0
Dohna M, Reincke M, Mincheva A, Allolio B, Solinas-Toldo S, Lichter P (2000) Adrenocortical carcinoma is characterized by a high frequency of chromosomal gains and high-level amplifications. Genes Chromosomes Cancer 28:145–152
Doghman M, Karpova T, Rodrigues GA, Arhatte M, De Moura J, Cavalli LR, Virolle V, Barbry P, Zambetti GP, Figueiredo BC, Heckert LL, Lalli E (2007) Increased steroidogenic factor-1 dosage triggers adrenocortical cell proliferation and cancer. Mol Endocrinol 21:2968–2987. https://doi.org/10.1210/me.2007-0120
Liu T, Brown TC, Juhlin CC, Andreasson A, Wang N, Bäckdahl M, Healy JM, Prasad ML, Korah R, Carling T, Xu D, Larsson C (2014) The activating TERT promoter mutation C228T is recurrent in subsets of adrenal tumors. Endocr Relat Cancer 21:427–434. https://doi.org/10.1530/ERC-14-0016
Ragazzon B, Libé R, Assié G, Tissier F, Barreau O, Houdayer C, Perlemoine K, Audebourg A, Clauser E, René-Corail F, Bertagna X, Dousset B, Bertherat J, Groussin L (2014) Mass-array screening of frequent mutations in cancers reveals RB1 alterations in aggressive adrenocortical carcinomas. Eur J Endocrinol 170:385–391. https://doi.org/10.1530/EJE-13-0778
Heppner C, Reincke M, Agarwal SK, Mora P, Allolio B, Burns AL, Spiegel AM, Marx SJ (1999) MEN1 gene analysis in sporadic adrenocortical neoplasms. J Clin Endocrinol Metab 84:216–219. https://doi.org/10.1210/jcem.84.1.5388
Fogt F, Vargas MP, Zhuang Z, Merino MJ (1998) Utilization of molecular genetics in the differentiation between adrenal cortical adenomas and carcinomas. Hum Pathol 29:518–521. https://doi.org/10.1016/s0046-8177(98)90069-7
Giordano TJ, Thomas DG, Kuick R, Lizyness M, Misek DE, Smith AL, Sanders D, Aljundi RT, Gauger PG, Thompson NW, Taylor JMG, Hanash SM (2003) Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol 162:521–531. https://doi.org/10.1016/S0002-9440(10)63846-1
de Fraipont F, El Atifi M, Cherradi N, Le Moigne G, Defaye G, Houlgatte R, Bertherat J, Bertagna X, Plouin P-F, Baudin E, Berger F, Gicquel C, Chabre O, Feige J-J (2005) Gene expression profiling of human adrenocortical tumors using complementary deoxyribonucleic Acid microarrays identifies several candidate genes as markers of malignancy. J Clin Endocrinol Metab 90:1819–1829. https://doi.org/10.1210/jc.2004-1075
Fernandez-Ranvier GG, Weng J, Yeh R-F, Khanafshar E, Suh I, Barker C, Duh QY, Clark OH, Kebebew E (2008) Identification of biomarkers of adrenocortical carcinoma using genomewide gene expression profiling. Arch Surg 143:841–846; discussion 846. https://doi.org/10.1001/archsurg.143.9.841
de Reyniès A, Assié G, Rickman DS, Tissier F, Groussin L, René-Corail F, Dousset B, Bertagna X, Clauser E, Bertherat J (2009) Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol 27:1108–1115. https://doi.org/10.1200/JCO.2008.18.5678
Laurell C, Velázquez-Fernández D, Lindsten K, Juhlin C, Enberg U, Geli J, Höög A, Kjellman M, Lundeberg J, Hamberger B, Larsson C, Nilsson P, Bäckdahl M (2009) Transcriptional profiling enables molecular classification of adrenocortical tumours. Eur J Endocrinol 161:141–152. https://doi.org/10.1530/EJE-09-0068
Ragazzon B, Libé R, Gaujoux S, Assié G, Fratticci A, Launay P, Clauser E, Bertagna X, Tissier F, de Reyniès A, Bertherat J (2010) Transcriptome analysis reveals that p53 and {beta}-catenin alterations occur in a group of aggressive adrenocortical cancers. Cancer Res 70:8276–8281. https://doi.org/10.1158/0008-5472.CAN-10-2014
Fonseca AL, Kugelberg J, Starker LF, Scholl U, Choi M, Hellman P, Åkerström G, Westin G, Lifton RP, Björklund P, Carling T (2012) Comprehensive DNA methylation analysis of benign and malignant adrenocortical tumors. Genes Chromosomes Cancer 51:949–960. https://doi.org/10.1002/gcc.21978
Rechache NS, Wang Y, Stevenson HS, Killian JK, Edelman DC, Merino M, Zhang L, Nilubol N, Stratakis CA, Meltzer PS, Kebebew E (2012) DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J Clin Endocrinol Metab 97:E1004-1013. https://doi.org/10.1210/jc.2011-3298
Barreau O, Assié G, Wilmot-Roussel H, Ragazzon B, Baudry C, Perlemoine K, René-Corail F, Bertagna X, Dousset B, Hamzaoui N, Tissier F, de Reynies A, Bertherat J (2013) Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J Clin Endocrinol Metab 98:E174-184. https://doi.org/10.1210/jc.2012-2993
Chehade M, Bullock M, Glover A, Hutvagner G, Sidhu S (2020) Key MicroRNA’s and Their Targetome in Adrenocortical Cancer. Cancers (Basel) 12. https://doi.org/10.3390/cancers12082198
Soon PSH, Tacon LJ, Gill AJ, Bambach CP, Sywak MS, Campbell PR, Yeh MW, Wong SG, Clifton-Bligh RJ, Robinson BG, Sidhu SB (2009) miR-195 and miR-483-5p Identified as Predictors of Poor Prognosis in Adrenocortical Cancer. Clin Cancer Res 15:7684–7692. https://doi.org/10.1158/1078-0432.CCR-09-1587
Hao H-X, Jiang X, Cong F (2016) Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers (Basel) 8. https://doi.org/10.3390/cancers8060054
Gaujoux S, Grabar S, Fassnacht M, Ragazzon B, Launay P, Libé R, Chokri I, Audebourg A, Royer B, Sbiera S, Vacher-Lavenu M-C, Dousset B, Bertagna X, Allolio B, Bertherat J, Tissier F (2011) β-catenin activation is associated with specific clinical and pathologic characteristics and a poor outcome in adrenocortical carcinoma. Clin Cancer Res 17:328–336. https://doi.org/10.1158/1078-0432.CCR-10-2006
Mohan DR, Lerario AM, Else T, Mukherjee B, Almeida MQ, Vinco M, Rege J, Mariani BMP, Zerbini MCN, Mendonca BB, Latronico AC, Marie SKN, Rainey WE, Giordano TJ, Fragoso MCBV, Hammer GD (2019) Targeted Assessment of G0S2 Methylation Identifies a Rapidly Recurrent, Routinely Fatal Molecular Subtype of Adrenocortical Carcinoma. Clin Cancer Res 25:3276–3288. https://doi.org/10.1158/1078-0432.CCR-18-2693
Papathomas TG, Oudijk L, Zwarthoff EC, Post E, Duijkers FA, van Noesel MM, Hofland LJ, Pollard PJ, Maher ER, Restuccia DF, Feelders RA, Franssen GJH, Timmers HJ, Sleijfer S, de Herder WW, de Krijger RR, Dinjens WNM, Korpershoek E (2014) Telomerase reverse transcriptase promoter mutations in tumors originating from the adrenal gland and extra-adrenal paraganglia. Endocr Relat Cancer 21:653–661. https://doi.org/10.1530/ERC-13-0429
Svahn F, Paulsson JO, Stenman A, Fotouhi O, Mu N, Murtha TD, Korah R, Carling T, Bäckdahl M, Wang N, Juhlin CC, Larsson C (2018) TERT promoter hypermethylation is associated with poor prognosis in adrenocortical carcinoma. Int J Mol Med 42:1675–1683. https://doi.org/10.3892/ijmm.2018.3735
Jouinot A, Lippert J, Fassnacht M, de La Villeon B, Septier A, Neou M, Perlemoine K, Appenzeller S, Sibony M, Gaujoux S, Dousset B, Libe R, Groussin L, Ronchi CL, Assié G, Bertherat J (2020) Intratumor heterogeneity of prognostic DNA-based molecular markers in adrenocortical carcinoma. Endocr Connect 9:705–714. https://doi.org/10.1530/EC-20-0228
Else T, Rodriguez-Galindo C (2016) 5th International ACC Symposium: Hereditary Predisposition to Childhood ACC and the Associated Molecular Phenotype: 5th International ACC Symposium Session: Not Just for Kids! Horm Cancer 7:36–39. https://doi.org/10.1007/s12672-015-0244-z
Domènech M, Grau E, Solanes A, Izquierdo A, Del Valle J, Carrato C, Pineda M, Dueñas N, Pujol M, Lázaro C, Capellà G, Brunet J, Navarro M (2020) Characteristics of adrenocortical carcinoma associated with Lynch Syndrome. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgaa833
Else T, Lerario AM, Everett J, Haymon L, Wham D, Mullane M, Wilson TL, Rainville I, Rana H, Worth AJ, Snyder NW, Blair IA, McKay R, Kilbridge K, Hammer G, Barletta J, Vaidya A (2017) Adrenocortical carcinoma and succinate dehydrogenase gene mutations: an observational case series. Eur J Endocrinol 177:439–444. https://doi.org/10.1530/EJE-17-0358
Author information
Authors and Affiliations
Contributions
Concept and Design: O.M and C.C.J; Literature Search, Writing, and Illustrations: C.C.J and O.M; Critical Reviews and Approval of the Final version of the manuscript: O.M, C.C.J, J.B, T.J., G.H and H.S.
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Juhlin, C.C., Bertherat, J., Giordano, T.J. et al. What Did We Learn from the Molecular Biology of Adrenal Cortical Neoplasia? From Histopathology to Translational Genomics. Endocr Pathol 32, 102–133 (2021). https://doi.org/10.1007/s12022-021-09667-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12022-021-09667-0