
Abstract
Purpose
Patients with familial hyperparathyroidism and low urinary calcium excretion may have familial hypocalciuric hypercalcemia (FHH) with mutations in one of three genes: the calcium-sensing receptor (CaSR) defining FHH-type 1, the adaptor-related protein complex 2 (AP2S1) related to FHH-type 3 or the G-protein subunit alpha11 (GNA11) associated with FHH-type 2. We aimed to evaluate the presence of mutations in these genes and to identify phenotypic specificities and differences in these patients.
Subjects and methods
Selected patients were recruited for genetic evaluation. After informed consent was signed, blood for DNA extraction was obtained and genetic sequencing of CaSR was done. In negative cases, we further performed sequencing of AP2S1 and GNA11.
Results
A total of 10 index cases were recruited. CaSR sequencing yielded three missense heterozygous mutations (30%): c.554G > A (p.I32V) previously characterized by our team, c.1394 G > A (p.R465Q) and a novel expected disease-causing mutation c.2479 A > C (p.S827R). We identified 2 additional patients (20%) carrying the deleterious recurrent mutation c.44G > T (p.R15L) in the AP2S1 gene. No GNA11 mutation was found. Clinically, patients with AP2S1 mutations had significant cognitive and behavioral disorders, and higher blood calcium and magnesium levels than patients with FHH1.
Conclusion
CaSR and AP2S1 sequencing is worthwhile in patients with familial hyperparathyroidism and phenotype suggesting FHH as it can diagnose up to 50% of cases. GNA11 mutations seem much rarer. Learning disabilities in these patients, associated with higher serum calcium and magnesium levels may suggest the presence of AP2S1 rather than CaSR mutation and may guide the first step in the genetic evaluation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Primary hyperparathyroidism is a heterogeneous phenotypic disease with around 10% of cases related to a genetic germline mutation [1, 2]. In these cases, the mutation may be sporadic or familial, and may be isolated or associated with a tumor syndrome as in multiple endocrine neoplasia (MEN) 1, 2A and 4 or hyperparathyroidism jaw-tumor (HPT-JT) syndrome [1, 2]. Non-syndromic familial hyperparathyroidism may be related to familial hypocalciuric hypercalcemia (FHH) and neonatal severe hyperparathyroidism (NSHPT), or familial isolated hyperparathyroidism (FIHP) [1, 2]. FHH was initially found to be caused by inactivating mutations in the calcium sensing receptor (CaSR), a G-protein-coupled receptor, localized on chromosome 3q13.3–21.3. CaSR mutations define FHH-type 1 (FHH1), but up to 30 % of cases with a typical phenotype do not harbor any CaSR mutations; in the latter cases, segregation analyses could identify mutations either on chromosome 19p or on 19q.13, defining FHH-type 2 (FHH2) and FHH-type 3 (FHH3), respectively [3]. Recently, loss-of-function mutations of the G-protein subunit alpha11 (GNA11), which regulates CaSR activity, were recently shown to cause FHH2 in rare cases [4] whereas a hotspot missense mutation in codon 15 (residue 15R) of the adaptor protein-2 σ subunit (AP2S1) was shown to cause FHH3 [5]. Inheritance pattern of the disease is autosomal dominant, but some cases may be sporadic.
In a previous study [6] we described the yield of sequencing CaSR in a cohort of patients with idiopathic parathyroid disease whose phenotype was consistent with CaSR defect but underlined the pitfalls in selecting patients for this evaluation. One patient with familial hyperparathyroidism had a novel mutation c.94A > G (p.I32V) but five other with familial hyperparathyroidism had a negative CaSR sequencing. We proposed to the latter to extend their genetic evaluation with AP2S1 and GNA11 sequencing, and recruited four new patients who were evaluated with stepwise sequencing of the three genes. In the present study we report clinical and laboratory features as well as results of stepwise CaSR, AP2S1 and, GNA11 sequencing in overall 10 patients with familial history of hyperparathyroidism and phenotype compatible with FHH.
Patients and methods
Patients
Patients were recruited from endocrine, internal medicine, pediatric, and nephrology clinics throughout Israel. We included patients diagnosed with hyperparathyroidism and a first degree familial history of hyperparathyroidism, without personal or familial history by anamnesa and without clinical signs by physical examination of associated tumors which would have been compatible with MEN 1 (pituitary adenoma, pancreatic mass, carcinoid syndrome), MEN 2A (medullary thyroid carcinoma, pheochromocytoma) or hyperparathyroidism-jaw tumor syndrome (jaw, uterine or renal mass). Blood and urine tests were performed by routine laboratory techniques. Urine calcium-to-creatinine clearance ratio (CCCR) was calculated on the basis of 24 h urine collection. The research was approved by the local institutional review committees and the Israeli National Helsinki Commissions. All participating patients signed an informed consent form.
Preparation of genomic DNA and CaSR, AP2S1, and GNA11 sequencing
A venous blood sample was obtained from each patient. DNA was extracted from leukocytes and amplified by conventional methods. For sequencing, DNA was PCR-amplified using a set of primers encompassing the exonic regions and splice junctions of CaSR as previously described [6]. We used specific primers for exon 2 of the AP2S1 gene [5] and the whole sequencing of the GNA11 gene [4] as published in the literature. PCR products were prepared for sequencing, which was done with an ABI prism 310 machine. Results of the sequencing were compared to the normal base sequence of CaSR (NM_000388), AP2S1 (NM_004069.3) and GNA11 (NM_002067) sequence alignment was performed with BLAST NCBI software. CaSR sequencing was performed first. If normal, AP2S1 sequencing was done, followed lastly by GNA11 sequencing.
Bioinformatics
To predict the damaging potential of mutations, we used web servers PolyPhen2 (Polymorphism Phenotype v2) at http://genetics.bwh.harvard.edu/pph2/ [7] and Mutation Taster at http://www.mutationtaster.org [8].
Results
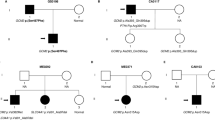
We recruited 10 patients with familial hyperparathyroidism without obvious familial syndrome by anamnesa and clinical examination. Clinical and laboratory features are presented in Table 1. Six of these patients were included in our previous study which consisted in CaSR sequencing only, and four new cases were recruited. They all had a phenotype compatible with FHH. Sequence analyses revealed CaSR mutations in three patients (30%) and their family members and R15 AP2S1 mutations in two other families (20%). Regarding the three CaSR mutations, we found one novel missense mutation c.2479 A > C (p. S827R) in proband 193 (Fig. 1) who was a young healthy 20 year old woman complaining about constipation, and had mild hypercalcemia with mildly elevated parathyroid hormone (PTH) levels and low CCCR (Table 1). She had a mother, a grand-mother, an aunt, and two cousins with hypercalcemia. PolyPhen-2 and Mutation Taster predicted damaging potential for this mutation. This variant was not identified in data obtained from the exome project (Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/) (09.2016). The second CaSR missense mutation observed in proband 202 (Table 1) was the known mutation c.1394 G > A (p. R465Q) (Fig. 2): the patient was a 43 y.o. man with a family history of hyperparathyroidism with negative imaging studies in his mother, two brothers, and a son. The other mutation was the missense mutation c.94A > G (p.I32V), in proband 153 and her father. Interestingly, the father had a relatively elevated CCCR (1.7%) whereas the proband had a typically low CCCR (<1%). We also found two AP2S1 mutations on residue 15R c.44G > T (p.R15L) in proband 151 and in proband 163 (Table 1). The former who was included in our previous study, had a father with known hyperparathyroidism, and a sister with hypercalcemia, all of them having learning disabilities and scholar retardations, whereas three other brother and sisters were normocalcemic with strictly normal cognitive function. Depression was noticed in the two sisters. Two children of the affected proband’s sister with learning disabilities and language skills deficit were also found to be hypercalcemic, and all had AP2S1 mutations, whereas the proband’s mother and all other family members with normal cognitive capacity and without depression were normocalcemic and had no AP2S1 mutation (Fig. 3). Proband 163 was a newborn male of a mother who had a failed three-and a half parathyroidectomy at age 20 year old. His psychomotor development was marked for severe attention deficit hyperactivity disorder (ADHD) medically treated since age 3, and skill languages retardation, whereas his mother also had ADHD, depression and cognitive impairment (Fig. 4). In the five remaining patients with normal CaSR and AP2S1 sequencing, GNA11 sequencing was normal.

Novel CaSR mutation S827R in patient 193 and her family

CaSR mutation R465Q in patient 202 and his family

a Pedigree showing proband 151 (arrow) and her family, showing a strong segregation with learning disabilities. Arrow showing proband. b Mutation p. R15L in proband 151

a Proband 163 (arrow) and his mother, both with marked cognitive disorders. b Mutation p. R15L in proband 163 and his mother
Discussion
In this study we investigated the presence of mutations in CaSR followed by AP2S1 and GNA11 in 10 patients with familial hyperparathyroidism and phenotype compatible with FHH. Two patients (157 and 198, Table 1) had urine CCCR above 1% which is not a typical feature of FHH. However, such a finding is described in up to 20% of patients with FHH [3], as observed in proband 153’s father with p. I32V CaSR mutation [6]; thus, some authors recommend a urine CCCR cut-off value of 2% to select patients for CaSR sequencing [9].
Five patients who had negative CaSR sequencing in our previous study completed AP2S1 evaluation and one of them (proband 151) was found to harbor the typical mutation 15 c.44G > T (p.R15L). Overall, we identified three patients (153, 193, and 202) with CaSR mutations (30%) presenting FHH1, and two other patients (20%) with AP2S1 mutations in residue 15 c.44G > T (p.R15L) presenting FHH3. Loss-of-function mutations of the G-protein subunit alpha11, which regulates CaSR activity and could cause FHH2 [4], were not identified. They are found in less than 5% of patients with FHH and normal CaSR sequencing [2]. In patient 153 (Table 1) the p. I32V CaSR mutation was a missense mutation that we previously described [6]. The missense CaSR mutation p.R465Q observed in proband 202 was previously described [10]; to note, the latter (Table 1) harbored osteoporosis, which is an unusual finding in FHH1 patients [11], possibly because of associated low vitamin 25(OH)D levels. The third observed CaSR mutation in our patients was a novel missense mutation p.S827R, localized in the transmembrane domain (TMD). Missense mutations in the CaSR count for more than 90% of cases in FHH1; most are localized in the extracellular domain but only 26–28% in the TMD [12]. Bio-informatic programs polyphen-2 and Mutation Taster predicted a damaging amino-acid change. The extension of the genetic evaluation with AP2S1 sequencing made the diagnosis in 20% of the tested patients. This finding is in accordance with the literature, showing that up to 20% of patients with suspected FHH but negative CaSR sequencing are found to have one of three rare specific mutations in the residue 15R of the AP2S1 gene [5]: R15L, R15C, and R15H. Frequency of CaSR mutation in appropriately selected patients with isolated familial hyperparathyroidism and phenotype compatible with FHH encourages the physician to start the genetic evaluation with CaSR sequencing, however, there may be clinical and laboratory features suggesting AP2S1 rather than CaSR mutation. If clinicians can identify these specific phenotypic findings, they can choose to start the genetic evaluation by sequencing the exon 2 of AP2S1 instead of performing the whole CaSR sequencing. Some case-reports were published [13–16], and 2 important series evaluated genotype–phenotype correlation [17, 18]. Around 40 index-cases from different kindreds were identified till today and our study adds clinical and biochemical description of 2 more families with FHH3.
In the first series published by Hannan et al. [17], specific clinical features of FHH3 patients included: more hypercalcemia-related symptoms (constipation, lethargy, muskuloskeletal pain and polydipsia) and lower bone mineral density. In both series [17, 18], similar biochemical specificities were found: patients with FHH3 had higher blood calcium levels, higher calcium tubular reabsorption, and higher magnesium levels. In the series published by Hannan et al. [17], R15L mutations was associated with most severe clinical and biochemical abnormalities and the use of a calcium–magnesium–calcium clearance ratio (sCa × sMg/100 × CCCR) above five distinguished patients with FHH3 from those with FHH1 with a specificity of 86% and a sensitivity of 83%. The clinical and biochemical features of our two patients with FHH3 corroborate the previous published data showing that these patients have higher calcium and magnesium levels (Table 1). The strong association of learning disabilities and AP2S1 mutations (pedigrees in Figs. 3 and 4) is in our eyes a particular phenotypic feature observed in FHH3 patients, which is not observed in FHH1 patients. Undoubtedly, patients with FHH3 harbor more neuropsychiatric disorders and learning disabilities than usual patients with primary hyperparathyroidism or FHH1. Despite not emphasized in the series published by Hannan et al. [17], there were also more cognitive dysfunction and behavioral disorders in their patients: 7 among the 19 described (37%) had learning disabilities. The French series [18] did not describe patients’ clinical features, but Hendy et al. [14] reported two cases with depression or psychiatric condition, one with R15L and the other with R15C mutation whereas a third patient with R15L mutation had cerebral palsy and a global development delay.
It is fascinating to understand if the higher prevalence of these cognitive disorders in patients with FHH3 are related to the higher blood calcium levels (particularly in association with the R15L mutation), or if it is caused by a specific molecular involvement of AP2S1: on one side, hypercalcemia and hyperparathyroidism are associated with neuropsychological symptoms [19], cognitive dysfunctions (concentration capacity, verbal resources, non-verbal learning abilities, visual memory) [20], depression and anxiety [21]; on the other side, the Adaptor protein-2 σ subunit is included in the clathrin-coated vesicles and thus participates to the clathrin-mediated endocytosis leading to internalization of plasma membrane constituents and modification of plasma membrane activity (as CaSR in the parathyroid) [5], but is also essential to neurotransmission and signal transduction [22]. It is clearly involved in the regulation of AMPA-glutamate receptors in the brain, playing a pivotal role in post-synaptic plasticity and neuronal trafficking [23], and may be involved in psychiatric diseases [24] and depression [25]. Further systematic clinical and maybe experimental evaluations are needed to verify the association and the potential causative relationship between R15 AP2S1 mutations and neuropsychiatric and cognitive disorders. If it turns to be confirmed, patients with isolated familial hyperparathyroidism, phenotype compatible with FHH and learning disabilities or psychiatric disorder should be evaluated first with sequencing of the second exon of AP2S1 which is easier, cheaper and less time-consuming than performing the whole CaSR sequencing. In other patients, the latter remains the initial work-up genetic test to perform.
Five patients in our study remain without genetic diagnosis. Typical FHH phenotype can be observed in primary hyperparathyroidism [1] without any mutation in CaSR, AP2S1 or GNA11. However, the familial feature of the disease raises the possibility of another hereditary disease. Occult MEN 1, MEN 2A or HPT-JT syndrome were not ruled-out by specific biochemical and imaging tests. Only patient 157 (Table 1) had more extensive evaluation. FIHP, if not associated with CaSR [26, 27], AP2S1 or GNA11 mutations, may be related to mutations in MEN1 [27, 28], HRPT2 (CDC73) responsible of HPT-JT syndrome [26], CDKN1A, 1B (associated with MEN-4), 2B, 2C [2] and RET (responsible of MEN2). Recently, activating mutations in GCM2 were identified in patients with FIHP [29].
We conclude that CaSR and AP2S1 sequencing has a high diagnostic yield in patients with familial hyperparathyroidism and typical phenotype of hypocalciuric hypercalcemia. Learning disabilities in these patients, associated with higher serum calcium and magnesium levels may suggest the presence of AP2S1 rather than CaSR mutation. Patients’ clinical and biochemical features may suggest initiating the genetic work-up with one rather than the other gene.
References
R. Eastell, M.L. Brandi, A.G. Costa, P. D’Amour, D.M. Shoback, R.V. Thakker, Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J. Clin. Endocrinol. Metab. 99(10), 3570–3579 (2014)
R.V. Thakker, Genetics of parathyroid tumours. J. Intern. Med. (2016) 280(6), 574–583
J. Varghese, T. Rich, C. Jimenez, Benign familial hypocalciuric hypercalcemia. Endocr. Pract. 17(Suppl 1), 13–17 (2011)
M.A. Nesbit, F.M. Hannan, S.A. Howles, V.N. Babinsky, R.A. Head, T. Cranston, N. Rust, M.R. Hobbs, H. Heath 3rd, R.V. Thakker, Mutations affecting G-protein subunit alpha11 in hypercalcemia and hypocalcemia. N. Eng. J. Med. 368(26), 2476–2486 (2013)
M.A. Nesbit, F.M. Hannan, S.A. Howles, A.A. Reed, T. Cranston, C.E. Thakker, L. Gregory, A.J. Rimmer, N. Rust, U. Graham, P.J. Morrison, S.J. Hunter, M.P. Whyte, G. McVean, D. Buck, R.V. Thakker, Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat. Genet. 45(1), 93–97 (2013)
A. Szalat, M. Shahar, S. Shpitzen, B. Nachmias, G. Munter, D. Gillis, R. Durst, D. Mevorach, E. Leitersdorf, V. Meiner, H. Rosen, Calcium-sensing receptor sequencing in 21 patients with idiopathic or familial parathyroid disorder: pitfalls and characterization of a novel I32 V loss-of-function mutation. Endocrine 48(2), 444–453 (2014)
I.A. Adzhubei, S. Schmidt, L. Peshkin, V.E. Ramensky, A. Gerasimova, P. Bork, A.S. Kondrashov, S.R. Sunyaev, A method and server for predicting damaging missense mutations. Nat. Methods 7(4), 248–249 (2010)
J.M. Schwarz, C. Rodelsperger, M. Schuelke, D. Seelow, Mutation Taster evaluates disease-causing potential of sequence alterations. Nat. Methods 7(8), 575–576 (2010)
S.E. Christensen, P.H. Nissen, P. Vestergaard, L. Heickendorff, K. Brixen, L. Mosekilde, Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: a follow-up study on methods. Clin. Endocrinol. (Oxf). 69(5), 713–720 (2008)
C. Leech, P. Lohse, V. Stanojevic, A. Lechner, B. Goke, C. Spitzweg, Identification of a novel inactivating R465Q mutation of the calcium-sensing receptor. Biochem. Biophys. Res. Commun. 342(3), 996–1002 (2006)
N.F. Jakobsen, L. Rolighed, E. Moser, P.H. Nissen, L. Mosekilde, L. Rejnmark, Increased trabecular volumetric bone mass density in Familial Hypocalciuric Hypercalcemia (FHH) type 1: a cross-sectional study. Calcif. Tissue Int. 95(2), 141–152 (2014)
F.M. Hannan, M.A. Nesbit, C. Zhang, T. Cranston, A.J. Curley, B. Harding, C. Fratter, N. Rust, P.T. Christie, J.J. Turner, M.C. Lemos, M.R. Bowl, R. Bouillon, C. Brain, N. Bridges, C. Burren, J.M. Connell, H. Jung, E. Marks, D. McCredie, Z. Mughal, C. Rodda, S. Tollefsen, E.M. Brown, J.J. Yang, R.V. Thakker, Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites. Hum. Mol. Genet. 21(12), 2768–2778 (2012)
Y. Fujisawa, R. Yamaguchi, E. Satake, K. Ohtaka, T. Nakanishi, K. Ozono, T. Ogata, Identification of AP2S1 mutation and effects of low calcium formula in an infant with hypercalcemia and hypercalciuria. J. Clin. Endocrinol. Metab. 98(12), E2022–E2027 (2013)
G.N. Hendy, L. Canaff, R.S. Newfield, L. Tripto-Shkolnik, B.Y. Wong, B.S. Lee, D.E. Cole, Codon Arg15 mutations of the AP2S1 gene: common occurrence in familial hypocalciuric hypercalcemia cases negative for calcium-sensing receptor (CASR) mutations. J. Clin. Endocrinol. Metab. 99(7), E1311–E1315 (2014)
S. Tenhola, G.N. Hendy, H. Valta, L. Canaff, B.S. Lee, B.Y. Wong, M.J. Valimaki, D.E. Cole, O. Makitie, Cinacalcet treatment in an adolescent with Concurrent 22q11.2 Deletion Syndrome and Familial Hypocalciuric Hypercalcemia Type 3 caused by AP2S1 mutation. J. Clin. Endocrinol. Metab. 100(7), 2515–2518 (2015)
S.A. Howles, F.M. Hannan, V.N. Babinsky, A. Rogers, C.M. Gorvin, N. Rust, T. Richardson, M.J. McKenna, M.A. Nesbit, R.V. Thakker, Cinacalcet for symptomatic hypercalcemia caused by AP2S1 mutations. N. Eng. J. Med. 374(14), 1396–1398 (2016)
F.M. Hannan, S.A. Howles, A. Rogers, T. Cranston, C.M. Gorvin, V.N. Babinsky, A.A. Reed, C.E. Thakker, D. Bockenhauer, R.S. Brown, J.M. Connell, J. Cook, K. Darzy, S. Ehtisham, U. Graham, T. Hulse, S.J. Hunter, L. Izatt, D. Kumar, M.J. McKenna, J.A. McKnight, P.J. Morrison, M.Z. Mughal, D. O’Halloran, S.H. Pearce, M.E. Porteous, M. Rahman, T. Richardson, R. Robinson, I. Scheers, H. Siddique, W.G. Van’t Hoff, T. Wang, M.P. Whyte, M.A. Nesbit, R.V. Thakker, Adaptor protein-2 sigma subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype-phenotype correlations, codon bias and dominant-negative effects. Hum. Mol. Genet. 24(18), 5079–5092 (2015)
R. Vargas-Poussou, L. Mansour-Hendili, S. Baron, J.P. Bertocchio, C. Travers, C. Simian, C. Treard, V. Baudouin, S. Beltran, F. Broux, O. Camard, S. Cloarec, C. Cormier, X. Debussche, E. Dubosclard, C. Eid, J.P. Haymann, S.R. Kiando, J.M. Kuhn, G. Lefort, A. Linglart, B. Lucas-Pouliquen, M.A. Macher, G. Maruani, S. Ouzounian, M. Polak, E. Requeda, D. Robier, C. Silve, J.C. Souberbielle, I. Tack, D. Vezzosi, X. Jeunemaitre, P. Houillier, Familial Hypocalciuric Hypercalcemia Types 1 and 3 and primary hyperparathyroidism: similarities and differences. J. Clin. Endocrinol. Metab. 101(5), 2185–2195 (2016)
H. Kahal, M. Aye, A.S. Rigby, T. Sathyapalan, R.J. England, S.L. Atkin, The effect of parathyroidectomy on neuropsychological symptoms and biochemical parameters in patients with asymptomatic primary hyperparathyroidism. Clin. Endocrinol. (Oxf). 76(2), 196–200 (2012)
D. Babinska, M. Barczynski, T. Stefaniak, T. Oseka, A. Babinska, D. Babinski, K. Sworczak, A.J. Lachinski, W. Nowak, Z. Sledzinski, Evaluation of selected cognitive functions before and after surgery for primary hyperparathyroidism. Langenbecks. Arch. Surg. 397(5), 825–831 (2012)
T. Weber, J. Eberle, U. Messelhauser, L. Schiffmann, C. Nies, J. Schabram, A. Zielke, K. Holzer, E. Rottler, D. Henne-Bruns, M. Keller, J. von Wietersheim, Parathyroidectomy, elevated depression scores, and suicidal ideation in patients with primary hyperparathyroidism: results of a prospective multicenter study. JAMA Surg. 148(2), 109–115 (2013)
H.T. McMahon, E. Boucrot, Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12(8), 517–533 (2011)
J.D. Shepherd, R.L. Huganir, The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643 (2007)
K.O. Schubert, M. Focking, J.H. Prehn, D.R. Cotter, Hypothesis review: are clathrin-mediated endocytosis and clathrin-dependent membrane and protein trafficking core pathophysiological processes in schizophrenia and bipolar disorder? Mol. Psychiatry 17(7), 669–681 (2012)
S. Matsuda, W. Kakegawa, T. Budisantoso, T. Nomura, K. Kohda, M. Yuzaki, Stargazin regulates AMPA receptor trafficking through adaptor protein complexes during long-term depression. Nat. Commun. 4, 2759 (2013)
W.F. Simonds, L.A. James-Newton, S.K. Agarwal, B. Yang, M.C. Skarulis, G.N. Hendy, S.J. Marx, Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore) 81(1), 1–26 (2002)
J. Warner, M. Epstein, A. Sweet, D. Singh, J. Burgess, S. Stranks, P. Hill, D. Perry-Keene, D. Learoyd, B. Robinson, P. Birdsey, E. Mackenzie, B.T. Teh, J.B. Prins, J. Cardinal, Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J. Med. Genet. 41(3), 155–160 (2004)
F.M. Hannan, M.A. Nesbit, P.T. Christie, C. Fratter, N.E. Dudley, G.P. Sadler, R.V. Thakker, Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat. Clin. Pract. Endocrinol. Metab. 4(1), 53–58 (2008)
B. Guan, J.M. Welch, J.C. Sapp, H. Ling, Y. Li, J.J. Johnston, E. Kebebew, L.G. Biesecker, W.F. Simonds, S.J. Marx, S.K. Agarwal, GCM2-Activating mutations in familial isolated hyperparathyroidism. Am. J. Hum. Genet. 99(5), 1034–1044 (2016)
Acknowledgements
This study was supported by the Research Foundation of the Chief Scientist at the Israel Ministry of Health and by the Israel Endocrine Society.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Szalat, A., Shpitzen, S., Tsur, A. et al. Stepwise CaSR, AP2S1, and GNA11 sequencing in patients with suspected familial hypocalciuric hypercalcemia. Endocrine 55, 741–747 (2017). https://doi.org/10.1007/s12020-017-1241-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-017-1241-5