
Abstract
Purpose
No genomic data have been put forth that prove beyond a shadow of doubt that sporadic medullary thyroid cancer (MTC) occurs in infancy, childhood, and/or adolescence.
Methods
This was a retrospective comparative study of consecutive patients with MTC who had neck surgery at a tertiary center over a 30-year period.
Results
Included were 1252 patients with MTC (337 hereditary and 915 sporadic), of whom 107 (8.5%) were operated before the age of 18 yrs. Only 4 (3.7%) of the 107 pediatric patients, aged 14, 16, 17 and 17 years, had sporadic MTC. These 4 patients, 3 of whom had been referred for completion surgery, revealed much larger thyroid tumors (medians of 20 mm vs. 1.5–5 mm) than the 103 pediatric patients with hereditary MTC. As for extrathyroid extension and nodal metastases, the 4 patients with sporadic MTC were more comparable to the 37 carriers of highest-risk mutations, 31 (84%) of whom were index patients with de novo disease, than to the 66 carriers of high-risk, intermediate-risk, or low-risk RET mutations (25–38% vs. 0–8%, and medians of 9–9.5 vs. 0 node metastases after dissection of more (medians of 72–91.5 vs. 4.5–9) nodes).
Conclusion
Sporadic MTC, arising rarely, if ever, below the age of 14 years, is exceptional in infancy and childhood, and infrequent in adolescence. At diagnosis, it is almost as widely metastatic as hereditary MTC of the highest-risk category which almost always, like sporadic MTC, presents as de novo disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Medullary thyroid cancer (MTC), a calcitonin-secreting neuroendocrine malignancy characterized by early nodal spread, comes in a hereditary (~25%) and a sporadic (~75%) form. The mean age-standardized annual incidence and prevalence are estimated at 0.06 and 1.3 per 100 000 inhabitants for hereditary MTC, and at 0.13 and 2.5 per 100 000 inhabitants for sporadic MTC [1]. Both forms of MTC, exhibiting similar tumor biology [2], differ primarily in tumor onset, but not in disease-specific survival and clinical outcomes when comparative tumor stages are compared with each other [3].
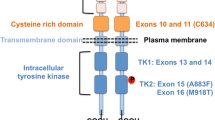
The advent of the molecular genetic era in the 1990s [4] has enabled unambiguous distinction between sporadic and hereditary MTC, requiring no more than a venous blood draw for a gene test. Patients with hereditary MTC feature predominantly heterozygous REarranged during Transfection (RET) germline mutation that cluster in a limited number of ‘hot spots’ on chromosome 10 (10q11.2). These RET germline mutations, which are distributed fairly evenly around the globe barring a few founder mutations [5], result in activation of the transmembrane RET tyrosine kinase receptor in the absence of substrate [6]. This constitutive RET receptor activation is believed to drive neoplastic C cell hyperplasia and eventually malignant progression to early hereditary MTC in RET carriers, all of whom are at an increased risk of multiple endocrine neoplasia type 2 (MEN 2). Because the age-dependent intensity of receptor activation can vary tremendously, pathogenic RET mutations are grouped into 4 RET risk categories that give rise to MEN 2B invariably, or to MEN 2A frequently, occasionally, or exceptionally [7].
Sporadic MTC, defined by the absence of pathogenic RET germline mutations, is thought to come into being upon accrual of RET, RAS or, less commonly, other somatic mutations by parafollicular C cells [8, 9]. This process, which obviously needs more time to take place, explains why most sporadic MTC manifest some 20–30 years later than hereditary MTC [2].
No genomic data have been put forth that prove beyond a shadow of doubt that sporadic MTC occurs in infancy (≤1–2 years of age), childhood (up to age 12–13 years) and/or adolescence (up to age 17 years). The present study was undertaken to address this important topic.
Patients and methods
Included in this retrospective comparative study were all patients with MTC who had been referred for prophylactic or therapeutic initial thyroidectomy or reoperative neck surgery between November 1994 and April 2024 to the Department of Visceral, Vascular, and Endocrine Surgery, a surgical referral center for thyroid cancer. All surgical procedures, as well as preoperative or postoperative genetic counseling and screening for RET germline mutations under informed consent, complied with national and international clinical standards relevant at the time of surgery, including the Declaration of Helsinki, and other applicable regulations (University of Halle-Wittenberg ethics committee approval reference 2020–237).
Surgical specimens had been analyzed at histopathological examination, separating thyroid vs. extrathyroid tissue and nodes. Paraffin embedded thyroid specimens had been stained with hematoxylin and eosin and calcitonin, as appropriate.
MTC had been diagnosed according to the histopathologic criteria set forth by the World Health Organization [10]. Thyroid tumor size and tumor extension had been measured directly on the surgical specimens at the time of histopathological examination.
The numbers of involved and dissected nodes were calculated by summing up all involved and all dissected nodes cleared at the authors’ institution and elsewhere.
The diagnosis of distant metastases required unequivocal evidence on computed tomography, magnetic resonance imaging, and/or positron emission tomography.
Biochemical cure was assumed when previously raised calcitonin serum levels declined postoperatively below the upper normal limit of the calcitonin assay or became undetectable.
Patients with MTC had undergone RET screening after genetic counseling under separate informed consent. For screening of relevant RET exons, DNA had been purified from peripheral blood leukocytes according to standard procedures.
For descriptive statistical analysis, the software package SPSS® version 28 (IBM, Armonk, New York, USA) was used.
Categorical variables were represented as absolute and relative frequencies. Continuous data were expressed as medians with interquartile ranges. Missing data were not replaced, and the number of individuals was specified for each analysis.
Results
A total of 1252 patients with MTC (337 hereditary and 915 sporadic) were identified, of whom 107 (3.7%) had been operated below the age of 18 years.
Table 1 details the pediatric population of 107 patients who had undergone thyroidectomy before reaching adulthood.
Altogether, 103 of 107 infants, children or adolescents had hereditary MTC, whereas only 4 adolescents, aged 14, 16, 17 and 17 years, had sporadic MTC. All 4 patients with sporadic MTC had negative RET tests in which the most important RET exons 8 (except for 1 patient), 10, 11, 13, 14, 15, and 16 had been scanned for pathogenic mutations.
Congruous with the excellent sensitivity and specificity of molecular genetic screening, none of these 4 RET-negative pediatric patients had personal or family histories of MEN2 or biochemical evidence of pheochromocytoma or primary hyperparathyroidism.
One (25%) of the 4 pediatric patients with sporadic MTC was found to have a 25 mm extrathyroid thyroid tumor ipsilaterally together with a 2 mm small thyroid tumor deposit contralaterally in conjunction with 40 node metastases, possibly reflecting intense lymphatic dissemination within the thyroid gland.
Development of MTC before the age of 18 years differed greatly by RET risk category, varying from 79% (37 of 47 patients) for highest-risk mutations to 41.7% (48 of 115 patients) for high-risk mutations, 19% (13 of 69 patients) for intermediate-risk mutations, and 4.7% (5 of 106 patients) for low-risk mutations. This percentage was much lower for pediatric sporadic MTC, which had emerged in no more than 0.4% (4 patients) of the 915 patients with sporadic MTC and 0.3% (4 patients) of the 1252 patients with MTC, respectively.
Absent pathognomonic stigmata and/or genetic screening, the 4 adolescents with sporadic MTC, 3 of whom had been referred for completion surgery, revealed much larger thyroid tumors (medians of 20 mm vs. 1.5–5 mm) than the 103 pediatric patients with hereditary MTC (Table 1).
As for extrathyroid extension and nodal metastases, the 4 pediatric patients with sporadic MTC were more comparable to the 37 pediatric carriers of highest-risk mutations, 31 (84%) of whom were index patients with de novo disease, than to the 66 pediatric carriers of high-risk, intermediate-risk, or low-risk RET mutations (25–38% vs. 0–8%, and medians of 9–9.5 vs. 0 node metastases after dissection of more (medians of 72–91.5 vs. 4.5–9) nodes).
All in all, the 4 pediatric patients with sporadic MTC attained biochemical cure less frequently than the 103 pediatric patients with hereditary MTC (25% vs. 39–91%).
Discussion
Owing to its rarity, pediatric sporadic MTC remains a severely understudied medical condition. The present investigation of 915 consecutive patients with sporadic MTC clarifies that pediatric MTC almost always has a hereditary background and rarely, if ever, arises sporadically below the age of 14 years.
A recent series of 144 patients diagnosed with MTC between 1961 and 2019 at an age of ≤21 years, using a mixture of clinical and genomic data to identify hereditary MTC, identified 20 patients (14%) with what appeared to be sporadic MTC, of whom 20% had ipsilateral and at least 27% bilateral multifocal thyroid tumors [11]. This 14% ‘pediatric’ estimate was 10 times greater than the 1.4% estimate in the present series, in which 13 of 915 patients with sporadic MTC were aged ≤21 years at thyroidectomy and in which 3 patients (0.2%) had multifocal MTC (data not shown).
The 20 patients who were referred to as ‘pediatric’ in the above publication [11] under the reasonable, but unproven assumption that their thyroid tumors had already developed before the age of 18 years, had been operated at a median age of 19 (range 10–21) years. It remained unclear, though, how many of these 20 patients diagnosed with ‘sporadic’ MTC, in particular the 10-year-old patient, had ruled out hereditary MTC through genomic scanning of RET exons 8, 10, 11, 13, 14, 15 and 16, as currently advised by the revised ATA management guidelines for MTC [6].
Comparison of thyroid glands, which are known to differ a lot in size among infants (very small), children (small) and adolescents (more similar to adults) [12], is not always straightforward, in particular when it comes to extrathyroid tumor extension and nodal spread. Thyroid size disparities may help explain, at least in part, why carriers of high-risk RET mutations developed tumors that were more similar to patients with sporadic MTC in terms of nodal spread than to carriers of high-risk, intermediate-risk, or low-risk RET mutations.
Routine workup of patients with thyroid nodular disease for MTC, hinging on high-resolution ultrasound and serum calcitonin screening, has been crucial in advancing the diagnosis of MTC [13,14,15,16]. The present comprehensive body of combined clinical and genomic data provides additional evidence that all infants, children and adolescents newly diagnosed with MTC must have hereditary disease ruled out by a DNA test that scans all relevant RET exons for pathogenic mutations. It may perhaps also be prudent to have a negative test result double-checked in an infant or child with MTC by sending another venous blood sample to the same or another certified laboratory. Clearly, there is a dire need for excluding hereditary MTC when primary thyroid tumors are present in both thyroid lobes.
In conclusion, sporadic MTC is exceptional in infancy and childhood, and infrequent in adolescence. This makes it imperative to rule out heredity in the pediatric population by a genetic test that scans exons 8, 10, 11, 13, 14, 15, and 16 for pathogenic RET germline mutations.
References
J.S. Mathiesen, J.P. Kroustrup, P. Vestergaard, K. Stochholm, P.L. Poulsen, Å.K. Rasmussen, U. Feldt-Rasmussen, S. Schytte, S.C. Londero, H.B. Pedersen, C.H. Hahn, B.D. Djurhuus, J. Bentzen, S. Möller, M. Gaustadnes, M. Rossing, F.C. Nielsen, K. Brixen, A.L. Frederiksen, C. Godballe; Danish Thyroid Cancer Group (DATHYRCA), Incidence and prevalence of sporadic and hereditary MTC in Denmark 1960-2014: a nationwide study. Endocr. Connect. 7, 829–839 (2018)
A. Machens, K. Lorenz, F. Weber, H. Dralle, Exceptionality of distant metastasis in node-negative hereditary and sporadic medullary thyroid cancer: lessons learned. J. Clin. Endocrinol. Metab. 106, e2968–e2979 (2021)
F. Raue, T. Bruckner, K. Frank-Raue, Similar stage-dependent survival and outcome in sporadic and hereditary medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 106, e3582–e3591 (2021)
A. Machens, K. Lorenz, E.M. Huessler, A. Stang, F. Weber, H. Dralle, Temporal trends in referrals of RET gene carriers for neck surgery to a tertiary surgical center in the era of international management guidelines. Endocrine 80, 100–110 (2023)
R.M.B. Maciel, A.L. Maia, GLOBAL ENDOCRINOLOGY: Geographical variation in the profile of RET variants in patients with medullary thyroid cancer: a comprehensive review. Eur. J. Endocrinol. 186, R15–R30 (2022)
S.A. Wells Jr, S.L. Asa, H. Dralle, R. Elisei, D.B. Evans, R.F. Gagel, N. Lee, A. Machens, J.F. Moley, F. Pacini, F. Raue, K. Frank-Raue, B. Robinson, M.S. Rosenthal, M. Santoro, M. Schlumberger, M. Shah, S.G. Waguespack; American Thyroid Association Guidelines task force on medullary thyroid carcinoma, Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25, 567–610 (2015)
A. Machens, K. Lorenz, F. Weber, T. Brandenburg, D. Führer, H. Dralle, Genotype-specific development of MEN 2 constituent components in 683 RET carriers. Endocr. Relat. Cancer. 31, e240038 (2024)
N. Agrawal, Y. Jiao, M. Sausen, R. Leary, C. Bettegowda, N.J. Roberts, S. Bhan, A.S. Ho, Z. Khan, J. Bishop, W.H. Westra, L.D. Wood, R.H. Hruban, R.P. Tufano, B. Robinson, H. Dralle, S.P. Toledo, R.A. Toledo, L.G. Morris, R.A. Ghossein, J.A. Fagin, T.A. Chan, V.E. Velculescu, B. Vogelstein, K.W. Kinzler, N. Papadopoulos, B.D. Nelkin, D.W. Ball, Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J. Clin. Endocrinol. Metab. 98, E364–E369 (2013)
B. Xu, K. Viswanathan, M.S. Ahadi, S. Ahmadi, B. Alzumaili, M.A. Bani, E. Baudin, D.B. Behrman, M. Capelletti, N.G. Chau, F. Chiarucci, A. Chou, R. Clifton-Bligh, S. Coluccelli, D. de Biase, A. De Leo, S. Dogan, J.A. Fagin, T.L. Fuchs, A.R. Glover, J. Hadoux, L. Lacroix, L. Lamartina, D.J. Lubin, C. Luxford, K. Magliocca, T. Maloberti, A.S. Mohanty, F. Najdawi, A. Nigam, A.J. Papachristos, A. Repaci, B. Robinson, J.Y. Scoazec, Q. Shi, S. Sidhu, E. Solaroli, M. Sywak, R.M. Tuttle, B. Untch, J.A. Barletta, A. Al Ghuzlan, A.J. Gill, R. Ghossein, G. Tallini, I. Ganly, Association of the genomic profile of medullary thyroid carcinoma with tumor characteristics and clinical outcomes in an international multicenter study. Thyroid 34, 167–176 (2024)
R.V. Lloyd, R.Y. Osamura, G. Klöppel, J. Rosai (eds.), WHO Classification of Tumours of Endocrine Organs, WHO Classification of Tumours, 4th edn, vol 10, IARC Press, Lyon, 2017.
S.G. Hensley, M.I. Hu, R.L. Bassett, A.K. Ying, M.E. Zafereo, N.D. Perrier, N.L. Busaidy, S.M. Hyde, E.G. Grubbs, S.G. Waguespack, Pediatric medullary thyroid carcinoma: clinical presentations and long-term outcomes in 144 patients over six decades. J. Clin. Endocrinol. Metab. (2024). https://doi.org/10.1210/clinem/dgae133
J. Farahati, C. Reiners, E.P. Demidchik, Is the UICC/AJCC classification of primary tumor in childhood thyroid carcinoma valid? J. Nucl. Med. 40, 2125 (1999)
A. Machens, H. Dralle, Surgical cure rates of sporadic medullary thyroid cancer in the era of calcitonin screening. Eur. J. Endocrinol. 175, 219–228 (2016)
R.W. Randle, C.J. Balentine, G.E. Leverson, J.A. Havlena, R.S. Sippel, D.F. Schneider, S.C. Pitt, Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 years. Surgery 161, 137–146 (2017)
A. Machens, K. Lorenz, T. Brandenburg, D. Führer-Sakel, F. Weber, H. Dralle, The changing face of multiple endocrine neoplasia 2A: from symptom-based to preventative medicine. J. Clin. Endocrinol. Metab. 108, e734–e742 (2023)
T. Weber, R. Hummel, C. Vorländer, A. Zielke, M. Hermann, A. Krappitz, T. Negele, C. Dotzenrath, A. Trupka, J. Schabram, I. Schmidtmann, C. Klinger, K. Lorenz, Thyroid surgery in children and adolescents: results from a multi-institutional German and Austrian database. Br. J. Surg. 110, 1808–1814 (2023)
Author contributions
A.M.: conceptualization, methodology, validation, formal analysis, investigation, writing - original draft. K.L.: investigation, writing - review & editing. F.W.: investigation, writing -review & editing. H.D.: conceptualization, methodology, investigation, writing - review & editing, supervision.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from each patient and/or legal guardian as applicable before each RET gene test and each operation, all of which represented standard of care.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Machens, A., Lorenz, K., Weber, F. et al. Oncological features of sporadic vs. hereditary pediatric medullary thyroid cancer. Endocrine 85, 1091–1095 (2024). https://doi.org/10.1007/s12020-024-03959-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-024-03959-1