
Abstract
Medullary thyroid carcinoma (MTC) occurring as part of the dominantly inherited multiple endocrine neoplasia (MEN) type 2 syndromes is a rare but notable endocrine disease. Since the discovery of the role of activating mutations in the RET proto-oncogene in MEN2A and MEN2B, considerable advances in management have occurred, and most of the asymptomatic RET carriers who have undergone early thyroidectomy can lead full lives unencumbered by the presence of metastatic MTC. Strong genotype–phenotype correlations have enabled the development of guidelines regarding the timing of thyroidectomy, and current approaches to decisions about the age of surgery for MEN2A incorporate data from ultrasonography and measurement of calcitonin in addition to the results of genotyping. MEN2B is associated with the most aggressive clinical presentation of MTC and thus requires earlier surgical intervention. To optimize care and to enable ongoing research, patients with hereditary MTC in the context of the MEN2 syndromes are optimally treated at tertiary centers with multidisciplinary expertise.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Hereditary medullary thyroid carcinoma
- Early thyroidectomy
- Prophylactic thyroidectomy
- Pediatric thyroid carcinoma
- Multiple endocrine neoplasia 2A
- Multiple endocrine neoplasia 2B
Introduction to Inherited Medullary Thyroid Carcinoma
When diagnosed during childhood and early adulthood, medullary thyroid carcinoma (MTC) most commonly results from a dominantly inherited or de novo gain-of-function mutation in the REarranged during Transfection (RET) proto-oncogene [1–4] that is associated with either multiple endocrine neoplasia (MEN) syndrome type 2A or type 2B (Fig. 8.1). Previously, there was a third distinct category of familial MTC, which is now considered a phenotypic variant of MEN2A with decreased penetrance and/or delayed onset of the other MEN2A-defining manifestations [5, 6]. MTC diagnosed in older adulthood, while more likely to be sporadic in origin, still has the potential to be secondary to an inherited RET mutation, even in the absence of a family history [6–8]. This observation has led to guideline recommendations for all individuals diagnosed with MTC, regardless of age, to undergo germline RET testing, preferably in the setting of formal genetic counseling [6, 9, 10]. If a mutation is found, first-degree relatives should be counseled to undergo testing as well, likely resulting in the diagnosis of additional family members with a RET mutation who may not exhibit any clinical or biochemical evidence of MTC or other MEN2-associated diseases. These individuals are called asymptomatic carriers and in this chapter, we will discuss their management in the context of their risk for developing clinically relevant MTC.

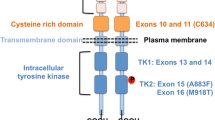
The RET receptor (illustration adapted from Ref. [28], with permission) and the commonly mutated codons and associated phenotypes in the MEN2 syndromes, including the 2015 ATA risk stratification (from Ref. [6]). Age at surgery denotes the age at which thyroidectomy is recommended with “Ctn/pref” denoting that timing can be determined by calcitonin (Ctn) and parent/patient preference in moderate-risk (MOD) patients. Abbreviations: MTC, medullary thyroid carcinoma; PHEO, pheochromocytoma; PHPT, primary hyperparathyroidism; CLA, cutaneous lichen amyloidosis; HSCR, Hirschsprung’s disease; ATA, American Thyroid Association; MOD, moderate risk; H, high risk; HST, highest risk; and Ctn/pref, calcitonin/preference
The original description of MEN2A is credited to Sipple, who reported the case of a 33-year-old man who died of intracranial hemorrhage and was found on a postmortem examination to have bilateral MTC, bilateral pheochromocytomas, and possible parathyroid hyperplasia [11]. Cushman subsequently proposed a relationship between these endocrine tumors [12] and this distinct clinical syndrome was thereafter named MEN2 by Steiner in 1968 [13]. Successively, several large kindreds of MEN2A were identified, which led to linkage analysis studies and the localization of the putative gene to chromosome 10 [14, 15]. In due course, mutations in the gene, RET (originally cloned as an oncogene [16]), were reported in 1993 to cause MEN2A [1, 2]. The MEN2B phenotype was originally characterized in the English literature in 1966 by Williams and Pollock [17], and MEN2B was further discerned as a variant of hereditary MTC with a mucosal neuroma phenotype [18]. In 1994, MEN2B was also identified to be caused by a germline-activating RET mutation [3, 4].
Because of the initial collaborative efforts of the International RET Mutation Consortium, it became quickly apparent that MEN2 was associated with a limited number of mutations and that convincing genotype–phenotype correlations were present (Fig. 8.1) [19]. The high penetrance of hereditary MTC and evolving knowledge regarding the age of MTC onset in MEN2 led to the rapid integration of genetic testing into management algorithms for patients with or at risk for MTC and this, in turn, marshaled in the era of performing total prophylactic thyroidectomy for asymptomatic RET carriers [20]. In the contemporary era, in which genetic testing is routinely incorporated into clinical care, children and young adults with hereditary MTC rarely present with clinical disease, outside of the rare sporadic case, newly identified MEN2A kindred or MTC associated with MEN2B. Thus, in the twenty-first century, the presymptomatic identification of a positive RET mutation is the predominate route to an MEN2A diagnosis in young patients. Additionally, an increasing number of asymptomatic adult carriers have been identified as testing for RET mutations has become more widespread [21]. The utilization of genetic testing in the management of MEN2 has become increasingly complex due to greater recognition of the capricious aggressiveness of hereditable MTC, even among members of a kindred with the same RET mutation [22, 23]. Furthermore, the mutational landscape continues to evolve as novel and uncommon RET DNA variants are identified but remain incompletely characterized as pathogenic mutations and are thus deemed “variants of unknown significance” [8, 21, 24–27]. These factors, plus the utilization of routine calcitonin screening and ultrasonography in asymptomatic RET carriers [6, 28, 29], have led to the present quandary of determining the optimal timing of early thyroidectomy , with the goal of diminishing potential medical and surgical morbidity while simultaneously ensuring an excellent oncologic outcomes by removing the thyroid before MTC metastasis occurs.
MTC is usually the first clinical manifestation of MEN2, and it is most commonly multifocal, bilateral, and located in the middle to upper regions of the thyroid lobes, an area where the calcitonin-positive C-cells are the most highly concentrated [20, 30–32]. There is a well-described, age-related progression of malignant disease with lymph node and distant metastases usually occurring years after the onset of tumorigenesis [33]. C-cell hyperplasia represents the initial stage in the oncologic evolution to microscopic, noninvasive MTC and ultimately to frankly invasive carcinoma that gives rise to metastatic disease [28, 30, 32, 33]. The cervical and mediastinal lymph nodes are the most common sites of metastatic disease; typical distant sites for hematogenous spread include the lungs, liver, and bone/bone marrow.
In MTC, older age at diagnosis, larger tumor size, positive lymph node disease, and the presence of distant metastases predict lower disease-free survival and higher mortality [34–40]. Patients with MTC arising as part of the MEN2 syndromes have a better prognosis than patients with sporadic MTC [38, 39], and individuals with MEN2B have the worst outcome [38]. However, it remains unclear that MTC arising in the context of MEN2B is inherently more biologically aggressive [41], as overall survival may be more impacted by the extremely early onset of MTC in MEN2B coupled with its frequently delayed diagnosis [42, 43].
MTC in MEN2A
MEN2A accounts for around 95 % of MEN2 cases and is characterized by the variable development of MTC in >90 % of RET mutation carriers during the course of their lifetimes [6, 19, 21, 26, 28, 44–46] (Fig. 8.2). Mutations associated with MEN2A are primarily located in the extracellular cysteine-rich domain of the RET proto-oncogene, usually in exon 10 (codons 609, 611, 618 or 620) or exon 11 (codon 634) but can also be found in the intracellular tyrosine kinase domain in exon 13 (codons 768 or 790), exon 14 (codon 804), or exon 15 (codon 891) [6, 8, 19, 47]. Whereas in past decades mutations in codon 634 (exon 11) accounted for the vast majority of MEN2A cases, more recently the prevalence of other mutations has increased [8, 48]. Individuals with a RET codon 634 mutation represent the prototypical MEN2A patient and have the greatest risk of developing early MTC followed by those with mutations in codons 609, 611, 618, 620, or 630, whereas mutations in codons 768, 790, 804, or 891 impart the lowest risk for clinically aggressive MTC [6, 21, 28, 45]. Although genotype–phenotype correlations are typically strong in MEN2A, it is important to remember that the age of MTC onset and disease aggressiveness may not always hold true to the predicted phenotype [22, 23].

Earliest age of medullary thyroid carcinoma (MTC) onset varies depending on the specific RET mutation present. The age of onset of MTC is decreasing for mutations in most codons over time probably because of more widespread genetic testing and earlier surgical intervention. Thus, a well-defined age determination for early thyroidectomy is becoming less clear based upon genotype alone. The mutations are color-coded by the ATA risk stratification Ref. [6]. Data compiled from the ARUP MEN2 RET online database [26] and an exhaustive literature review
MTC in MEN2B
MTC occurring in the clinical context of MEN2B is a completely penetrant disease with a uniformly early age of onset, even within the first few months of life [49]. The tempo of disease progression is accelerated compared with MEN2A, and metastatic lymph node disease has been documented within the first twelve months of life [50]. The average age of onset of MTC is in the second decade of life, about 10 years earlier than that seen in individuals with MEN2A [37, 43, 51–53]. MEN2B is due to a de novo RET mutation in >90 % of cases [8, 49], and thus, the diagnosis is usually delayed, leading to the likelihood that MTC will already have metastasized and become incurable. However, recent data would suggest that, even in de novo cases, surgery may still be curative if performed before age 4 years [49]. The M918T RET mutation is identified in >95 % of MEN2B patients. Rarer MEN2B mutations include double RET mutations involving codon 804 and the A883F mutation, which may be associated with a less aggressive MTC phenotype [54].
Early Thyroidectomy
After it metastasizes beyond the thyroid gland, MTC is most often an incurable disease and one that may ultimately lead to the death of the patient. However, given our ability to identify RET mutation carriers before they develop clinically relevant disease, hereditary MTC has become one of the few malignancies that can be prevented (via prophylactic thyroidectomy ) or cured (via early thyroidectomy ) before it becomes clinically apparent. In the hands of experienced surgeons, children with MEN2 who have a total thyroidectomy performed prior to the onset of metastatic disease have an excellent chance of remaining disease-free with minimal morbidity [6, 28, 36, 37, 49, 55–59].
Commonly accepted is the notion that early thyroidectomy is curative for MTC and ultimately will become necessary for most individuals with a RET mutation, but debate remains as to the optimal timing of surgery for those MEN2 patients with mutations in codons other than 918 and 634, particularly as rarer and less virulent RET mutations are becoming more prevalent in the genetic testing era. In general, the primary oncologic goal of early intervention is to render the patient free of MTC and the potential risk of death from metastatic disease. A true prophylactic thyroidectomy will prevent malignancy from occurring in the first place, but most important is to remove the thyroid before metastasis occurs (early thyroidectomy ). As more children undergo thyroidectomy earlier than in the past, it is anticipated that the earliest age of diagnosis of MTC for any given RET mutation will continue to drop (Fig. 8.2), but the diagnosis of isolated cases of microscopic non-metastatic disease at an extremely young age should not justify a broad prescription for similar at-risk patients to have surgery at that age.
Before the identification of RET and the incorporation of routine genetic testing into the management of kindreds with MTC/MEN2, the calcitonin response to the intravenous administration of a calcitonin secretagogue (calcium, pentagastrin) [60] was used to identify individuals at risk for MTC and to help establish the timing of surgery. This practice was supplanted by genetic testing, and the first recommendations regarding the appropriate age for thyroidectomy were generated on the basis of the specific RET mutation present and the earliest age at which MTC had been diagnosed for that particular mutation [20, 51].
Evolving from the 7th International Workshop on MEN in 1999, a consensus statement issued in 2001 was the first to categorize RET proto-oncogene mutations into one of three separate risk levels (Levels 1–3, level 3 being the highest risk) [51]. Subsequently, the American Thyroid Association (ATA) published guidelines in 2009 that further developed the risk categories, assimilating updated data on RET mutations and phenotypes and placing codon 634 mutations within a separate risk level [61]. The 2009 ATA guidelines stratified all known RET mutations into one of four risk levels (ATA risk levels A–D, level D being the highest risk), and the idea of safely deferring early thyroidectomy while offering careful, expectant monitoring in individuals with lower risk RET mutations was introduced. This new concept of monitoring children in whom the chance of identifying MTC is low (those with normal calcitonin levels, normal neck ultrasonography, and a less aggressive MTC family history) was shown to be a reasonable approach to the care of low-risk MEN2 patients [56, 62]. However, a subsequent study demonstrated the low sensitivity of ultrasonography in predicting microscopic MTC in the asymptomatic MEN2A carrier [29]. In 2015, the ATA guidelines were further refined to simplify the risk levels into “highest risk” (the previous level D, which includes MEN2B patients with a RET codon M918T mutation), “high risk” (includes patients with RET codon C634 mutations, the previous level C, and the RET codon A883F mutation, formerly level D), and “moderate risk” (previous levels A and B) [6] (Figs. 8.1 and 8.2).
The latest guidelines suggest performing a total thyroidectomy in the first year of life in asymptomatic carriers with the highest risk mutation and at or before age 5 for those with a high-risk mutation. With all other RET mutations, the timing of surgery can be determined by the detection of an elevated serum calcitonin level, recognizing that the ultimate decision should be made by the multidisciplinary team in consultation with the child’s parents or guardian, who may opt for an earlier intervention. Although largely unstudied, the approach of cautious surveillance may even be appropriate in select children with high-risk mutations, who may still be cured of their disease even if surgery is not undertaken at the currently prescribed ages [36, 37, 41, 43, 49, 54, 56, 58, 59, 62–65].
Children over the age of 36 months who have basal serum calcitonin levels <30–40 pg/ml and thyroid nodules <0.5 cm on a high-quality ultrasound are unlikely to have metastatic MTC [37, 61, 62, 66]. Thus, in MEN2 patients who have a normal basal serum calcitonin level and a normal thyroid ultrasound and therefore very little chance of having MTC, the benefits of postponing surgical intervention most likely outweigh the associated risks of early thyroidectomy , particularly if access to a high-volume multidisciplinary care center is unavailable. Nonetheless, the use of calcitonin monitoring must be undertaken with knowledge regarding normal serum calcitonin levels in children, which are highest in infancy and decline to adult levels after age 3 years [67, 68], and understanding that large studies validating normal calcitonin ranges in young children are not available for all commercial assays. Moreover, an elevated serum calcitonin level does not always indicate the presence of malignant C-cell disease [37, 66] and can also be found in non-neoplastic conditions, most notably chronic kidney disease, autoimmune thyroiditis, and hyperparathyroidism [6]. Conversely, MTC can also be present pathologically even in the presence of a normal serum calcitonin level [29].
It is essential for clinicians who treat children with MEN2 to distinguish those who clearly require early thyroidectomy to prevent MTC morbidity and mortality and not to overtreat those RET mutation carriers who are unlikely to develop clinically relevant disease over the short term. Although surgical intervention in a child by a high-volume surgeon should be as safe as it is in an adult, the regrettable truth is that many children with MEN2 do not have access to such experts [35, 69], and their complication rates may be higher for that reason [69, 70]. Treating permanent hypoparathyroidism in a child is quite challenging, in addition to the lifelong impact it has on the patient and family. Parental guilt and psychological distress may also occur after the identification of a child with a RET mutation [71], and reassuring parents that their child can be safely monitored instead of steering him or her swiftly to surgery may have positive effects on the family. Additionally, early surgical and medical intervention may also negatively impact the child, stressing the need for initial and ongoing psychological and genetic counseling support [28, 51, 61, 71]. Finally, it is apparent that many children with MEN2 have inadequate thyroid hormone replacement on follow-up [57, 72, 73], and the potential sequelae of iatrogenic hypothyroidism remain poorly studied in this population. Thus, a frank discussion regarding the need to adhere to lifelong levothyroxine therapy should also have when the timing of early thyroidectomy is discussed.
Although the timing of surgery is most often considered in the context of the pediatric age group, adult asymptomatic carriers are becoming more common as RET testing expands in at-risk cohorts. Considerations for surgical intervention in this population are similar to their younger counterpart with the exceptions that the risk of hypocalcemia is less and the patients are able to make well-informed decisions for themselves.
Surgical Management
Surgery remains the primary treatment and only curative intervention for MTC [6, 9, 10]. In MEN2A and MEN2B, early thyroidectomy , performed either in a prophylactic or therapeutic fashion, can alter the natural history of disease and improve long-term oncologic outcomes [36, 37, 55–59]. When discussing early thyroidectomy in asymptomatic RET mutation carriers, notable is the fact that surgical complication rates are higher in children compared with adults [70]. Therefore, as with any rare disease and to lessen the likelihood of iatrogenic injury, children requiring surgery should be operated on by a high-volume thyroid surgeon familiar with the MEN2 syndromes [6, 28]. Optimizing outcomes by utilizing high-volume surgeons necessitates a multidisciplinary approach and advocacy by parents, pediatricians, endocrinologists, surgeons, and third-party payors [69].
In addition to the decision of when to perform an early thyroidectomy , the surgeon must also understand the patient’s genotype and clinical data and incorporate this information into the decision-making process to determine the approach to lymph node and parathyroid gland management. For surgical planning, a thorough preoperative cervical ultrasound to identify nodular thyroid disease and lymph node metastases is essential. The goal of thyroidectomy in the MEN2 syndromes is the complete and safe removal of all thyroid tissue, including the posterior capsule (i.e., not a near-total or subtotal thyroidectomy) [74]. A central compartment (Level VI) neck dissection [75] is not required in the setting of an early thyroidectomy without clinical evidence of MTC, as lymph node metastases are exceedingly rare in that setting [6, 33, 36, 37, 47, 62, 66, 74]. However, in a child with MEN2B, central compartment neck dissection should be considered if the parathyroid glands can be readily identified and safely preserved or if previously unidentified lymph node metastases are identified intraoperatively. If the surgical intervention is therapeutic for a clinically evident tumor (i.e., evidence of lymph node metastases, serum calcitonin level >40 pg/ml), total thyroidectomy and a concomitant central compartment neck dissection should be performed [6]. Compartment-oriented dissection of the lateral cervical lymph node compartments (levels IIA–V) is indicated in cases in which there is clear evidence of lateral neck involvement and can be considered in MEN2B patients who have clinically apparent disease and significantly elevated serum calcitonin levels. In the rare presence of a high burden of distant metastatic disease, less aggressive neck surgical intervention may be appropriate [6].
Though highly unlikely, if primary hyperparathyroidism is diagnosed at the time of early thyroidectomy in patients with MEN2A, concomitant parathyroidectomy of the affected gland(s) should be performed [6, 10]. In the absence of primary hyperparathyroidism, normal parathyroid glands should be left in situ to offer the greatest chance at maintaining function, though there is some controversy in the literature [6, 74, 76, 77]. In the event that a normal parathyroid gland is devascularized during the course of the operation, the gland should be autotransplanted into either the sternocleidomastoid muscle or non-dominant forearm, depending on the specific RET mutation present and the inherent risk for the future development of primary hyperparathyroidism [6, 28]. RET mutations associated with primary hyperparathyroidism include those located on Exon 10, 11, 14, and 15.
Conclusions
Hereditary MTC and the MEN2 syndromes are rare endocrine disorders that are increasingly being managed during an asymptomatic phase secondary to the increased utilization of genetic testing in patients with MTC and at-risk family members. Despite the discovery of RET and its role in MTC, remarkable advances in our understanding and clinical care have occurred. It remains important to recognize the well-established MEN2 genotype–phenotype correlations while also acknowledging our limitations in predicting the appropriate timing of surgery in the asymptomatic, low-risk MEN2 patient. Future research should focus on the long-term oncologic and quality-of-life outcomes after early thyroidectomy , better delineation of genotype–phenotype correlations (especially as more RET DNA variants are characterized), and how to predict more accurately which individuals would benefit from timely thyroidectomy based upon clinical data and which patients can be safely monitored without early intervention.
References
Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2(7):851–6.
Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363(6428):458–60.
Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367(6461):375–6.
Carlson KM, Dou S, Chi D, Scavarda N, Toshima K, Jackson CE, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci USA. 1994;91(4):1579–83.
Jimenez C, Hu MI, Gagel RF. Management of medullary thyroid carcinoma. Endocrinol Metab Clin North Am. 2008;37(2):481–96, x–xi.
Wells SA Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid Official J Am Thyroid Assoc. 2015;25(6):567–610.
Wohllk N, Cote GJ, Bugalho MM, Ordonez N, Evans DB, Goepfert H, et al. Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab. 1996;81(10):3740–5.
Romei C, Tacito A, Molinaro E, Agate L, Bottici V, Viola D, et al. Twenty years of lesson learning: how does the RET genetic screening test impact the clinical management of medullary thyroid cancer? Clin Endocrinol. 2015;82(6):892–9.
Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39(6):775–83.
Tuttle RM, Ball DW, Byrd D, Daniels GH, Dilawari RA, Doherty GM, et al. Medullary carcinoma. J Natl Compr Canc Netw. 2010;8(5):512–30.
Sipple JH. The association of pheochromocytoma with carcinoma of the thyroid gland. Am J Med. 1961;31:163–6.
Cushman P Jr. Familial endocrine tumors; report of two unrelated kindred affected with pheochromocytomas, one also with multiple thyroid carcinomas. Am J Med. 1962;32:352–60.
Steiner AL, Goodman AD, Powers SR. Study of a kindred with pheochromocytoma, medullary thyroid carcinoma, hyperparathyroidism and Cushing’s disease: multiple endocrine neoplasia, type 2. Medicine (Baltimore). 1968;47(5):371–409.
Simpson NE, Kidd KK. Where is the locus for multiple endocrine neoplasia type 2A? Henry Ford Hosp Med J. 1987;35(2–3):168–71.
Mathew CG, Chin KS, Easton DF, Thorpe K, Carter C, Liou GI, et al. A linked genetic marker for multiple endocrine neoplasia type 2A on chromosome 10. Nature. 1987;328(6130):527–8.
Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42(2):581–8.
Williams ED, Pollock DJ. Multiple mucosal neuromata with endocrine tumours: a syndrome allied to von Recklinghausen’s disease. J Pathol Bacteriol. 1966;91:71–80.
Chong GC, Beahrs OH, Sizemore GW, Woolner LH. Medullary carcinoma of the thyroid gland. Cancer. 1975;35(3):695–704.
Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. Int RET Mutat Consortium Anal JAMA. 1996;276(19):1575–9.
Wells SA Jr, Chi DD, Toshima K, Dehner LP, Coffin CM, Dowton SB, et al. Predictive DNA testing and prophylactic thyroidectomy in patients at risk for multiple endocrine neoplasia type 2A. Ann Surg 1994;220(3):237–47, discussion 47–50.
Rich TA, Feng L, Busaidy N, Cote GJ, Gagel RF, Hu M, et al. Prevalence by age and predictors of medullary thyroid cancer in patients with lower risk germline RET proto-oncogene mutations. Thyroid Official J Am Thyroid Assoc. 2014;24(7):1096–106.
Frohnauer MK, Decker RA. Update on the MEN 2A c804 RET mutation: is prophylactic thyroidectomy indicated? Surgery 2000;128(6):1052–7, discussion 7–8.
Mukherjee S, Zakalik D. RET codon 804 mutations in multiple endocrine neoplasia 2: genotype-phenotype correlations and implications in clinical management. Clin Genet. 2011;79(1):1–16.
Erlic Z, Hoffmann MM, Sullivan M, Franke G, Peczkowska M, Harsch I, et al. Pathogenicity of DNA variants and double mutations in multiple endocrine neoplasia type 2 and von Hippel-Lindau syndrome. J Clin Endocrinol Metab. 2010;95(1):308–13.
Crockett DK, Piccolo SR, Ridge PG, Margraf RL, Lyon E, Williams MS, et al. Predicting phenotypic severity of uncertain gene variants in the RET proto-oncogene. PLoS ONE. 2011;6(3):e18380.
Margraf RL, Crockett DK, Krautscheid PM, Seamons R, Calderon FR, Wittwer CT, et al. Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat. 2009;30(4):548–56.
Frank-Raue K, Rondot S, Schulze E, Raue F. Change in the spectrum of RET mutations diagnosed between 1994 and 2006. Clin Lab. 2007;53(5–6):273–82.
Waguespack SG, Rich TA, Perrier ND, Jimenez C, Cote GJ. Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol. 2011;7(10):596–607.
Morris LF, Waguespack SG, Edeiken-Monroe BS, Lee JE, Rich TA, Ying AK, et al. Ultrasonography should not guide the timing of thyroidectomy in pediatric patients diagnosed with multiple endocrine neoplasia syndrome 2A through genetic screening. Ann Surg Oncol. 2013;20(1):53–9.
Wolfe HJ, Melvin KE, Cervi-Skinner SJ, Saadi AA, Juliar JF, Jackson CE, et al. C-cell hyperplasia preceding medullary thyroid carcinoma. N Engl J Med. 1973;289(9):437–41.
Gagel RF, Melvin KE, Tashjian AH Jr, Miller HH, Feldman ZT, Wolfe HJ, et al. Natural history of the familial medullary thyroid carcinoma-pheochromocytoma syndrome and the identification of preneoplastic stages by screening studies: a five-year report. Trans Assoc Am Phys. 1975;88:177–91.
Baloch ZW, LiVolsi VA. C-cells and their associated lesions and conditions: a pathologists perspective. Turk patoloji dergisi. 2015;31(Suppl 1):60–79.
Machens A. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2004;350(9):943.
de Groot JW, Plukker JT, Wolffenbuttel BH, Wiggers T, Sluiter WJ, Links TP. Determinants of life expectancy in medullary thyroid cancer: age does not matter. Clin Endocrinol (Oxf). 2006;65(6):729–36.
Raval MV, Sturgeon C, Bentrem DJ, Elaraj DM, Stewart AK, Winchester DJ, et al. Influence of lymph node metastases on survival in pediatric medullary thyroid cancer. J Pediatr Surg. 2010;45(10):1947–54.
Skinner MA, Moley JA, Dilley WG, Owzar K, Debenedetti MK, Wells SA Jr. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med. 2005;353(11):1105–13.
Rohmer V, Vidal-Trecan G, Bourdelot A, Niccoli P, Murat A, Wemeau JL, et al. Prognostic factors of disease-free survival after thyroidectomy in 170 young patients with a RET germline mutation: a multicenter study of the Groupe Francais d’Etude des Tumeurs Endocrines. J Clin Endocrinol Metab. 2011;96(3):E509–18.
Modigliani E, Cohen R, Campos JM, Conte-Devolx B, Maes B, Boneu A, et al. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: results in 899 patients. The GETC study group. Groupe d’etude des tumeurs a calcitonine. Clin Endocrinol (Oxf) 1998;48(3):265–73.
Cupisti K, Wolf A, Raffel A, Schott M, Miersch D, Yang Q, et al. Long-term clinical and biochemical follow-up in medullary thyroid carcinoma: a single institution’s experience over 20 years. Ann Surg. 2007;246(5):815–21.
Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006;107(9):2134–42.
Leboulleux S, Travagli JP, Caillou B, Laplanche A, Bidart JM, Schlumberger M, et al. Medullary thyroid carcinoma as part of a multiple endocrine neoplasia type 2B syndrome: influence of the stage on the clinical course. Cancer. 2002;94(1):44–50.
Wray CJ, Rich TA, Waguespack SG, Lee JE, Perrier ND, Evans DB. Failure to recognize multiple endocrine neoplasia 2B: more common than we think? Ann Surg Oncol. 2008;15(1):293–301.
Brauckhoff M, Machens A, Hess S, Lorenz K, Gimm O, Brauckhoff K, et al. Premonitory symptoms preceding metastatic medullary thyroid cancer in MEN 2B: an exploratory analysis. Surgery 2008;144(6):1044–50, discussion 50–3.
Machens A, Gimm O, Hinze R, Hoppner W, Boehm BO, Dralle H. Genotype-phenotype correlations in hereditary medullary thyroid carcinoma: oncological features and biochemical properties. J Clin Endocrinol Metab. 2001;86(3):1104–9.
Frank-Raue K, Rondot S, Raue F. Molecular genetics and phenomics of RET mutations: impact on prognosis of MTC. Mol Cell Endocrinol. 2010;322(1–2):2–7.
Kouvaraki MA, Shapiro SE, Perrier ND, Cote GJ, Gagel RF, Hoff AO, et al. RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid Official J Am Thyroid Assoc. 2005;15(6):531–44.
Machens A, Lorenz K, Dralle H. Constitutive RET tyrosine kinase activation in hereditary medullary thyroid cancer: clinical opportunities. J Intern Med. 2009;266(1):114–25.
Grubbs EG, Gagel RF. My, how things have changed in multiple endocrine neoplasia type 2A! J Clin Endocrinol Metab. 2015;100(7):2532–5.
Brauckhoff M, Machens A, Lorenz K, Bjoro T, Varhaug JE, Dralle H. Surgical curability of medullary thyroid cancer in multiple endocrine neoplasia 2B: a changing perspective. Ann Surg. 2014;259(4):800–6.
Zenaty D, Aigrain Y, Peuchmaur M, Philippe-Chomette P, Baumann C, Cornelis F, et al. Medullary thyroid carcinoma identified within the first year of life in children with hereditary multiple endocrine neoplasia type 2A (codon 634) and 2B. Eur J Endocrinol. 2009;160(5):807–13.
Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86(12):5658–71.
O’Riordain DS, O’Brien T, Weaver AL, Gharib H, Hay ID, Grant CS, et al. Medullary thyroid carcinoma in multiple endocrine neoplasia types 2A and 2B. Surgery. 1994;116(6):1017–23.
Yip L, Cote GJ, Shapiro SE, Ayers GD, Herzog CE, Sellin RV, et al. Multiple endocrine neoplasia type 2: evaluation of the genotype-phenotype relationship. Arch Surg. 2003;138(4):409–16, discussion 16.
Jasim S, Ying AK, Waguespack SG, Rich TA, Grubbs EG, Jimenez C, et al. Multiple endocrine neoplasia type 2B with a RET proto-oncogene A883F mutation displays a more indolent form of medullary thyroid carcinoma compared with a RET M918T mutation. Thyroid Official J Am Thyroid Assoc. 2011;21(2):189–92.
Waguespack SG, Rich TA. Multiple endocrine neoplasia [corrected] syndrome type 2B in early childhood: long-term benefit of prophylactic thyroidectomy. Cancer. 2010;116(9):2284.
Grubbs EG, Waguespack SG, Rich TA, Xing Y, Ying AK, Evans DB, et al. Do the recent American Thyroid Association (ATA) guidelines accurately guide the timing of prophylactic thyroidectomy in MEN2A? Surgery. 2010;148(6):1302–10.
Frank-Raue K, Buhr H, Dralle H, Klar E, Senninger N, Weber T, et al. Long-term outcome in 46 gene carriers of hereditary medullary thyroid carcinoma after prophylactic thyroidectomy: impact of individual RET genotype. Eur J Endocrinol. 2006;155(2):229–36.
Etit D, Faquin WC, Gaz R, Randolph G, DeLellis RA, Pilch BZ. Histopathologic and clinical features of medullary microcarcinoma and C-cell hyperplasia in prophylactic thyroidectomies for medullary carcinoma: a study of 42 cases. Arch Pathol Lab Med. 2008;132(11):1767–73.
Dralle H, Gimm O, Simon D, Frank-Raue K, Gortz G, Niederle B, et al. Prophylactic thyroidectomy in 75 children and adolescents with hereditary medullary thyroid carcinoma: German and Austrian experience. World J Surg. 1998;22(7):744–50, discussion 50–1.
Costante G, Durante C, Francis Z, Schlumberger M, Filetti S. Determination of calcitonin levels in C-cell disease: clinical interest and potential pitfalls. Nat Clin Pract Endocrinol Metab. 2009;5(1):35–44.
Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid Official J Am Thyroid Assoc. 2009;19(6):565–612.
Elisei R, Romei C, Renzini G, Bottici V, Cosci B, Molinaro E, et al. The timing of total thyroidectomy in RET gene mutation carriers could be personalized and safely planned on the basis of serum calcitonin: 18 years experience at one single center. J Clin Endocrinol Metab. 2012;97(2):426–35.
Machens A, Niccoli-Sire P, Hoegel J, Frank-Raue K, van Vroonhoven TJ, Roeher HD, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2003;349(16):1517–25.
Waguespack SG. A perspective from pediatric endocrinology on the hereditary medullary thyroid carcinoma syndromes. Thyroid Official J Am Thyroid Assoc. 2009;19(6):543–6.
Rodriguez GJ, Balsalobre MD, Pomares F, Torregrosa NM, Rios A, Carbonell P, et al. Prophylactic thyroidectomy in MEN 2A syndrome: experience in a single center. J Am Coll Surg. 2002;195(2):159–66.
Machens A, Lorenz K, Dralle H. Individualization of lymph node dissection in RET (rearranged during transfection) carriers at risk for medullary thyroid cancer: value of pretherapeutic calcitonin levels. Ann Surg. 2009;250(2):305–10.
Samaan NA, Yang KP, Schultz P, Hickey RC. Diagnosis, management, and pathogenetic studies in medullary thyroid carcinoma syndrome. Henry Ford Hosp Med J. 1989;37(3–4):132–7.
Basuyau JP, Mallet E, Leroy M, Brunelle P. Reference intervals for serum calcitonin in men, women, and children. Clin Chem. 2004;50(10):1828–30.
Wang TS, Roman SA, Sosa JA. Predictors of outcomes following pediatric thyroid and parathyroid surgery. Curr Opin Oncol. 2009;21(1):23–8.
Sosa JA, Tuggle CT, Wang TS, Thomas DC, Boudourakis L, Rivkees S, et al. Clinical and economic outcomes of thyroid and parathyroid surgery in children. J Clin Endocrinol Metab. 2008;93(8):3058–65.
Grosfeld FJ, Beemer FA, Lips CJ, Hendriks KS, ten Kroode HF. Parents’ responses to disclosure of genetic test results of their children. Am J Med Genet. 2000;94(4):316–23.
Engiz O, Ocal G, Siklar Z, Erdogan M, Kologlu M, Percinel S, et al. Early prophylactic thyroidectomy for RET mutation-positive MEN 2B. Pediatr Int. 2009;51(4):590–3.
Morris LF, Waguespack SG, Warneke CL, Ryu H, Ying AK, Anderson BJ, et al. Long-term follow-up data may help manage patient and parent expectations for pediatric patients undergoing thyroidectomy. Surgery. 2012;152(6):1165–71.
Guillem JG, Wood WC, Moley JF, Berchuck A, Karlan BY, Mutch DG, et al. ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol. 2006;24(28):4642–60.
Carty SE, Cooper DS, Doherty GM, Duh QY, Kloos RT, Mandel SJ, et al. Consensus statement on the terminology and classification of central neck dissection for thyroid cancer. Thyroid Official J Am Thyroid Assoc. 2009;19(11):1153–8.
Spinelli C, Di Giacomo M, Costanzo S, Elisei R, Miccoli P. Role of RET codonic mutations in the surgical management of medullary thyroid carcinoma in pediatric age multiple endocrine neoplasm type 2 syndromes. J Pediatr Surg. 2010;45(8):1610–6.
Moley JF, Skinner M, Gillanders WE, Lairmore TC, Rowland KJ, Traugott AL, et al. Management of the parathyroid glands during preventive thyroidectomy in patients with multiple endocrine neoplasia type 2. Ann Surg. 2015;262(4):641–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Grubbs, E.G., Waguespack, S.G. (2016). Inherited Medullary Thyroid Carcinoma: Indications and Technique of Early Thyroidectomy . In: Wang, T., Evans, D. (eds) Medullary Thyroid Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-39412-1_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-39412-1_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39410-7
Online ISBN: 978-3-319-39412-1
eBook Packages: MedicineMedicine (R0)