
Abstract
Epidemiological studies indicate that patients with type 2 diabetes mellitus (T2DM) are at increased risk of developing dementia/Alzheimer’s disease (AD). This review, which is based on recent studies, presents a molecular framework that links the two diseases and explains how physical training could help counteract neurodegeneration in T2DM patients. Inflammatory, oxidative, and metabolic changes in T2DM patients cause cerebrovascular complications and can lead to blood–brain-barrier (BBB) breakdown. Peripherally increased pro-inflammatory molecules can then pass the BBB more easily and activate stress-activated pathways, thereby promoting key pathological features of dementia/AD such as brain insulin resistance, mitochondrial dysfunction, and accumulation of neurotoxic beta-amyloid (Aβ) oligomers, leading to synaptic loss, neuronal dysfunction, and cell death. Ceramides can also pass the BBB, induce pro-inflammatory reactions, and disturb brain insulin signaling. In a vicious circle, oxidative stress and the pro-inflammatory environment intensify, leading to further cognitive decline. Low testosterone levels might be a common risk factor in T2DM and AD. Regular physical exercise reinforces antioxidative capacity, reduces oxidative stress, and has anti-inflammatory effects. It improves endothelial function and might increase brain capillarization. Physical training can further counteract dyslipidemia and reduce increased ceramide levels. It might also improve Aβ clearance by up-regulating Aβ transporters and, in some cases, increase basal testosterone levels. In addition, regular physical activity can induce neurogenesis. Physical training should therefore be emphasized as a part of prevention programs developed for diabetic patients to minimize the risk of the onset of neurodegenerative diseases among this specific patient group.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Epidemiological associations between type 2 diabetes mellitus and dementia/Alzheimer’s disease
The epidemiology of dementia and type 2 diabetes mellitus (T2DM) and their possible relationship is a recurring subject of neuro-psychiatric research, dating back to at least three decades [1]. In a 2014 statement issued by the World Dementia Council, diabetes was classified as one of the leading preventable risk factors related to cognitive decline and dementia [2]. Recent meta-analyses conclude that T2DM is associated with decreased executive function and memory [3–5], and describe associations between T2DM and magnetic resonance imaging findings of hippocampal and total gray matter atrophy [6]. Conversely, T2DM was also found to increase the risk of amnestic mild cognitive impairment (MCI) progression towards dementia in numerous longitudinal studies [7, 8]. A conventional wisdom is that vascular damage can lead to cognitive decline [vascular dementia (VD)]. However, Moran et al. [6] did not observe that reductions in executive function and memory in T2DM patients are attenuated after adjusting for cerebrovascular lesions, suggesting that T2DM can lead to cognitive decline also in other ways.
Furthermore, meta-analyses of prospective cohort studies report that diabetes may increase the risk of Alzheimer’s disease (AD) when present during midlife [9] and contributes to the progression from amnestic MCI to AD [10]. A 2013 meta-analysis of 28 prospective observational studies involving a noteworthy number of 1,148,041 subjects across all studies found robust associations of T2DM with all-cause dementia, VD and AD, with a relative risk increase for AD being estimated at 73 % [11].
Public health impacts of reducing the incidence of diabetes are high, which alone considering its contribution to dementia/AD, are crucial: in an epidemiological investigation to assess the potential impact of primary prevention, Norton et al. [12] conclude that up to one-third of AD cases are attributable to preventable risk factors, diabetes being among them.
The available evidence points towards T2DM as a risk factor for dementia and AD. Following this line of thought, we will summarize the basal principles of dementia/AD pathophysiology and then explore some molecular connections between T2DM and neurodegeneration.
Vascular and cellular modifications: triggering dementia and Alzheimer’s disease
VD refers to any disease of the vasculature that causes or contributes to a state of dementia by reducing energy supply for cell function and cell survival. As such, this umbrella term includes various concepts. Cerebral small vessel disease (CSVD) is thought to be the most important subentity (for a review, see [13]). VD shares many of the same risk factors as AD (as discussed above), but differs in its pathophysiology. With the main focus of its pathogenesis within the blood vessels, VD as a result of CSVD involves phenomena such as subcortical leukencephalopathy caused by arteriosclerosis and thromboembolism in small white matter arterioles, lacunar strokes affecting penetrating small arteries supplying deeper brain regions, and cerebral microbleeds associated chiefly with hypertension and cerebral amyloid angiopathy [14]. Endothelial dysfunction is considered a key causative agent in many vascular diseases, including stroke [15] and may also be involved in CSVD [16].
By contrast, the fundamental mechanisms of AD are widely accepted to originate in nerve cells, independently of vascular pathology. Beta-amyloid (Aβ) peptides are generated from Aβ precursor protein (APP) through sequential cleavages and can form soluble Aβ oligomers which are thought to be causative as main mediators of the progression of the disease—either through intracellular toxicity or by interaction with synaptic proteins [17]. The notion of pathological insulin signaling being at the core of AD has been gaining traction for some time [18]: a fact that is especially pertinent to this review and will be addressed in detail below. It is important to note, however, that much research does not draw a dogmatic line between vascular and Aβ-mediated dementia/AD, instead illuminates the many points at which both diseases intersect and entwine in a vicious circle. Autopsy studies have found a significant rate of mixed VD and AD, suggesting additive or synergistic effects of both pathologies on cognitive decline [19]. Therefore, while we highlight the distinction on a theoretical level, the question of what exactly is responsible for cognitive decline remains nebulous in real-life diagnoses.
Several parallels and molecular links between the pathology of type 2 diabetes mellitus, vascular dementia, and Alzheimer’s disease
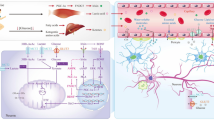
There are some parallels in the pathology of T2DM, VD, and AD and several molecular mechanisms which link these diseases and include inflammatory, oxidative, and metabolic changes (Fig. 1a).

a Crosstalk between type 2 diabetes and dementia/Alzheimer’s disease (hypothetical model). b Possible preventive effects of physical training. TNF-α tumor necrosis factor-α, IL-6 interleukin-6, ROS reactive oxygen species, AGEs advanced glycation end products, RAGE receptor for AGEs, NFTs neurofibrillary tangles, LRP-1 lipoprotein receptor-related protein-1
Type 2 diabetes mellitus-associated vascular complications and their contribution to the progression of dementia/Alzheimer’s disease
T2DM can lead to vascular complications as has been extensively reviewed elsewhere [20]. Hence, only some of the most important mechanisms are summarized in this section. Macro- and microvascular complications mostly occur due to the presence of clustered risk factors such as hyperglycemia, dyslipidemia, and hypertension. As a key event, metabolic abnormalities lead to an overproduction of reactive oxygen species (ROS) which restrict the bioavailability of nitric oxide (NO), a potent vasodilator, trigger vascular inflammation, and thus predispose endothelial dysfunction [21]. Hyperglycemia up-regulates endothelin-1, a potent vasoconstrictor, via protein kinase C and a lack of insulin (in insulin-dependent patients) can induce calcium accumulation in blood platelets, resulting in hyperactivity and platelet aggregation [22, 23].
Furthermore, comorbidities can intensify the risk for vascular complications. Changes in lipid profile, in particular elevated very low density lipoprotein levels, an increased release of pro-inflammatory adipocytokines in patients with obesity as well as hypertension-driven oxidative stress, inflammation, and vascular damage, can further favor arteriosclerosis [24–26].
Type 3 diabetes mellitus? Molecular evidence of central nervous system insulin/insulin-like growth factor resistance in AD
Insulin resistance is a likely candidate in the pathogenesis of AD because it is known that insulin acts as a neurotrophic factor that crosses the blood–brain barrier (BBB) via a receptor-mediated transport mechanism and is also synthesized locally within the brain [27]. There, insulin contributes to both synaptogenesis and synaptic remodeling [27]. Other central nervous system (CNS) functions mediated by insulin and insulin-like growth factor (IGF), that can also cross the BBB, may include modulation of neurotransmitter concentrations [28], the augmentation of long-term potentiation [29], hippocampal synaptic plasticity [30], and glucose uptake in cortical areas [31, 32]. Key downstream targets in these functions are the Akt/protein kinase B (PKB) pathway or the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase pathway [32–35].
Talbot et al. [36] sought to compare postmortem brain insulin/IGF-1 sensitivity in AD subjects with that in healthy controls. They tested insulin signaling [insulin receptor (IR) → IR substrate (IRS)-1 → phosphoinositide 3-kinase (PI3K) → Akt/PKB] and IGF-1 signaling (IGF-1 receptor → IRS-2 → PI3K) in the hippocampal formation and the cerebellar cortex using an ex vivo stimulation protocol. While they report no difference in the contents of signaling molecules at baseline, a comparison after stimulation with near physiological doses of insulin/IGF-1 demonstrated significant insulin/IGF-1 resistance in the AD cases.
Two candidates of brain insulin resistance, which have been identified by Talbot et al. [36], are elevated levels of IRS-1 phosphorylations in serine residues (IRS-1 pS616 and pS636/639) due to chronically active IRS-1 serine kinases. This corresponds to a key mechanism of peripheral insulin resistance, where IRS proteins are likewise phosphorylated at serine residues, decreasing their transmission activity [37]. It is notable that insulin resistance demonstrated in AD brains in Talbot et al.’s [36] study did not affect neuronal glucose uptake in neurons. They therefore consider the term “type 3 diabetes” inappropriate. However, reduced glucose utilization has been measured in AD patients based on 2-[18F]fluoro-2-deoxy-d-glucose positron emission tomography and even prior to the onset of AD symptoms [38].
Bomfim et al. [39] have demonstrated that Aβ oligomers can induce IRS-1 serine phosphorylation in hippocampal neuronal cells in vitro, as well as in the hippocampi of mice and monkeys in vivo. The interaction of IRs with interfering Aβ oligomers have been linked with activated c-Jun N-terminal kinase (JNK) [40]. JNK is a stress kinase which is already known to mediate IRS-1 serine phosphorylation in peripheral insulin resistance in type 2 diabetes, e.g., in response to lipotoxic stress [41]. In AD, Aβ oligomers have been shown to activate the JNK/tumor necrosis factor (TNF)-α pathway [42].
Reduced insulin/IGF-1 signaling in the brain has disastrous consequences. Insulin signaling has been shown to prevent Aβ formation and accumulation by dephosphorylation of APP (in the threonine 668 site) in human cortical neuronal cells. APP dephosphorylation prevents the translocation of the APP intracellular domain into the nucleus, normally leading to an increased transcription of anti-amyloidogenic proteins, such as insulin-degrading enzyme (IDE), involved in Aβ degradation, and α-secretase as well as to decreased transcription of pro-amyloidogenic proteins such as APP, β-secretase, or glycogen synthase kinase (GSK)-3β [32, 34, 43].
GSK-3β has been implicated in tau hyperphosphorylation in AD [33]. Tau is a neuronal protein found in axons. Its hyperphosphorylation leads to aggregation and the build-up of neurofibrillary tangles (NFTs). Although many studies have demonstrated that the amount and distribution of NFTs correlate well with the severity and the duration of AD, it is still controversial whether NFTs lead to neuronal death [33, 44, 45]. Insulin has also been demonstrated to inactivate GSK-3β through phosphorylation, which, without inactivation, increases tau phosphorylation [46, 47].
IDE catabolizes insulin, and is also directly involved in Aβ clearance [48]. IDE processes both insulin and Aβ, but has a higher affinity towards insulin [49]. Thus, insulin resistance and hyperinsulinemia may reduce the IDE degradation of Aβ and contribute to neuronal Aβ toxicity.
IGF-1 signaling normally leads to enhanced clearance of brain Aβ peptides by modulating transporters/carriers of Aβ at the blood–brain interface [50]. Thus, a vicious circle arises in which Aβ oligomers up-regulate their own formation. An accumulation of Aβ oligomers further deteriorates insulin resistance by decreasing the binding affinity of insulin to its receptor and by reducing the amount of cell surface receptors [51].
In conclusion, there are a number of similarities between T2DM and AD with regard to insulin signaling, and it has been suggested that some form of crosstalk between peripheral insulin resistance and the brain may be responsible for the association of the two diseases—for instance, inflammatory cytokines [52].
Possible crosstalk between type 2 diabetes mellitus and dementia/Alzheimer’s disease: chronic inflammation
Overweight/Obese T2DM patients show increased levels of pro-inflammatory cytokines and markers, e.g., interleukin (IL)-1β, IL-6, TNF-α—not only in adipose tissue, but also in blood [53–55]. These molecules are also increased in brain slices, spinal fluid, and in the blood of dementia/AD patients [56–59]. Pro-inflammatory signaling has been proven to promote peripheral insulin resistance in diabetes [60, 61] and to impair neuronal insulin signaling as well [39, 62]. The question arises, whether pro-inflammatory molecules, produced in peripheral tissues and released to the blood, can pass the BBB to exacerbate neuronal insulin resistance. Interestingly, diabetes and hypercholesterolemia increased BBB permeability in a pig model [63]. Takeda et al. [64] have reviewed the role of BBB in the progression of AD extensively. An overlap between the pathogenesis of AD and VD is evident. The BBB can become leaky due to damage from an accumulation of Aβ along brain–blood vessels and/or vascular inflammation/oxidative stress [65]. Other studies underline the assumption of an increased brain influx of pro-inflammatory molecules in diabetes. Sonnen et al. [66] found increased IL-6 levels in AD brains of diabetic subjects compared with AD brains of non-diabetic subjects post mortem. Alterations in BBB permeability allow for peripherally produced cytokines to cross the BBB more easily [67] and it may be that the “first hit” needed to kick off the feed-forward loop for the development of brain insulin resistance which is intensified by peripheral chronic inflammation.
Possible crosstalk between type 2 diabetes mellitus and dementia/Alzheimer’s disease: oxidative stress, mitochondrial dysfunction, and advanced glycation end products and their receptors
Oxidative stress is an imbalance between the presence of ROS and antioxidative capacity. In type 2 diabetes, several mechanisms can lead to excessive generation of ROS, e.g., the autoxidation of glucose or increased mitochondrial ROS production due to the excess supply of substrates (e.g., lipids) in skeletal muscle [68, 69]. ROS can damage lipids, proteins, or DNA and thus contribute to the progression of secondary complications in diabetes [70]. Markers of oxidative stress are also increased in AD brains and are present in spinal fluid even before dementia manifests [71, 72]. It has been suggested that an abnormal stimulation of excitatory N-methyl-d-aspartate receptors by Aβ oligomers is most responsible for brain ROS production in the context of calcium-related mitochondrial dysfunction [73]. Because Aβ-induced oxidative stress is blocked by insulin, brain insulin resistance seems to be directly linked to brain oxidative stress [73]. ROS can activate the JNK signaling pathway [74] leading to insulin resistance [75]. ROS can also induce mitochondrial dysfunction, and thus sustain a vicious circle in which mitochondrial dysfunction further increases ROS levels [76]. Mitochondrial dysfunction is a common condition of T2DM and AD [77]. Among others, mitochondrial dysfunction has also been found to be a factor in AD pathophysiology. ROS-induced damages can potentiate when increasing ROS levels overwhelm antioxidative capacity, and promote cell death [78].
The question is whether ROS in diabetes can contribute to the progression of dementia/AD. What we know is that oxidative stress is one of the key triggers in changing BBB permeability resulting in an increased influx of pro-inflammatory molecules, such as TNF-α, into the brain [79].
The generation of advanced glycation end products (AGEs) can also contribute to oxidative stress. AGEs are modifications of proteins or lipids that become glycated and oxidized [80]. AGEs bind to cell surface receptors for AGEs (RAGEs). AGE–RAGE interaction leads to ROS production via NADPH oxidase system which in turn activates the Ras-MAPK pathway and ultimately, nuclear factor kappa-light-chain-enhancer of activated B cell (NFκB) proteins which further contribute to inflammation [35, 81].
RAGE protein content is up-regulated when AGE ligands accumulate and RAGE levels are thus increased in T2DM [49, 82, 83]. It is known that RAGE can mediate transcytosis of Aβ into the brain [82]. APP is also expressed in several tissues other than the brain and soluble Aβ is detectable in blood [84]. RAGE-mediated Aβ influx results in increased IL-6 and TNF-α concentrations in brain [82]. RAGE is not only found in the BBB but also in neurons, astrocytes, and microglia, where it directly contributes to inflammatory responses in AD [85, 86]. It can be assumed that this mechanism presents yet another path from T2DM to AD [85].
Possible crosstalk between type 2 diabetes mellitus and dementia/Alzheimer’s disease: dyslipidemia and increased ceramide levels
Ceramides constitute a family of sphingolipids composed of a fatty acid and sphingosine, and serve a dual function as element of biological membranes, but also as molecules involved in cellular signaling [87]. Saturated fatty acids can induce the formation of sphingolipids [88]. Furthermore, insulin resistance in T2DM dysregulates lipid metabolism resulting in increased ceramide generation (e.g., in liver) [89].
Ceramides can interfere with insulin signaling by increasing pro-inflammatory cytokines thereby exacerbating insulin resistance [90]. Peripherally generated ceramides can cross the BBB and exert their pro-inflammatory and pro-diabetic effects in the brain, triggering the various cascades involved in the neurodegenerative process by inducing brain insulin resistance [90]. Ceramides can also change the environment of lipid rafts (membrane microdomains) favoring stress-related signaling and Aβ generation [77].
Studies in rats found an increase in circulating ceramides when feeding the animals a high-fat diet, and a mild neurodegenerative phenotype [91]. In a later experiment by the de la Monte Group, an intraperitoneal injection of ceramides induced inhibition of IRS-1 and Akt signaling in the brain and impaired neurocognitive function [92].
The neurotoxic potential of ceramides is thus another factor worth consideration in the study of AD as a metabolic disease.
Possible crosstalk between type 2 diabetes mellitus and dementia/Alzheimer’s disease: reduced Aβ clearance
We know that Aβ is usually moved from the brain at the BBB through a number of transport proteins. Lipoprotein receptor-related protein (LRP)-1 transports Aβ from the brain in order for Aβ to be released in blood and further processed in the liver [93]. It is known that Aβ inhibits its own clearance by reducing LRP-1 expression in the microvasculature and furthermore resists efflux by aggregation, as the transporters are most specific for the monomeric form of Aβ [85, 94].
Since it has been shown that insulin induces LRP-1 translocation to the plasma membrane in hepatic cells and facilitates LRP-1-mediated hepatic Aβ uptake, it is conceivable that Aβ clearance fails in type 2 diabetes due to attenuation of the insulin signaling response, thus favoring Aβ accumulation in the brain [95]. Additionally, there is some evidence that oxidative stress also plays a role in Aβ clearance, because AD patients show oxidative damage to LRP-1 [85].
Possible crosstalk between type 2 diabetes mellitus and dementia/Alzheimer’s disease: low testosterone levels
The assumption is that low testosterone levels are a further common link between T2DM and AD. On the one hand, low testosterone levels have been found in T2DM men in comparison with age-matched controls without diabetes [96], and testosterone therapy has been shown to improve insulin resistance and glycemic control as well as total blood cholesterol in hypogonadal diabetic men [97]. However, a recent meta-analysis did not indicate significant improvements in diabetes features in T2DM patients following the routine use of testosterone treatment [98].
On the other hand, low levels of testosterone have been proven a risk factor for the development of AD in men [99, 100]. Although positive effects of testosterone replacement therapy on brain glucose uptake have been demonstrated in case studies involving elderly patients with AD [101], further research is necessary to entirely clarify the role of testosterone deficiency in the progression of AD.
Beneficial effects of exercise training in type 2 diabetes mellitus to prevent dementia/Alzheimer’s disease
The beneficial effects of exercise training to prevent and treat T2DM-associated metabolic changes and vascular complications are well known. Physical exercise has been shown to reduce hyperglycemia and insulin resistance [102] and lead to a significant reduction in HbA1c [103, 104]. As the incidence of complications arising from diabetes is directly related to HbA1c, a reduction in HbA1c also reduces the incidence of vascular complications, myocardial infarction, and deaths related to diabetes [105].
Regular physical exercise could also prevent the development and progression of dementia/AD in T2DM by various mechanisms (Fig. 1b) which are discussed in detail below.
Physical training can improve endothelial function, blood pressure regulation, and might increase brain capillarization thus fighting vascular dementia
Endothelial dysfunction can predict cardiovascular events and mortality [106, 107]. Exercise has positive effects on vascular endothelial function by increasing the bioavailability of NO and restoring vasodilation response to shear stress. Possible mechanisms include transiently increased shear stress through higher blood velocity during exercise inducing endothelial gene expression, and a training-induced increase in anti-inflammatory/antioxidative molecules [108]. Maiorana et al. [109] found an improved endothelial function in T2DM patients (n = 16) following circuit training (combined cycling/walking and strength training) with submaximal intensities/loads [70–85 % maximum (peak) heart rate (HRmax), 55–65 % maximum voluntary contraction, 3 times per week, 60 min each session] for 8 weeks. Okada et al. [110] also found improvements in the endothelial function in T2DM patients (n = 21) following a combined training [submaximal intensity aerobic dance (20 min), cycling (20 min), and strength–endurance training (20 min) with submaximal loads (training heart rate was determined according to Karvonen’s formula with k = 0.6, strength training intensity not precisely specified, 3–5 times per week, 60 min each session + 10 min warm-up + 5 min cool-down] for 3 months. Cohen et al. [111] demonstrated that strength training with submaximal loads [50–85 % 1-repetition maximum (1-RM)] alone can also improve endothelial function in T2DM patients (n = 29, 2–3 times per week, 45 min each session + 5 min warm-up + 5 min cool-down, for 14 months).
Physical training also has a demonstrable effect on blood pressure (BP). The basis of exercise training-induced BP reduction appears to be a decrease in systemic vascular resistance, involving alterations to the sympathetic nervous system and the renin–angiotensin–aldosterone system, according to a 2005 meta-analysis of 72 trials. Studies have shown significant reductions in circulating norepinephrine and renin levels concomitant with BP reduction after submaximal endurance exercise-based interventions lasting at least 4 weeks [112]. In a meta-analysis involving 42 studies with submaximal intensity endurance training or strength training interventions with submaximal loads (for at least 8 weeks), the positive effect of regular physical activity in optimizing BP in type 2 diabetic patients is evident [113].
Intriguingly, there is also evidence from animal studies with rats that regular exercise directly enhances cerebral angiogenesis by promoting the growth of new capillaries in the motor cortex [114] and that it can increase total brain capillary volume [115]. Another study has demonstrated a direct effect of regular exercise in a rat model after occlusion of the medial cerebral artery (MCA): trained rats showed significantly reduced stroke damage and less neurological deficits [116]. The latter observation is consistent with an improved collateralization of the microvascular network in the MCA territory, as neighboring unoccluded vessels could have helped supply oxygen to the protected regions.
Based on the research outline above, it is clear that any exercise training-associated reduction in vascular complications should also lead to a reduction in the occurrence of VD. The most recent meta-analysis comprising five human longitudinal studies concludes that exercise training indeed is a preventive factor for VD, as a significant association between physical activity and a reduced risk of developing VD has been evaluated among more than 10,000 individuals [117].
In addition to these cardiovascular benefits of physical exercise and their overlap with brain health, the effects of exercise training on other links between T2DM and dementia/AD are also of considerable interest in dementia/AD research to create guidelines for the prevention of neurodegeneration or to develop medication that can mimic beneficial training effects.
Physical training can attenuate inflammatory state and oxidative stress
While acute intense exercise triggers a spike in inflammatory cells/molecules like leukocytes or C-reactive protein (CRP), repeated bouts of submaximal intensity exercise induce adaptive mechanisms that can counteract inflammation in the long term [118–120]. These changes are measureable as reductions in the inflammatory mediators CRP, IL-6, or TNF-α, while anti-inflammatory cytokines IL-4 and IL-10 increase [102, 121]. As we have outlined above, TNF-α in the brain can induce JNK-mediated brain insulin resistance and probably plays a key role in the crosstalk between T2DM and dementia/AD.
Numerous studies involving T2DM subjects have demonstrated that regular physical exercise can attenuate circulating TNF-α in T2DM patients (Table 1). However, some studies did not ascertain reductions in TNF-α levels [Balducci et al. [123]: n = 20, submaximal intensity walking/running or cycling, 70–80 % maximum (peak) oxygen uptake (VO2max), twice a week, 60 min each session, for 12 months; Giannopoulou et al. [125]: n = 11, submaximal intensity walking, 65–70 % VO2max, 3–4 times per week, 60 min each session, for 14 weeks; Kadoglou [126]: n = 30, submaximal intensity walking/running or cycling, 50–75 % VO2max, 4 times per week, 45 min each session + 10 min warm-up + 5 min cool-down + calisthenics, for 6 months]. The question of which training is most effective for T2DM patients in this regard cannot be clearly answered. Further studies are needed involving more subjects. However, Abd El-Kader [122] has shown that both endurance and strength training can potentially reduce inflammatory state (TNF-α) in T2DM patients.
Physical activity is also known to reduce the incidence of ROS and to enhance the antioxidative defense [102, 121]. In skeletal muscle in particular, repeated moderate intensity exercise improves antioxidative capacity by up-regulating endogenous antioxidative molecules [127, 128]. Cells respond to the stressful stimulus of exercise by activating pathways to abolish it—in this case, increasing protein expression of enzymes such as superoxide dismutase, glutathione peroxidase, or antioxidative peroxiredoxins. A likely mediator of this response are NFκB proteins, which can activate gene expression of antioxidative proteins and show enhanced DNA binding around 2 h after acute exercise [129, 130].
A large body of studies exists demonstrating that regular exercise can reduce markers of oxidative stress in the blood of T2DM patients (Table 1), while Kurban et al. [135] did not find significant changes in oxidative stress in T2DM patients (n = 30) following a submaximal intensity walking program (intensity not precisely specified, 3 times per week, 30 min each session + 10 min warm-up + 10 min cool-down) for 3 months. Reductions in oxidative stress could prevent the activation of redox-sensitive stress-induced signaling pathways, which are continuously activated under chronic inflammation in T2DM, and prevent alterations to the permeability of the BBB, thus curbing brain inflammation.
There is evidence from animal studies with rats that immobilization worsens memory performance by promoting oxidative damage to macromolecules in the hippocampus and that regular exercise can reduce oxidative damage and improves memory [136]. Other researchers have explored the antioxidative potency of different exercise protocols in the rodent hippocampus, finding a dose-dependent reduction in oxidative damage when submaximal intensity exercise frequency was increased from two to five times per week [137].
The literature shows that regular exercise also improves the function of the mitochondria in the mouse hippocampus, protecting against an age-associated decline in mitochondrial function. Hippocampal tissue studied in murine models of senescence showed increased mitochondrial function as measured by oxidative phosphorylation complex content and reduced oxidative stress when animals performed regular wheel running [138]. The adaptations were driven by the sirtuin-1/AMP-activated protein kinase pathway through the regulation of proliferator-activated receptor γ coactivator (PGC)-1α, the master controller of mitochondrial biogenesis [138]. Thus, regular exercise could also be useful in maintaining brain mitochondrial health and efficiency.
Physical training can improve the lipid profile and reduce ceramide levels
As outlined above, ceramides can disturb insulin signaling and favor Aβ generation in the brain.
Physical exercise has been shown to reduce ceramide levels and to improve the overall lipid profile [139, 140], which can serve to reduce the detrimental impact of lipotoxic mediators.
In a study by Dubé et al. [139] the authors showed that submaximal intensity walking or cycling (50–70 % VO2max, 4–5 times per week, 45 min each session) for 16 weeks, decreased intramyocellular ceramides in patients with impaired fasting glucose or impaired glucose tolerance (n = 8).
Kasumov et al. [140] have recently demonstrated that ceramide levels in the blood of T2DM patients can be reduced through physical training (Table 1).
Physical training might improve Aβ clearance
Up to this day, nothing is known about the influence of physical exercise on Aβ clearance in type 2 diabetic patients. However, there are some indices from animal studies that regular physical exercise and can improve Aβ clearance. Lin et al. [141] demonstrated that exercise training can increase LRP-1 levels in the amygdala and hippocampus of AD transgenic mice and that regular exercise reduces the levels of soluble Aβ in both brain regions. How far lifestyle interventions (exercise, diet) can influence LRP-1 levels and Aβ clearance in diabetic patients, requires further research. However, this might be an interesting therapeutic approach to prevent/delay the onset of dementia/AD in T2DM patients.
Physical training can increase testosterone levels
As mentioned above, low testosterone levels have been assumed to be a common risk factor for the development of T2DM and AD. Basal testosterone levels have shown to usually not change or even decrease in men and women following endurance training, but can be increased after strength training interventions [142]. Daly et al. [143] found that a 12-month strength training program with submaximal loads (Table 1) increases total testosterone levels in older T2DM patients, while a 16-week strength training program with submaximal loads (50–80 % 1-RM, twice a week, 45–60 min each session) did not lead to increases in the testosterone levels of older T2DM subjects (n = 9) in a study by Ibanez et al. [144]. It can be speculated that more frequent strength training sessions as opposed to less frequent ones can increase testosterone levels and/or that a longer training duration is necessary to induce changes in basal testosterone levels.
Physical training can induce neurogenesis and improve memory
Aside from counteracting numerous deleterious forces of T2DM that can affect brain health, exercise training also promotes brain health by enhancing the action of trophic factors in the brain as has been illuminated in excellent reviews elsewhere [145–147]. One of the most prominent mediators is the brain-derived neurotrophic factor (BDNF). BDNF is involved in neurogenesis, neuroprotection, and synaptogenesis [148]. It also promotes cell survival by inducing anti-apoptotic proteins and inhibits pro-apoptotic factors [149]. BDNF is expressed throughout the CNS, but also in skeletal muscle [150]. It has been shown to be depleted in AD patients and in a multitude of AD animal models, and exerts neuroprotective effects when administered in animal models of neurodegeneration [151].
It has been reviewed that acute endurance exercise can temporarily elevate BDNF concentrations in blood (2–3-fold) while strength exercises possibly cannot [147] and that physical training slightly increases or does not increase basal BDNF levels in healthy subjects [147, 152]. However, much more research is necessary on the impact of exercise and training on BDNF levels in patients with metabolic syndrome/diabetes and/or dementia/AD. It has further been demonstrated that brain produces and releases BDNF during acute exercise and contributes to increased circulating BDNF levels, while muscle-derived BDNF appears not to be released into circulation [150, 153, 154]. However, exercise-induced transiently increased BDNF levels seem to be important stimuli for the induction of neurogenesis in the hippocampus [155, 156]. Among BDNF, exercise may also stimulate neurogenesis via other trophic factors, such as IGF-1 and vascular endothelial growth factor (for a review, see [157]). While the time course of exercise-induced neurogenesis has not been directly verified in humans, size increases in both hippocampi could be induced by a 1-year submaximal intensity walking program [50-75 % heart rate reserve (HRR), frequency not given, 10–40 min each session + 5 min warm-up + 5 min cool-down] in 60 older adults in addition to improved memory function [158].
A further study suggests that a significant increase in hippocampus size in addition to improvements in memory performance is attainable even in elderly patients already suffering from probable MCI [159]. Compared with the hippocampal size of a control group (which performed balance and tone training, n = 11), hippocampal size was increased in the MCI patients (n = 10) following a 6-month submaximal intensity endurance training program (40–80 % HRR, twice a week, 40 min each session + 10 min warm-up + 10 min cool-down), but not in the MCI patients (n = 8) following a strength training program [Borg’s rating of perceived exertion (6–20) 13–15, same frequency and duration as in the endurance training program]. While the human data need to be considered with caution due to the overall small cohort sizes, a beneficial effect of regular exercise on neurogenesis can be assumed.
Training recommendations
It can be deduced from the training studies included in this review that both conventional endurance as well as strength training with submaximal intensities/submaximal loads or a combination of both types of training can have positive effects on dementia/AD-related biomarkers in T2DM patients (Table 1). Most of the training programs are in line with the general recommendations of the American College of Sports Medicine and the American Diabetes Association for physical activity of T2DM patients [160]: moderate–vigorous training should be performed at least 3 days per week and with a minimum of 150 min weekly. Patients with long-term complications from diabetes and/or cardiovascular abnormalities should be screened and seek physician approval before participating in any training programs.
Conclusions
Epidemiological studies provide evidence of a strong association between diabetes and dementia/AD and call for a closer look at the molecular links between the diseases. In this review, a theoretical framework on the crosstalk between diabetes and neurodegeneration was presented, which can guide future research and approaches to therapy. Regular physical exercise should be emphasized as a part of prevention programs for diabetic patients who are at increased risk of developing neurodegenerative diseases considering that exercise programs have been demonstrated to attenuate diabetes-related dementia/AD risk factors.
References
J. Li, Y.H. Shao, Y.P. Gong, Y.H. Lu, Y. Liu, C.L. Li, Diabetes mellitus and dementia—a systematic review and meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 18, 1778–1789 (2014)
M. Baumgart, H.M. Snyder, M.C. Carrillo, S. Fazio, H. Kim, H. Johns, Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimers Dement. 11, 718–726 (2015)
P. Palta, A.L. Schneider, G.J. Biessels, P. Touradji, F. Hill-Briggs, Magnitude of cognitive dysfunction in adults with type 2 diabetes: a meta-analysis of six cognitive domains and the most frequently reported neuropsychological tests within domains. J. Int. Neuropsychol. Soc. 20, 278–291 (2014)
S. Sadanand, R. Balachandar, S. Bharath, Memory and executive functions in persons with type 2 diabetes: a meta-analysis. Diabetes Metab. Res. Rev. 32, 132–142 (2016)
C. Vincent, P.A. Hall, Executive function in adults with type 2 diabetes: a meta-analytic review. Psychosom. Med. 77, 631–642 (2015)
C. Moran, T.G. Phan, J. Chen, L. Blizzard, R. Beare, A. Venn, G. Münch, A.G. Wood, J. Forbes, T.M. Greenaway, S. Pearson, V. Srikanth, Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care 36, 4036–4042 (2013)
C. Cooper, A. Sommerlad, C.G. Lyketsos, G. Livingston, Modifiable predictors of dementia in mild cognitive impairment: a systematic review and meta-analysis. Am. J. Psychiatry 172, 323–334 (2015)
R.O. Roberts, D.S. Knopman, Y.E. Geda, R.H. Cha, V.S. Pankratz, L. Baertlein, B.F. Boeve, E.G. Tangalos, R.J. Ivnik, M.M. Mielke, R.C. Petersen, Association of diabetes with amnestic and nonamnestic mild cognitive impairment. Alzheimers Dement. 10, 18–26 (2014)
X.F. Meng, J.T. Yu, H.F. Wang, M.S. Tan, C. Wang, C.C. Tan, L. Tan, Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimers Dis. 42, 1295–1310 (2014)
J.Q. Li, L. Tan, H.F. Wang, M.S. Tan, L. Tan, W. Xu, Q.F. Zhao, J. Wang, T. Jiang, J.T. Yu, Risk factors for predicting progression from mild cognitive impairment to Alzheimer’s disease: a systematic review and meta-analysis of cohort studies. J. Neurol. Neurosurg. Psychiatry. 87, 476–484 (2015)
K. Gudala, D. Bansal, F. Schifano, A. Bhansali, Diabetes mellitus and risk of dementia: a meta-analysis of prospective observational studies. J. Diabetes Investig. 4, 640–650 (2013)
S. Norton, F.E. Matthews, D.E. Barnes, K. Yaffe, C. Brayne, Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 13, 788–794 (2014)
J.T. O’Brien, A. Thomas, Vascular dementia. Lancet 386, 1698–1706 (2015)
C. Zhiyou, W. Chuanling, H. Wenbo, T. Hanjun, T. Zhengang, X. Ming, Y. Liang-Jun, Cerebral small vessel disease and Alzheimer’s disease. Clin. Interv. Aging 10, 1695–1704 (2015)
P. Rajendran, T. Rengarajan, J. Thangavel, Y. Nishigaki, D. Sakthisekaran, G. Sethi, I. Nishigaki, The vascular endothelium and human diseases. Int. J. Biol. Sci. 9, 1057–1069 (2013)
X.-L. Tan, Y.-Q. Xue, T. Ma, X. Wang, J.J. Li, L. Lan, K.U. Malik, M.P. McDonald, A.M. Dopico, F.-F. Liao, Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol. Neurodegener. 7, 273 (2015)
R.H. Swerdlow, Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiol. Aging 28, 1465–1480 (2007)
S.M. de la Monte, J.R. Wands, Alzheimer’s disease is type 3 diabetes—evidence reviewed. J. Diabetes Sci. Technol. 2, 1101–1113 (2008)
J. Attems, K.A. Jellinger, The overlap between vascular disease and Alzheimer’s disease—lessons from pathology. BMC Med. 11, 206 (2014)
F. Paneni, J.A. Beckman, M.A. Creager, F. Cosentino, Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur. Heart J. 34, 2436–2443 (2013)
M.A. Creager, T.F. Luscher, F. Cosentino, J.A. Beckman, Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 108, 1527–1532 (2003)
P. Geraldes, G.L. King, Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 106, 1319–1331 (2010)
A.L. Vinik, T. Erbas, T.S. Park, R. Nolan, G.L. Pittenger, Platelet dysfunction in type 2 diabetes. Diabetes Care 24, 1476–1485 (2001)
M. Lee, J.L. Saver, A. Towfighi, J. Chow, B. Ovbiagele, Efficacy of fibrates for cardiovascular risk reduction in persons with atherogenic dyslipidemia: a meta-analysis. Atherosclerosis 217, 492–498 (2011)
Z.Y. Li, P. Wang, C.Y. Miao, Adipokines in inflammation, insulin resistance and cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 38, 888–896 (2011)
A.C. Montezano, M. Dulak-Lis, S. Tsiropoulou, A. Harvey, A.M. Briones, R.M. Touyz, Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. Can. J. Cardiol. 31, 631–641 (2015)
L.P. van der Heide, G.M. Ramakers, M.P. Schmidt, Insulin signaling in the central nervous system: learning to survive. Prog. Neurobiol. 79, 2005–2021 (2006)
F.T. Boyd, D.W. Clarke, T.F. Muther, M.K. Raizada, Insulin receptors and insulin modulation of norepinephrine uptake in neuronal cultures from rat brain. J. Biol. Chem. 260, 15880–15884 (1985)
E.D. Martín, A. Sánchez-Perez, J.L. Trejo, J.A. Martin-Aldana, M. Cano-Jaimez, S. Pons, C. Acosta Umanzor, L. Menes, M.F. White, D.J. Burks, IRS-2 deficiency impairs NMDA receptor-dependent long-term potentiation. Cereb. Cortex 22, 1717–1727 (2012)
D.A. Costello, M. Claret, H. Al-Qassab, F. Plattner, E.E. Irvine, A.l. Choudhury, K.P. Giese, D.J. Withers, P. Pedarzani, Brain deletion of insulin receptor substrate 2 disrupts hippocampal synaptic plasticity and metaplasticity. PLoS One 7, e31124 (2012)
E.M. Bingham, D. Hopkins, D. Smith, A. Pernet, W. Hallett, L. Reed, P.K. Marsden, S.A. Amiel, The role of insulin in human brain glucose metabolism: an 18-fluoro-deoxyglucose positron emission tomography study. Diabetes 51, 3384–3390 (2002)
B. Cholerton, L.D. Baker, S. Craft, Insulin, cognition and dementia. Eur. J. Pharmacol. 719, 170–179 (2013)
G. Bedse, F. Di Domenico, G. Serviddio, T. Cassano, Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front. Neurosci. 16, 204 (2015)
J.M. Duarte, Metabolic alterations associated to brain dysfunction in diabetes. Aging Dis. 6, 304–321 (2015)
R. Sandhir, S. Gupta, Molecular and biochemical trajectories from diabetes to Alzheimer’s disease: a critical appraisal. World J. Diabetes 6, 1223–1242 (2015)
K. Talbot, H. Wang, H. Kazi, L. Han, K.P. Bakshi, A. Stucky, R.L. Fuino, K.R. Kawaguchi, A.J. Samoyedny, R.S. Wilson, Z. Arvanitakis, J.A. Schneider, B.A. Wolf, D.A. Bennett, J.Q. Trojanowski, S.E. Arnold, Demonstrated brain insulin resistance in AD patients is associated with IGF-1 resistance, IRS-1 dysregulation and cognitive decline. J. Clin. Investig. 122, 1316–1338 (2012)
K.D. Copps, M.F. White, Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55, 2565–2582 (2012)
L. Mosconi, Glucose metabolism in normal aging and Alzheimer’s disease: methodological and physiological considerations for PET studies. Clin. Transl. Imaging 1, 217–233 (2013)
T.R. Bomfim, L. Forny-Germano, L.B. Sathler, J. Brito-Moreira, J.C. Houzel, H. Decker, M.A. Silverman, H. Kazi, H.M. Melo, P.L. McClean, C. Holscher, S.E. Arnold, K. Talbot, W.L. Klein, D.P. Munoz, S.T. Ferreira, F.G. De Felice, An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J. Clin. Investig. 122, 1339–1353 (2012)
J. Freiherr, M. Hallschmid, W.H. Frey II, Y.F. Brünner, C.D. Chapman, C. Hölscher, S. Craft, F.G. De Felice, C. Benedict, Intranasal insulin as a treatment for Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs 27, 505–514 (2013)
C.R. Weston, R.J. Davis, The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149 (2007)
S. Chakrabarti, V.K. Khemka, A. Banerjee, G. Chatterjee, A. Ganguly, A. Biswas, Metabolic risk factors of sporadic Alzheimer’s disease: implications in the pathology, pathogenesis, and treatment. Aging Dis. 6, 282–299 (2015)
G. Pandini, V. Pace, A. Copani, S. Squatrito, D. Milardi, R. Vigneri, Insulin has multiple antiamyloidogenic effects on human neuronal cells. Endocrinology 154, 375–387 (2013)
H.W. Querfurth, F.M. LaFarla, Alzheimer’s disease. NEJM 362, 329–344 (2010)
A. Serrano-Pozo, M.P. Frosch, E. Masliah, B.T. Hyman, Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1(1), a006189 (2011)
M. Perluigi, G. Pupo, A. Tramutola, C. Cini, R. Coccia, E. Barone, E. Head, D.A. Butterfield, F. Di Domenico, Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim. Biophys. Acta 1842, 1144–1153 (2014)
W.Q. Zhao, P.N. Lacor, H. Chen, M.P. Lambert, M.J. Quon, G.A. Krafft, W.L. Klein, Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric a{beta}. J. Biol. Chem. 284, 18742–18753 (2009)
A. Perez, L. Morelli, J.C. Cresto, E.M. Castano, Degradation of soluble amyloid β-peptides 1-40, 1-42, and the Dutch variant 1-40q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem. Res. 25, 247–255 (2000)
A. Jayaraman, C.J. Pike, Alzheimer’s disease and type 2 diabetes: multiple mechanisms contribute to interactions. Curr. Diabetes Rep. 14, 476 (2014)
N.M. Ashpole, J.E. Sanders, E.L. Hodges, H. Yan, W.E. Sonntag, Growth hormone, insulin-like growth factor-1 and the aging brain. Exp. Gerontol. 68, 76–81 (2015)
P.N. Lacor, M.C. Buniel, P.W. Furlow, A.S. Clemente, P.T. Velasco, M. Wood, K.L. Viola, W.L. Klein, Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807 (2007)
F.G. De Felice, Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. J. Clin. Investig. 123, 531–539 (2013)
N. Rajkovic, M. Zamaklar, K. Lalic, A. Jotic, L. Lukic, T. Milicic, S. Singh, L. Stosic, N.M. Lalic, Relationship between obesity, adipocytokines and inflammatory markers in type 2 diabetes: relevance for cardiovascular risk prevention. Int. J. Environ. Res. Public Health 11, 4049–4065 (2014)
T. Reinehr, B. Karges, T. Meissner, S. Wiegand, B. Stoffel-Wagner, R.W. Holl, J. Woelfle, Inflammatory markers in obese adolescents with type 2 diabetes and their relationship to hepatokines and adipokines. J. Pediatr. (2016). doi:10.1016/j.jpeds.2016.02.055
J. Spranger, A. Kroke, M. Möhlig, K. Hoffmann, M.M. Bergmann, M. Ristow, H. Boeing, A.F. Pfeiffer, Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC) Potsdam Study. Diabetes 52, 812–817 (2003)
R. Cacabelos, M. Barguero, P. Garcia, X.A. Alvarez, E. Varela de Seijas, Cerebrospinal fluid interleukin-1 beta (IL-1 beta) in Alzheimer’s disease and neurological disorders. Methods Find. Exp. Clin. Pharmacol. 13, 455–458 (1991)
O.V. Forlenza, B.S. Diniz, L.L. Talib, V.A. Mendonca, E.B. Ojopi, W.F. Gattaz, A.L. Teixeira, Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 28, 507–512 (2009)
J.P. Jia, R. Meng, Y.X. Sun, W.J. Sun, X.M. Ji, L.F. Jia, Cerebrospinal fluid tau, abeta1-42 and inflammatory cytokines in patients with Alzheimer’s disease and vascular dementia. Neurosci. Lett. 383, 12–16 (2005)
K. Wada-Isoe, Y. Wakutani, K. Urakami, K. Nakashima, Elevated interleukin-6 levels in cerebrospinal fluid of vascular dementia patients. Acta Neurol. Scand. 110, 124–127 (2004)
M.C. Arkan, A.L. Hevener, F.R. Greten, S. Maeda, Z.W. Li, J.M. Long, A. Wynshaw-Boris, G. Poli, J. Olefsky, M. Karin, IKK-beta links inflammation to obesity induced-insulin-resistance. Nat. Med. 11, 191–198 (2005)
B. de Roos, V. Rungapamestry, K. Ross, G. Rucklidge, M. Reid, G. Duncan, G. Horgan, S. Toomey, J. Browne, C.E. Loscher, K.H. Mills, H.M. Roche, Attenuation of inflammation and cellular stress-related pathways maintains insulin sensitivity in obese type I interleukin-1 receptor knockout mice on a high fat diet. Proteomics 9, 3244–3256 (2009)
Q.L. Ma, F. Yang, E.R. Rosario, O.J. Ubeda, W. Beech, D.J. Gant, P.P. Chen, B. Hudspeth, C. Chen, X. Zhao, H.V. Vinters, S.A. Frautschy, G.M. Cole, Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun-N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J. Neurosci. 29, 9078–9089 (2009)
N.K. Acharya, E.C. Levin, P.M. Clifford, M. Han, R. Tourtellotte, D. Chamberlain, M. Pollaro, N.J. Coretti, M.C. Kosciuk, E.P. Nagele, C. Demarshall, T. Freeman, Y. Shi, C. Guan, C.H. Macphee, R.L. Wilensky, R.G. Nagele, Diabetes and hypercholesterolemia increase blood–brain barrier permeability and brain amyloid deposition: beneficial effects of the LpPLA2 inhibitor darapladib. J. Alzheimers Dis. 35, 179–198 (2013)
S. Takeda, N. Sato, R. Morishita, Systemic inflammation, blood–brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer’s disease: relevance to pathogenesis and therapy. Front. Aging Neurosci. 6, 171 (2014)
S. Takeda, N. Sato, K. Ikimura, H. Nishino, H. Rakugi, R. Morishita, Increased blood–brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol. Aging 34, 2064–2070 (2014)
J.A. Sonnen, E.B. Larson, K. Brickell, P.K. Crane, R. Woltjer, T.J. Montine, S. Craft, Different patterns of cerebral injury in dementia with or without diabetes. Arch. Neurol. 66, 315–322 (2009)
I.A.C. Arnoldussen, A.J. Kiliaan, D.R. Gustafson, Obesity and dementia. Eur. Neuropsychopharmacol. 23, 1982–1999 (2014)
E.M.A. Vasconcelos, G.R. Degasperi, H.C.F. de Oliveira, A.E. Vercesi, E.C. de Faria, L.N. Castilho, Reactive oxygen species generation in peripheral blood monocytes and oxidized LDL are increased in hyperlipidemic patients. Clin. Biochem. 42, 1222–1227 (2009)
S.P. Wolff, R.T. Dean, Glucose autoxidation and protein modification. Biochem. J. 245, 243–250 (1987)
X. Du, T. Matsumura, D. Edelstein, L. Rosetti, Z. Zsengeller, C. Szabo, M. Brownlee, Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 112, 1049–1057 (2003)
F. Di Domenico, G. Pupo, E. Giraldo, M.C. Badia, P. Monllor, A. Lloret, M. Eugenia Schinina, A. Giorgi, C. Cini, A. Tramutola, D.A. Butterfield, J. Vina, M. Perluigi, Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic. Biol. Med. 91, 1–9 (2016)
M.A. Pappolla, R.A. Omar, K.S. Kim, N.K. Robakis, Immunohistochemical evidence of oxidative stress in Alzheimer’s disease. Am. J. Pathol. 140, 621–628 (1992)
F.G. De Felice, M.N.N. Viera, T.R. Bomfirm, H. Decker, P.T. Velasco, M.P. Lambert, K.L. Viola, W. Zhao, S.T. Ferreira, W.L. Klein, protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc. Natl Acad. Sci. USA 106, 1971–1976 (2009)
J.V. Cross, D.J. Templeton, Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 381, 675–683 (2004)
V. Aguirre, T. Uchida, L. Yenush, R. Davis, M.F. White, The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser-307. J. Biol. Chem. 275, 9047–9054 (2000)
C. Bonnard, A. Durand, S. Peyrol, E. Chanseaume, M. Chauvin, B. Morio, H. Vidal, J. Rueusset, Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 118, 789–800 (2008)
F.G. De Felice, S.T. Ferreira, Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 63, 2262–2272 (2014)
A. Grimm, K. Friedland, A. Eckert, Mitochondrial dysfunction: the missing link between aging and sporadic Alzheimer’s disease. Biogerontology 17, 281–296 (2016)
G. Schreibelt, G. Kooij, A. Reijerkek, R. van Doorn, S.I. Gringhuis, S. van der Pol, B.B. Weksler, I.A. Romero, P. Couraud, J. Piontek, I.E. Blasig, C.D. Dijkstra, E. Ronken, H.E. de Vries, Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 21, 3666–3676 (2007)
A. Goldin, J.A. Beckman, A.M. Schmidt, M.A. Creager, Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114, 597–605 (2006)
M. Pansuria, H. Xi, L. Li, X.F. Yang, H. Wang, Insulin resistance, metabolic stress, and atherosclerosis. Front. Biosci. (Sch. Ed.) 4, 916–931 (2012)
R. Deane, S. Du Yan, R.K. Submamaryan, B. LaRue, S. Jovanovic, E. Hogg, D. Welch, L. Manness, C. Lin, J. Yu, H. Zhu, J. Ghiso, B. Frangione, A. Stern, A.M. Schmidt, D.L. Armstrong, B. Arnold, B. Liliensiek, P. Nawroth, F. Hofman, M. Kindy, D. Stern, B. Zlokovic, RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat. Med. 9, 907–913 (2003)
S.D. Yan, X. Chen, J. Fu, M. Chen, H. Zhu, A. Roher, T. Slattery, L. Zhao, M. Nagashima, J. Morser, A. Migheli, P. Nawroth, D. Stern, A.M. Schmidt, RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691 (1996)
P. Seubert, C. Vigo-Pelfrey, F. Esch, M. Lee, H. Dovey, D. Davis, S. Sinha, M. Schlossmacher, J. Whaley, C. Swindlehurst, R. McCormack, R. Wolfert, D. Selkoe, I. Lieberburg, D. Schenk, Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 359, 325–327 (1992)
M.A. Erickson, W.A. Banks, Blood–brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 33, 1500–1513 (2013)
L.F. Lue, D.G. Walker, L. Brachova, T.G. Beach, J. Rogers, A.M. Schmidt, D.M. Stern, S.D. Yan, Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp. Neurol. 171, 29–45 (2001)
E.E. Jones, S. Dworski, D. Canals, J. Casas, G. Fabrias, D. Schoenling, T. Levade, C. Denlinger, Y.A. Hannun, J.A. Medin, R.R. Drake, On-tissue localization of ceramides and other sphingolipids by MALDI mass spectrometry imaging. Anal. Chem. 86, 8303–8311 (2014)
W.L. Holland, B.T. Bikman, L.P. Wang, G. Yuguang, K.M. Sargent, S. Bulchand, T.A. Knotts, G. Shui, D.J. Clegg, M.R. Wenk, M.J. Pagliassotti, P.E. Scherer, S.A. Summers, Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 121, 1858–1870 (2011)
W.L. Holland, T.A. Knotts, J.A. Chavez, L.P. Wang, K.L. Hoehn, S.A. Summers, Lipid mediators of insulin clearance. Nutr. Rev. 65, 39–46 (2007)
S.M. de la Monte, Triangulated mal-signaling in Alzheimer’s disease: roles of neurotoxic ceramides, ER stress, and insulin resistance reviewed. J. Alzheimers Dis. 30, 231–249 (2012)
L.E. Lynn-Cook, M. Lawton, M. Tong, E. Silbermann, L. Longato, P. Jiao, P. Mark, J.R. Wands, H. Xu, S.M. de la Monte, Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J. Alzheimers Dis. 16, 715–729 (2009)
S.M. de la Monte, M. Tong, V. Nguyen, M. Setshedi, L. Longato, J.R. Wands, Ceramide-mediated insulin resistance and impairment of cognitive-motor functions. J. Alzheimers Dis. 21, 967–984 (2010)
J. Ghiso, M. Shayo, M. Calero, D. Ng, Y. Tomidokoro, S. Gandy, A. Rostagno, B. Frangione, Systemic catabolism of Alzheimer’s Abeta40 and Abeta42. J. Biol. Chem. 279, 45897–45908 (2004)
S. Ito, S. Ohtsuki, J. Kamiie, Y. Nezu, T. Terasaki, Cerebral clearance of human amyloid-beta peptide (1-40) across the blood–brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J. Neurochem. 103, 2482–2490 (2007)
C. Tamaki, S. Ohtsuki, T. Terasaki, Insulin facilitates the hepatic clearance of plasma amyloid beta-peptide (1-40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol. Pharmacol. 72, 850–855 (2007)
M. Grossmann, M.C. Thomas, S. Panagiotopoulos, K. Sharpe, R.J. Macisaac, S. Clarke, J.D. Zajac, G. Jerums, Low testosterone levels are common and associated with insulin resistance in men with diabetes. J. Clin. Endocrinol. Metab. 93, 1834–1840 (2008)
D. Kapoor, E. Goodwin, K.S. Channer, T.H. Jones, Testosterone replacement therapy improves insulin resistance, glycaemic control, visceral adiposity and hypercholesterolaemia in hypogonadal men with type 2 diabetes. Eur. J. Endocrinol. 154, 899–906 (2006)
M. Grossmann, R. Hoermann, G. Wittert, B.B. Yeap, Effects of testosterone treatment on glucose metabolism and symptoms in men with type 2 diabetes and the metabolic syndrome: a systematic review and meta-analysis of randomized controlled clinical trials. Clin. Endocrinol. (Oxf.) 83, 344–351 (2015)
E. Hogervorst, J. Williams, M. Budge, L. Barnetson, M. Combrinck, A.D. Smith, Serum total testosterone is lower in men with Alzheimer’s disease. Neuroendocrinol. Lett. 22, 163–168 (2001)
E.R. Rosario, L. Chang, F.Z. Stanczyk, C.J. Pike, Age-related testosterone depletion and the development of Alzheimer disease. JAMA 292, 1431–1432 (2004)
R.S. Tan, Testosterone effect on brain metabolism in elderly patients with Alzheimer’s disease: comparing two cases at different disease stages. Aging Clin. Exp. Res. 25, 343–347 (2013)
E. Teixeira-Lemos, S. Nunes, F. Teixeira, F. Reis, Regular physical exercise training assists in preventing type 2 diabetes development: focus on its antioxidant and anti-inflammatory properties. Cardiovasc. Diabetol. 28(10), 12 (2011)
N.G. Boulè, E. Haddad, G.P. Kenny, G.A. Wells, R.J. Sigal, Effects of exercise on glycemic control and body mass in type 2 diabetes mellitus: a metaanalysis of controlled clinical trials. JAMA 286, 1218–1227 (2001)
N.G. Boulè, G.P. Kenny, E. Hadda, G.A. Wells, R.J. Sigal, Meta-analysis of the effect of structured exercise training on cardiorespiratory fitness in Type 2 diabetes mellitus. Diabetologia 46, 1071–1081 (2003)
I.M. Stratton, A.I. Adler, H.A. Neil, D.R. Matthews, S.E. Manley, C.A. Cull, D. Hadden, R.C. Turner, R.R. Holman, Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35) prospective observational study. BMJ 321, 405–412 (2000)
R. Rubinshtein, J.T. Kuvin, M. Soffler, R.J. Lennon, S. Lavi, R.E. Nelson, G.M. Pumper, L.O. Lerman, A. Lerman, Assessment of endothelial function by non-invasive peripheral arterial tonometry predicts late cardiovascular adverse events. Eur. Heart J. 31, 1142–1148 (2010)
J. Yeboah, A.R. Folsom, G.L. Burke, C. Johnson, J.F. Polak, W. Post, J.A. Lima, J.R. Crouse, D.M. Herrington, Perdictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation 120, 502–509 (2009)
B.C.S. Boa, M.C. Souza, R.D. Leite, S.V. da Silva, T.C. Barja-Fidalgo, L.G. Kraemer-Aguiar, E. Bouskela, Chronic aerobic exercise associated to dietary modification improve endothelial function and eNOS expression in high fat fed hamsters. PLoS One 18, e102554 (2014)
A. Maiorana, G. O’Driscoll, C. Cheetham, L. Dembo, K. Stanton, C. Goodman, R. Taylor, D. Green, The effect of combined aerobic and resistance exercise training on vascular function in type 2 diabetes. J. Am. Coll. Cardiol. 38, 850–856 (2001)
S. Okada, A. Hiuge, H. Makino, A. Nagumo, H. Takaki, H. Konishi, Y. Goto, Y. Yoshimasa, Y. Myamoto, Effect of exercise intervention on endothelial function and incidence of cardiovascular disease in patients with type 2 diabetes. J. Atheroscler. Thromb. 17, 828–833 (2010)
N.D. Cohen, D.W. Dunstan, C. Robinson, E. Vulikh, P.Z. Zimmet, J.E. Shaw, Improved endothelial function following a 14-month resistance exercise training program in adults with type 2 diabetes. Diabetes Res. Clin. Pract. 79, 405–411 (2008)
V.A. Cornelissen, R.H. Fagard, Effects of endurance training on blood pressure, blood pressure-regulating mechanisms, and cardiovascular risk factors. Hypertension 46, 667–675 (2005)
Y. Hayashino, J.L. Jackson, N. Fukumori, F. Nakamura, S. Fukuhara, Effects of supervised exercise on lipid profiles and blood pressure control in people with type 2 diabetes mellitus: a meta-analysis of randomized controlled trials. Diabetes Res. Clin. Pract. 98, 349–360 (2012)
R.A. Swain, A.B. Harris, E.C. Wiener, M.V. Dutka, H.D. Morris, B.E. Theien, S. Konda, K. Engberg, P.C. Lauterbur, W.T. Greenough, Prolonged exercise induces angiogenesis and increases cerebral blood volume in primary motor cortex of the rat. Neuroscience 117, 1037–1046 (2003)
S. Wang, L. Chen, L. Zhang, C. Huang, Y. Xiu, F. Wang, C. Zhou, Y. Luo, Q. Xiao, Y. Tang, Effects of long-term exercise on spatial learning, memory ability, and cortical capillaries in aged rats. Med. Sci. Monit. 21, 945–954 (2015)
Y. Ding, J. Li, Y.H. Ding, Q. Lai, J.A. Rafols, J.W. Phillis, J.C. Clark, F.G. Diaz, Exercise pre-conditioning reduces brain damage in ischemic rats that may be associated with regional angiogenesis and cellular overexpression of neurotrophin. Neuroscience 124, 538–591 (2004)
D. Aarsland, F.S. Sardahaee, S. Anderssen, C. Ballard, Alzheimer’s Society Systematic Review Group: Is physical activity a potential preventive factor for vascular dementia? A systematic review. Aging Ment. Health 14, 386–395 (2010)
J. Andersson, J.H. Jansson, G. Hellsten, T.K. Nilsson, G. Hallmans, K. Boman, Effects of heavy endurance physical exercise on inflammation markers in non-athletes. Atherosclerosis 209, 601–605 (2010)
C. Kasapis, P.D. Thompson, The effects of physical activity on serum C-reactive protein and inflammatory markers: a systematic review. J. Am. Coll. Cardiol. 45, 1563–1569 (2005)
C.B. Papini, P.M. Nakamura, L.P. Zorzetto, J.L. Thompson, A.C. Phillips, E. Kokubun, The effect of a community-based, primary health care exercise program on inflammatory biomarkers and hormone levels. Mediat. Inflamm 2014, 185707 (2014)
A.C. McKee, D.H. Daneshvar, V.E. Alvarez, T.D. Stein, The neuropathology of sport. Acta Neuropathol. 127, 29–51 (2014)
S.M. Abd El-Kader, Aerobic versus resistance exercise training in modulation of insulin resistance, adipocytokines and inflammatory cytokine levels in obese type 2 diabetic patients. J. Adv. Res. 2, 179–183 (2011)
S. Balducci, S. Zanuso, A. Nicolucci, F. Fernando, S. Cavallo, P. Cardelli, S. Fallucca, E. Alessi, C. Letizia, A. Jimenez, F. Fallucca, G. Pugliese, Anti-inflammatory effect of exercise training in subjects with type 2 diabetes and the metabolic syndrome is dependent on exercise modalities and independent of weight loss. Nutr. Metab. Cardiovasc. Dis. 20, 608–617 (2010)
M. Straczkowski, I. Kowalska, S. Dzienis-Straczkowska, A. Stepién, E. Skibinska, M. Szelachowska, I. Kinalska, Changes in tumor necrosis factor-alpha system and insulin sensitivity during an exercise training program in obese women with normal and impaired glucose tolerance. Eur. J. Endocrinol. 145, 273–280 (2001)
I. Giannopoulou, B. Fernhall, R. Carhart, R.S. Weinstock, T. Baynard, A. Figueroa, J.A. Kanaley, Effects of diet and/or exercise on the adipocytokine and inflammatory cytokine levels of postmenopausal women with type 2 diabetes. Metabolism 54, 866–875 (2005)
N.P. Kadoglou, F. Iliadis, N. Angelopoulou, D. Perrea, G. Ampaatzidis, C.D. Liapis, M. Alevizos, The anti-inflammatory effects of exercise training in patients with type 2 diabetes mellitus. Eur. J. Cardiovasc. Prev. Rehabil. 14, 837–843 (2007)
A. Nguyen, N. Duquette, M. Mamarbachi, E. Thorin, Epigenetic regulatory effect of exercise on glutathione peroxidase 1 expression in the skeletal muscle of severely dyslipidemic mice. PLoS One 17(10), e0142287 (2016)
Z. Qi, J. He, Y. Zhang, Y. Shao, S. Ding, Exercise training attenuates oxidative stress and decreases p53 protein content in skeletal muscle of type 2 diabetic Goto–Kazaki rats. Free Radic. Biol. Med. 50, 794–800 (2011)
M.C. Gomez-Cabrera, E. Domenech, J. Vina, Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic. Biol. Med. 44, 126–131 (2008)
L.L. Ji, Modulation of skeletal muscle antioxidant defense by exercise: role of redox signaling. Free Radic. Biol. Med. 44, 142–152 (2008)
R.T. Iborra, I.C. Ribeiro, M.Q. Neves, A.M. Charf, S.A. Lottenberg, C.E. Negrao, E.R. Nakandakare, M. Passarelli, Aerobic exercise training improves the role of high-density lipoprotein antioxidant and reduces plasma lipid peroxidation in type 2 diabetes mellitus. Scand. J. Med. Sci. Sports 18, 742–750 (2008)
G. Lazarevic, S. Antic, T. Cvetkovic, V. Djordjevic, P. Vlahovic, V. Stefanovic, Effects of regular exercise on cardiovascular risk factors profile and oxidative stress in obese type 2 diabetic patients in regard to SCORE risk. Acta Cardiol. 63, 485–491 (2008)
C.K. Roberts, D. Won, S. Pruthi, S.S. Lin, R.J. Barnard, Effect of a diet and exercise intervention on oxidative stress, inflammation and monocyte adhesion in diabetic men. Diabetes Res. Clin. Pract. 73, 249–259 (2006)
T.P. Wycherley, G.D. Brinkworth, M. Noakes, J.D. Buckley, P.M. Clifton, Effect of caloric restriction with and without exercise training on oxidative stress and endothelial function in obese subjects with type 2 diabetes. Diabetes Obes. Metab. 10, 1062–1073 (2008)
S. Kurban, I. Mehmetoglu, J.F. Yerlikaya, S. Gönen, S. Erdem, Effect of chronic regular exercise on serum ischemia-modified albumin levels and oxidative stress in type 2 diabetes mellitus. Endocr. Res. 36, 116–123 (2011)
Z. Radak, M. Sasvari, C. Nyakas, T. Kaneko, S. Tahara, H. Ohno, S. Goto, Single bout of exercise eliminates the immobilization-induced oxidative stress in rat brain. Neurochem. Int. 39, 33–38 (2001)
A.E. Speck, C.B. Tromm, B.G. Pozzi, C.S. Paganini, T. Tuon, P.C. Silveira, A.S. Aguiar Jr., R.A. Pinho, The dose-dependent antioxidant effects of physical exercise in the hippocampus of mice. Neurochem. Res. 39, 1496–1501 (2014)
S. Bayod, C. Guzmán-Brambila, S. Sanchez-Roige, J.F. Lalanza, P. Kaliman, D. Ortuno-Sahagun, R.M. Escorihuela, M. Pallàs, Voluntary exercise promotes beneficial anti-aging mechanisms in SAMP8 female brain. J. Mol. Neurosci. 55, 525–532 (2015)
J.J. Dubé, F. Amati, F.G.S. Toledo, M. Stefanovic-Racic, A. Rossi, P. Coen, B.H. Goodpaster, Effect of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 54, 1147–1156 (2011)
T. Kasumov, T.P.J. Solomon, C. Hwang, H. Huang, J.M. Haus, R. Zhang, J.P. Kirwan, Improved insulin sensitivity after exercise training is linked to reduced plasma C14:0 ceramide in obesity and type 2 diabetes. Obesity 23, 1414–1421 (2015)
T.W. Lin, Y.H. Shih, S.J. Chen, C.H. Lien, C.Y. Chang, T.Y. Huang, S.H. Chen, C.J. Jen, Y.M. Kuo, Running exercise delays neurodegeneration in amygdala and hippocampus of Alzheimer’s disease (APP/PS1) transgenic mice. Neurobiol. Learn. Mem. 118, 189–197 (2015)
L.A. Consitt, J.L. Copeland, M.S. Tremblay, Endogenous anabolic hormone responses to endurance versus resistance exercise and training in women. Sports Med. 32, 1–22 (2002)
R.M. Daly, D.W. Dunstan, N. Owen, D. Jolley, J.E. Shaw, P.Z. Zimmet, Does high-intensity resistance training maintain bone mass during moderate weight loss in older overweight adults with type 2 diabetes? Osteoporos. Int. 16, 1703–1712 (2005)
J. Ibanez, M. Izquierdo, I. Arguelles, L. Forga, J.L. Larrión, M. García-Unciti, F. Idoate, E.M. Gorostiaga, Twice-weekly progressive resistance training decreases abdominal fat and improves insulin sensitivity in older men with type 2 diabetes. Diabetes Care 28, 662–667 (2005)
C.W. Cotman, N.C. Berchtold, L.A. Christie, Exercise builds brain health. Key roles of growth factor cascades and inflammation. Trends Neurosci. 30, 464–472 (2007)
C.H. Hillman, K.I. Erickson, A.F. Kramer, Be smart, exercise your heart. Exercise effects on brain and cognition. Nat. Rev. Neurosci. 9, 58–65 (2008)
T. Huang, K.T. Larsen, M. Ried-Larsen, N.C. Møller, L.B. Andersen, The effects of physical activity and exercise on brain-derived neurotrophic factor in healthy humans. A review. Scand. J Med. Sci. Sports 24, 1–10 (2014)
D.K. Binder, H.E. Scharfman, Brain-derived neurotrophic factor. Growth Factors 22, 123–131 (2004)
K. Marosi, M.P. Mattson, BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol. Metab. 25, 89–98 (2014)
B.K. Pedersen, Muscles and their myokines. J. Exp. Biol. 214, 337–346 (2010)
C. Zuccato, E. Cattaneo, Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 5, 311–322 (2009)
K. Knaepen, M. Goekint, E.M. Heyman, R. Meeusen, Neuroplasticity—exercise-induced response of peripheral brain-derived neurotrophic factor: a systematic review of experimental studies in human subjects. Sports Med. 40, 765–801 (2010)
V.B. Matthews, M.-B. Åström, M.H.S. Chan, C.R. Bruce, K.S. Krabbe, O. Prelovsek, T. Åkerström, C. Yfanti, C. Broholm, O.H. Mortensen, M. Penkowa, P. Hojman, A. Zankari, M.J. Watt, H. Bruunsgaard, B.K. Pedersen, M.A. Febbraio, Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 52, 1409–1418 (2009)
P. Rasmussen, P. Brassard, H. Adser, M.V. Pedersen, L. Leick, E. Hart, N.H. Secher, B.K. Pedersen, H. Pilegaard, Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 94, 1062–1069 (2009)
G.S. Griesbach, D.A. Hovda, F. Gomez-Pinilla, Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Res. 1288, 105–115 (2009)
S. Vayman, Z. Ying, F. Gomez-Pinilla, Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur. J. Neurosci. 20, 2580–2590 (2004)
K.A. Intlekofer, C.W. Cotman, Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiol. Dis. 57, 47–55 (2013)
K.I. Erickson, M.W. Voss, R.S. Prakash, C. Basak, A. Szabo, L. Chaddock, J.S. Kim, S. Heo, H. Alves, S.M. White, T.R. Wojcicki, E. Mailey, V.J. Vieira, S.A. Martin, B.D. Pence, J.A. Woods, E. McAuley, A.F. Kramer, Exercise training increases size of hippocampus and improves memory. Proc. Natl Acad. Sci. USA 108, 3017–3022 (2011)
L.F. ten Brinke, N. Bolandzadeh, L.S. Nagamatsu, C.L. Hsu, J.C. Davis, K. Miran-Khan, T. Liu-Ambrose, Aerobic exercise increases hippocampal volume in older women with probable mild cognitive impairment: a 6-month randomised controlled trial. Br. J. Sports Med. 49, 248–254 (2015)
S.R. Colberg, A.L. Albright, B.J. Blissmer, B. Braun, L. Chasan-Taber, B. Fernhall, J.G. Regensteiner, R.R. Rubin, R.J. Sigal, American College of Sports Medicine; American Diabetes Association. Exercise and type 2 diabetes: American College of Sports Medicine and the American Diabetes Association: joint position statement. Exercise and type 2 diabetes. Med. Sci. Sports Exerc. 42, 2282–2303 (2010)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bertram, S., Brixius, K. & Brinkmann, C. Exercise for the diabetic brain: how physical training may help prevent dementia and Alzheimer’s disease in T2DM patients. Endocrine 53, 350–363 (2016). https://doi.org/10.1007/s12020-016-0976-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-016-0976-8