
Abstract
Insulin resistance in skeletal muscle is a key feature in the pathogenesis of type 2 diabetes (T2D) that often manifests early in its development. Pharmaceutical and dietary strategies have targeted insulin resistance to control T2D, and many natural products with excellent pharmacological properties are good candidates for the control or prevention of T2D. Dihydromyricetin (DHM) is a natural flavonol which provides a wide range of health benefits including anti-inflammatory and anti-tumor effects. However, little information is available regarding the effects of DHM on skeletal muscle insulin sensitivity as well as the underlying mechanisms. In the present study, we found that DHM activated insulin signaling and increased glucose uptake in skeletal muscle in vitro and in vivo. The expression of light chain 3, the degradation of sequestosome 1, and the formation of autophagosomes were also upregulated by DHM. DHM-induced insulin sensitivity improvement was significantly abolished in the presence of 3-methyladenine, bafilomycin A1, or Atg5 siRNA in C2C12 myotubes. Furthermore, DHM increased the levels of phosphorylated AMP-activated protein kinase (AMPK), peroxisome proliferator-activated receptor coactivator-1α (PGC-1α), and Sirt3 in skeletal muscle in vitro and in vivo. Autophagy was inhibited in the presence of Sirt3 siRNA in C2C12 myotubes and in skeletal muscles from Sirt3−/− mice. Additionally, PGC-1α or AMPK siRNA transfection attenuated DHM-induced Sirt3 expression, thereby abrogating DHM-induced autophagy in C2C12 myotubes. In conclusion, DHM improved skeletal muscle insulin sensitivity by partially inducing autophagy via activation of the AMPK-PGC-1α-Sirt3 signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes (T2D) is the most common endocrine disorder worldwide. Patients with T2D generally suffer from both reduced insulin secretion and resistance to the actions of insulin. Muscle tissue is the major site for insulin-stimulated glucose uptake in vivo. Accumulating evidence indicates that insulin resistance often begins with decreasing insulin sensitivity in skeletal muscle in T2D. Pharmaceutical and dietary strategies have targeted insulin insufficiencies to control T2D, and many natural products with excellent pharmacological properties are good candidates for the control or prevention of T2D. Dihydromyricetin (DHM) is a natural flavonol from fruits and vegetables and provides a wide range of health benefits including anti-oxidant, anti-inflammatory, and anti-tumor effects [1, 2]. Moreover, several flavonoids with similar chemical structure to DHM, such as quercetin and naringenin, have been shown to improve insulin sensitivity [3, 4]. However, little information is available regarding the effects of DHM on skeletal muscle insulin sensitivity as well as the underlying mechanism.
Autophagy, an evolutionarily conserved catabolic process, has been identified as a homeostasis regulator through removing excessive or unnecessary proteins and damaged or aged organelles [5]. A recent study showed that autophagy deficiency promoted T2D, indicating its protective function. Both basal and glucose-stimulated insulin secretion were significantly decreased in β-cells of Atg7△β−cell mice. And autophagy activity and expression of some key autophagy genes were suppressed in the presence of insulin resistance in hepatocytes [6]. Moreover, it has also been demonstrated that autophagy is required for exercise-induced muscle glucose homeostasis in mice [7]. Additionally, previous studies have found that DHM can suppress many types of cancer cells through autophagy induction [8]. Therefore, in the present study, we investigated whether DHM could improve skeletal muscle insulin sensitivity through autophagy induction.
Silent mating-type information regulation 2 homolog 3 (Sirt3), a member of the sirtuin family of NAD+-dependent deacetylases, plays a key role in T2D through regulation of insulin resistance in skeletal muscle [9]. Notably, Sirt3 expression is significantly decreased in the muscles of mice with insulin-deficient diabetes [10]. Moreover, it has been demonstrated that Sirt3 functions as a downstream target of peroxisome proliferator-activated receptor (PPARγ) coactivator-1α (PGC-1α), which is directly regulated by adenosine monophosphate-activated protein kinase (AMPK) and exerts multiple cellular functions by inducing autophagy, especially mitophagy, through deacetylating mitochondrial proteins [10]. Our previous study highlights that DHM supplementation improves physical performance under acute hypoxic conditions partially by activating AMPK in skeletal muscles [11]. Thus, we hypothesized that DHM could improve skeletal muscle insulin sensitivity by inducing autophagy through the AMPK-PGC-1α-Sirt3 signaling pathway. To verify this hypothesis, we tested the effects of DHM on insulin sensitivity and the role of autophagy in skeletal muscle in vitro and in vivo as well as the potential involvement of the AMPK-PGC-1α-Sirt3 signaling pathway. Our findings showed that DHM improved skeletal muscle insulin sensitivity through the induction of autophagy in vivo and in vitro, and the autophagy was in part mediated by the activation of the AMPK-PGC-1α-Sirt3 signaling pathway.
Methods and materials
Antibodies and reagents
3-Methyladenine (3-MA; M9281), Bafilomycin A1 (BafA1; B1793), Compound C (CC; P5499), Palmitate acid (PA; P5585), antibodies against light chain 3 (LC3; L7543), and minimum essential medium Eagle (MEM; M4526) were purchased from Sigma-Aldrich. 2-[1,2-3H]-Deoxy-d-glucose (NET328A001MC) was purchased from PerkinElmer. A Cell Counting Kit (CCK-8; CK04) was purchased from Dojindo Laboratories. Lipofectamine™ 2000 transfection reagent (11668-019) was purchased from Invitrogen. Fetal bovine serum (FBS; SH30370.03) was purchased from Hyclone Laboratories. Antibodies against p-PRKAA1/2 (p-AMPKα1/2; sc-33524), PRKAA1/2 (AMPKα1/2; sc-74461), P-IRS-1(sc-17199-R) and IRS-1(sc-599), siRNAs for autophagy-related gene 5 (Atg5; sc-41445), PGC-1α (sc-62784), AMPK (sc-45312), and the control (sc-44230) were obtained from Santa Cruz Biotechnology. The GFP-LC3 plasmid was kindly provided by ADDGENE. Antibodies against Atg5 (8540), Akt (9272), p-Akt (9271), and sequestosome 1 (p62; 5114) were obtained from Cell Signaling Technology. DHM (msat-120131108, HPLC ≥98 %) was purchased from Chengdu MUST Bio-Technology Co., Ltd.
Animals and treatments
Male 129/SvJ (wild type, WT) and SIRT3−/− 129/SvJ mice (Jackson Laboratory, Bar Harbor, ME, USA) at 3 months of age were maintained on a standard chow diet (5053 PicoLab diet, Ralston Purina Company, St. Louis, MO). DHM (50 mg/kg/day) was given by gavage once a day for 3 months. At the end of the experiment, all mice were fasted for 12 h and then anesthetized with pentobarbital sodium prior to the surgical procedures. All efforts were made to minimize suffering. Skeletal samples were collected for subsequent measurements. Animals were maintained under a regular 12-h light period at a controlled temperature (22 ± 2 °C) and received chow and water ad libitum. Animal care and treatments were conducted according to established guidelines and protocols approved by the Animal Care and Use Committee of the Third Military Medical University (Chongqing, China).
Insulin tolerance test
All rats with free access to water and food were injected intraperitoneally with insulin (1 IU/kg.BW) and blood glucose was quantified in tail blood samples collected at 0 (prior to insulin administration), 30, 60, 90, and 120 min after the administration of insulin.
Cell culture and treatments
Mouse skeletal muscle C2C12 myoblast cells were maintained in MEM containing 10 % FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified atmosphere with 5 % CO2. For differentiation into myotubes, the medium was replaced with MEM containing 2 % FBS for 5 days. These differentiated C2C12 myotubes were used for the different treatments.
PA preparation
PA was dissolved in 40 mL of 0.1 M NaOH at 70 °C. BSA solution (1 %) was prepared in distilled water. Different concentrations of palmitate were used for treatment of C2C12 myotubes after conjugation with 1 % BSA on a magnetic stirrer set at 40 °C.
Cell viability measurement
The CCK-8 detection kit was used to measure cell viability as described before [12]. Briefly, C2C12 myotubes were seeded in a 96-well microplate (Corning Life Sciences; 3650) at a density of 8000 cells/well and then treated with PA for 16 h at a series of concentrations (0.15, 0.25, 0.5, 0.75, and 1 mM) or pretreated with DHM for 2 h at a series of concentrations (0.1, 0.5, 1, and 5 μM), then myotubes were treated with PA (0.75 mM) for another 16 h. The control group was treated with 0.2 % DMSO. Subsequently, CCK-8 solution (20 μL/well) was added to the wells and the plate was incubated at 37 °C for 2 h. Viable myotubes were counted by absorbance measurements with a monochromator microplate reader (Safire II; Tecan Group Ltd., Männedorf, Switzerland) at a wavelength of 450 nm. The optical density value at 450 nm was reported as the percentage of cell viability in relation to the control group (set as 100 %).
Cell transfection
Control non-targeted siRNA or siRNA against ATG5, PGC-1α, or AMPK and Lipofectamine 2000 were diluted in reduced serum MEM according to the manufacturer’s protocol. The final siRNA concentration was 100 nM and plasmid concentration was 4 μg. C2C12 myotubes were incubated with the transfection mixture for 10 h and then supplemented with fresh medium for an additional 24 h, after which they were exposed to the indicated treatments. The same method was used for transfection of GFP-LC3 plasmids.
Transmission electron microscopy
C2C12 myotubes were fixed in 3 % glutaraldehyde in 0.1 M MOPS buffer (pH 7.0) for 8 h at room temperature, followed by incubation in 3 % glutaraldehyde/1 % paraformaldehyde in 0.1 M MOPS buffer (pH 7.0) for 16 h at 4 °C. Myotubes were then postfixed in 1 % osmium tetroxide for 1 h, embedded in Spurr’s resin, sectioned, double stained with uranyl acetate and lead citrate, and analyzed using a Zeiss EM 10 transmission electron microscope.
3H-2-deoxyglucose uptake assays
At the end of treatment, C2C12 myotubes were washed with glucose-free Krebs–Ringer phosphate HEPES (KRPH) buffer containing 0.1 % fatty acid-free BSA (w/v) followed by incubation in the same buffer for 30 min at 37 °C. C2C12 myotubes were stimulated with insulin (10 nM) for 15 min followed by the addition of 0.5 μCi/well of 3H-2-deoxyglucose in KRPH buffer for 45 min at 37 °C, and then washed with KRPH and lysed using 0.1 % SDS. The lysates were analyzed in a scintillation counter.
Western blotting
The protein samples extracted from C2C12 myotubes or skeletal muscle of high-fat diet-fed rats were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. Membranes were blocked with TBST containing 5 % milk and incubated with the different primary antibodies, as indicated, overnight at 4 °C. The membranes were then incubated with horseradish peroxidase conjugated to secondary antibodies and visualized using an enhanced chemiluminescence system. Densitometric analysis was performed using Scion Image software.
Statistical analysis
Quantitative data are presented as mean ± standard deviation (SD) of three experiments. Statistical analyses were performed with Student’s t test and one-way analysis of variance using SPSS 13.0 statistical software (SPSS Inc., Chicago, IL, USA). A p value <0.05 was considered to be statistically significant and the Tukey–Kramer post hoc test was applied if p < 0.05.
Results
DHM improved skeletal muscle insulin sensitivity in vitro and in vivo
As shown in Fig. 1a–d DHM significantly increased p-IRS-1 and p-AKT expressions as well as glucose uptake in a dose-dependent manner in C2C12 myotubes. As insulin plays an important role in the transport of glucose from the blood into the cells, the insulin-sensitizing potential of DHM was elucidated in Fig. 1e and f. Myotubes were stimulated with or without 100 nM insulin for 20 min after DHM treatment. DHM treatment markedly improved the glucose uptake in C2C12 myotubes both in the presence or absence of insulin stimulation. Moreover, single-DHM treatment stimulated glucose uptake to a similar extent to insulin, which was not significantly different from that in DHM and insulin co-treated C2C12 myotubes. Additionally, we also investigated the effect of DHM on insulin sensitivity in skeletal muscle in mice. As shown in Fig. 1g and h, DHM at 50 mg/kg per day markedly upregulated p-IRS-1 and p-AKT expressions with or without insulin stimulation. Simultaneously, improved insulin sensitivity was also observed in DHM-fed mice as measured by the insulin tolerance test (Fig. 1i). These results suggested that DHM improved skeletal muscle insulin sensitivity in vitro and in vivo.

DHM improved skeletal muscle insulin sensitivity in vitro and in vivo. a Differentiated C2C12 myotubes were pretreated with different concentrations (0.1, 0.5, and 1 μM) of DHM for 2 h. a The expression of indicated proteins was detected by Western blotting. b The bar graphs show the quantification of the indicated proteins. c Differentiated C2C12 myotubes were pretreated with DHM (1 μM) for 2 h and stimulated with or without 100 nM insulin for 20 min. The expression of indicated proteins was detected by Western blotting. d The bar graphs show the quantification of the indicated proteins. e Glucose uptake by C2C12 myotubes was assessed as indicated in the “Methods and Materials” section. f Glucose uptake by C2C12 myotubes was assessed as indicated in the “Methods and Materials” section. Mice were treated with or without 50 mg/kg/day of DHM for 12 weeks then injected intraperitoneally with or without insulin (1 IU/kg BW). g The expression of indicated proteins was detected by Western blotting. h The bar graphs show the quantification of the indicated proteins. i Insulin tolerance was assessed as indicated in the “Methods and Materials” section. Values are presented as mean ± SD. n = 3, a p < 0.05 versus the control group; b p < 0.01 versus the control group; c p < 0.01 versus DHM-treated group. A.U. arbitrary units
DHM improved skeletal muscle insulin sensitivity by inducing autophagy in vitro and in vivo
The effect of DHM on autophagy in C2C12 myotubes was examined by measuring the following two autophagy-related markers via Western blotting: LC3-II and p62. C2C12 myotubes were pretreated with different concentrations (0.1, 0.5, and 1 μM) of DHM for 2 h. DHM dose-dependently increased the expression of LC3-II and the degradation of p62 (Fig. 2a, b). We also assessed the effect of DHM on autophagy in mice. As shown in Fig. 2c and d, DHM treatment markedly upregulated the expression of LC3-II and the degradation of p62 in skeletal muscle tissues compared with the control group.

DHM improved insulin sensitivity by inducing autophagy in skeletal muscle myotubes in vitro and in vivo. a Differentiated C2C12 myotubes were pretreated with different concentrations (0.1, 0.5, and 1 μM) of DHM for 2 h. Total C2C12 cell lysates were immunoblotted with anti-p62, anti-LC3, and anti-β-actin antibodies. b The bar graphs show the quantification of the indicated proteins. c Mice were treated with or without 50 mg/kg/day of DHM for 12 weeks. Muscle tissue lysates were then immunoblotted with the antibodies described in (A). d The bar graphs show the quantification of the indicated proteins. e C2C12 myotubes were pretreated with 3-MA (5 mM) followed by incubation with or without DHM (1 µM) for an additional 2 h. Representative TEM depicting the ultrastructure of myotubes. Arrows indicate autophagosomes. f The bar graphs show the number of autophagosomes. g Representative confocal images of GFP fluorescent puncta in C2C12 myotubes transfected with GFP-LC3 plasmids for 24 h. h The number of GFP-LC3 dots in each cell was counted. i Differentiated C2C12 myotubes were pretreated with 3-MA (5 mM) or BafA1 (10 nM) for 1 h, followed by incubation with or without DHM (1 µM) for an additional 2 h. Total cell lysates were detected by Western blot. j The bar graphs show the quantification of the indicated proteins. k C2C12 myotubes were transfected with Atg5 siRNA and then treated with or without DHM (1 µM) for 2 h. The expressions of the indicated proteins were detected by Western blot. l The bar graphs show the quantification of the indicated proteins. m Myotubes treated as described in (i). The uptake of glucose by C2C12 myotubes was measured as indicated in the “Methods and Materials” section. n Myotubes treated as described in (h). The uptake of glucose by C2C12 myotubes was measured by 3H-2-deoxyglucose uptake assays. o Myotubes were treated as described in (i), then stimulated with 100 nM insulin for 20 min. The expressions of indicated proteins were detected by Western blotting. p The bar graphs show the quantification of the indicated proteins. Values are presented as mean ± SD. n = 3, a p < 0.05 versus the control group; b p < 0.01 versus the control group; c p < 0.01 versus DHM-treated group; * p < 0.01 versus the insulin-stimulated control group; # p < 0.01 versus DHM and insulin co-treated group. A.U. arbitrary units
To further confirm DHM-induced autophagy in skeletal muscles, transmission electron microscopy (TEM), the most valid method for both qualitative and quantitative analysis of autophagy, [25] showed that more vacuoles were present in DHM-treated myotubes, but the number decreased when the cells were pretreated with 3-MA (Fig. 2e, f). Moreover, myotubes were transfected with GFP-LC3 plasmid and visualized by fluorescence microscopy. DHM significantly increased the number of autophagic structures (GFP-LC3 dots), which then decreased with pre-treatment of 3-MA (Fig. 2g, h). These results suggested that DHM treatment upregulated autophagy in myotubes.
Additionally, when autophagy was inhibited with 3-MA (5 mM), a well-known inhibitor of the early autophagy stages and BafA1 (10 nM), which inhibits the acidification of organelles and, subsequently, autophagosome–lysosome fusion as well as transfection of ATG5 siRNA, DHM-induced improvement of insulin sensitivity was notably attenuated in myotubes (Fig. 2i–n). We also find that that when autophagy was inhibited by 3-MA and BafA1, DHM-induced improvement of insulin sensitivity was abolished in myotubes under insulin stimulation (Fig. 2o, p). These results indicated that autophagy was required for the favorable effect of DHM on skeletal muscle insulin sensitivity.
DHM-induced autophagy through regulating Sirt3 in skeletal muscle in vitro and in vivo
C2C12 myotubes were pretreated with different concentrations (0.1, 0.5, and 1 μM) of DHM for 2 h. DHM dose-dependently increased the expression of sirt3 (Fig. 3a, b). To elucidate the role of Sirt3 in the metabolic effect of DHM, we used siRNA-mediated knockdown (KD) in cultured C2C12 myotubes. As shown in Fig. 3c, the Western blotting result revealed an over 90 % decrease of Sirt3 protein in the Sirt3 KD myotubes. Suppression of Sirt3 significantly abrogated DHM-induced upregulation of LC3-II and down-regulation of P62 (Fig. 3c, d), thereby decreasing the induction of p-IRS-1, p-Akt expressions, and glucose uptake in response to DHM in C2C12 myotubes (Fig. 3e and g).

DHM-induced autophagy by regulating Sirt3 in C2C12 myotubes. a Differentiated C2C12 myotubes were pretreated with different concentrations (0.1, 0.5, and 1 μM) of DHM for 2 h. Total lysates detected by Western blot. b The bar graphs show the quantification of the indicated proteins. c C2C12 myotubes were transiently transfected with Sirt3 siRNA or a scrambled control. Then, cells were treated with or without DHM (1 µM) for 2 h, the expressions of indicated proteins were detected by Western blot. d The bar graphs show the quantification of the indicated proteins. e C2C12 myotubes were treated as described in (c), then stimulated with or without 100 nM insulin for 20 min. Total lysates were detected by Western blot. f The bar graphs show the quantification of the indicated proteins. g C2C12 myotubes treated as described in (e), the uptake of glucose by C2C12 myotubes was measured by 3H-2-deoxyglucose uptake assays. Values are presented as mean ± SD. n = 3, a p < 0.05 versus the control group; b p < 0.01 versus the control group; c p < 0.01 versus DHM-treated group; * p < 0.01 versus the insulin-stimulated control group; # p < 0.01 versus DHM and insulin co-treated group. A.U. arbitrary units
We next assessed the effects of DHM on Sirt3 in mice. As expected, DHM increased the expression of Sirt3 in vivo (Fig. 4a, b). To further clarify the role of Sirt3 in the effect of DHM on skeletal muscle insulin sensitivity, Sirt3−/− mice were used. As shown in Fig. 4c–f, the effect of DHM on autophagy and insulin signaling was abolished in skeletal muscle of Sirt3−/− mice. Moreover, DHM could not significantly improve insulin sensitivity in Sirt3−/− mice as measured by the insulin tolerance test (Fig. 4g). Altogether, these data indicated that DHM-induced autophagy and improved insulin sensitivity in a Sirt3-dependent manner in skeletal muscle in vitro and in vivo.

DHM-induced autophagy by regulating Sirt3 in skeletal muscle in mice. WT or Sirt3−/− mice were treated with or without 50 mg/kg/day of DHM for 12 weeks then injected intraperitoneally with or without insulin (1 IU/kg BW). a Total tissue lysates from skeletal muscle of WT mice were immunoblotted with anti-sirt3 and anti-β-actin antibodies. b The bar graphs show the quantification of the indicated proteins. c Muscle tissue lysates from WT and Sirt3−/− mice were measured by Western blot. d The bar graphs show the quantification of the indicated proteins. e The expressions of indicated proteins were detected by Western blot. f The bar graphs show the quantification of the indicated proteins. g Insulin tolerance was assessed as indicated in the “Methods and materials” section. Values are presented as mean ± SD. n = 3, a p < 0.05 versus the control group; b p < 0.01 versus the control group; c p < 0.01 versus DHM-treated group; * p < 0.01 versus the insulin-stimulated control group; # p < 0.01 versus DHM and insulin co-treated group. A.U. arbitrary units
DHM regulated Sirt3 through the AMPK-PGC-1α signaling pathway in skeletal muscle in vitro and in vivo
It has been found that Sirt3 functions as a downstream target of PGC-1α, which is directly regulated by AMPK. Thus, we investigated the possible role of the AMPK-PGC-1α signaling pathway as a moderator for the effect of DHM on Sirt3 regulation. As shown in Fig. 5a and b, DHM significantly upregulated p-AMPK and PGC-1α expressions in a dose-dependent manner in C2C12 myotubes. Similar results were observed in skeletal muscle tissues in mice (Fig. 5c, d). These results indicate that DHM activated the AMPK-PGC-1α signaling pathways in vitro and in vivo. The correlation between the activation of AMPK signaling and Sirt3 induction by DHM was further evaluated using the AMPK siRNA and PGC-1α siRNA. As shown in Fig. 5e and f, PGC-1α siRNA abrogated DHM-induced Sirt3 expression. In addition, AMPK siRNA decreased the expression of PGC-1α and subsequent Sirt3 expression induced by DHM in C2C12 myotubes (Fig. 5g, h). These results suggested that the activation of the AMPK/PGC-1α signaling pathway played a key role in the effect of DHM on Sirt3 regulation.

DHM-induced Sirt3 expression through activating AMPK/PGC-1α in skeletal muscle myotubes in vitro and in vivo. a Differentiated C2C12 myotubes were pretreated with different concentrations (0.1, 0.5, and 1 μM) of DHM for 2 h. The expressions of indicated proteins were determined by Western blot. b The bar graphs show the quantification of the indicated proteins. c Mice were treated with or without 50 mg/kg.d of DHM for 12 weeks. Muscle tissue lysates were measured by Western blot. d The bar graphs show the quantification of the indicated proteins. Differentiated C2C12 myotubes were transfected with PGC-1α (e) or AMPK (g) siRNA as described in the “Methods and Materials” section before the addition of DHM (1 μM) for another 2 h. And the expression of indicated proteins was detected by Western blot. f and h The bar graphs show the quantification of the indicated proteins. Values are presented as mean ± SD. n = 3, a p < 0.05 versus the control group; b p < 0.01 versus the control group; c p < 0.01 versus DHM-treated group. A.U. arbitrary units
DHM improved PA-induced insulin resistance in skeletal muscle myotubes
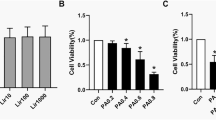
We also investigated the effect of DHM on skeletal muscle insulin sensitivity in a PA-induced insulin resistance model in C2C12 myotubes. To induce insulin resistance in muscle cells, we treated differentiated C2C12 myotubes with different concentrations of PA (0.15, 0.25, 0.5, 0.75, and 1 mM) for 16 h. As shown in Fig. 6a, lower than 1 mM of PA did not markedly suppress the cell viability of C2C12 myotubes, which was in line with the published data before [13–16]. And our previous results found that treatment with 0.75 mM PA for 16 h could markedly reduce insulin sensitivity in C2C12 myotubes (data not shown). Thus, 0.75 mM PA and 16 h were selected for the subsequent experiments. As expected, DHM and PA co-treatment also had no significant effect on the cell viability of C2C12 myotubes. Moreover, DHM pre-treatment markedly reversed the PA-induced decrease in glucose uptake and p-IRS-1 and p-AKT expressions in C2C12 myotubes under insulin-stimulated condition (Fig. 6b–d).

DHM improved PA-induced insulin resistance by inducing autophagy in skeletal muscle myotubes. a C2C12 myotubes were treated with PA for 16 h at a series of concentrations (0.15, 0.25, 0.5, 0.75, and 1 mM) or pretreated with DHM for 2 h at a series of concentrations (0.1, 0.5, 1, and 5 μM), then myotubes were treated with PA (0.75 mM) for another 16 h. The cell viability was measured by CCK-8 detection kit. b The glucose uptake by C2C12 myotubes was assessed as indicated in the “Methods and Materials” section. c Differentiated C2C12 myotubes were pretreated with 1 μM of DHM for 2 h, then myotubes were treated with PA (0.75 mM) for another 16 h. Finally, myotubes were stimulated with or without 100 nM insulin for 20 min. The expressions of indicated proteins were detected by Western blot. (d The bar graphs show the quantification of the indicated proteins. e Representative TEM images depicting the ultrastructure of myotubes. Arrows indicate autophagosomes. f The bar graphs show the number of autophagosomes. g Representative confocal images of GFP fluorescent puncta in C2C12 myotubes transfected with GFP-LC3 plasmids for 24 h. h The number of GFP-LC3 dots in each cell was counted. i Total C2C12 cell lysates were immunoblotted with anti-p62, anti-LC3, and anti-β-actin antibodies. j The bar graphs show the quantification of the indicated proteins. k The expressions of AMPK, p-AMPK, PGC-1α, sirt3, and β-actin were determined by Western blot. l The bar graphs show the quantification of the indicated proteins. Values are presented as mean ± SD, n = 3. b p < 0.01 versus vehicle-treated control group; c p < 0.01 versus PA-treated group; * p < 0.01 versus the insulin-stimulated control group; # p < 0.01 versus PA and the insulin-stimulated group. A.U. arbitrary units
Assessment of autophagosome formation by TEM revealed that DHM induced the formation of vacuoles in PA-treated C2C12 myotubes (Fig. 6e and f). As shown in Fig. 6g and h, C2C12 myotubes co-treated with DHM (1 µM) and PA (0.75 mM) displayed a significant increase in the number of autophagic structures (GFP-LC3 dots) compared with C2C12 myotubes treated with PA alone. LC3-II and p62 immunoblotting showed significant induction of autophagy in DHM-treated myotubes (Fig. 6i, j). Furthermore, we saw concomitant upregulation of p-AMPK, PGC-1α, and Sirt3 expressions in PA-treated C2C12 myotubes (Fig. 6k, l). Collectively, our results strongly suggested that the inverse association between DHM intake and skeletal muscle insulin sensitivity is a result of DHM’s pro-autophagic action through the AMPK-PGC-1α-Sirt3 signaling pathway.
Discussion
In the current study, we found that DHM could improve insulin sensitivity in skeletal muscle in vitro and in vivo. To the best of our knowledge, this is the first report demonstrating that DHM could improve skeletal muscle insulin sensitivity and that DHM could be developed as a novel therapeutic agent for the prevention and treatment of T2D.
Autophagy is a lysosome-dependent mechanism by which dysfunctional or damaged intracellular organelles are broken down and recycled through the lysosomes. As a putative adaptive catabolic process, autophagy plays an important role in many human diseases [17]. Despite the protective actions of autophagy in several human pathologies, the role of autophagy in T2D, especially in insulin resistance, is poorly understood. Recently, several studies have shown that autophagy is activated in insulin-resistant pancreatic β-cells [18, 19]. In contrast, hepatic autophagy was reported to be suppressed in insulin resistance [20, 21] and autophagy was also inhibited in adipose tissue from high-fat diet (HFD)-induced insulin-resistant mice [22]. Furthermore, autophagy in the muscle, hypothalamus, and kidney of HFD-induced insulin-resistant mice was decreased [23–25]. Overall, although the mechanism for reduced autophagy is not clear, autophagy seems to be suppressed in the insulin-resistant state. However, Kim and his colleagues found that autophagy deficiency in skeletal muscle could attenuate HFD-induced insulin resistance, probably owing to leanness [26]. These results are in contrast to the previous suggestion that autophagy deficiency would worsen glucose and lipid metabolism, indicating that the role of autophagy in insulin resistance might be more complicated than expected. Herein, we found that autophagy was triggered in skeletal muscle after treatment with DHM in vitro and in vivo, and inhibition of autophagy significantly abolished the DHM-induced improvement of insulin sensitivity. These results seemed different from those of Kim’s, but they were still an important complement to the previous work, which provided new insight into the potential mechanism underlying the anti-insulin resistance effects of flavonoids including DHM, in which autophagy may play an important role.
Furthermore, we examined the potential mechanisms of DHM-induced autophagy in this study. The sirtuins are a family of nicotinamide adenine dinucleotide (NAD +)-dependent protein deacetylases [27] and regulate multiple cellular processes including metabolic homeostasis. Sirt3 is a mitochondrial sirtuin and regulates energy homeostasis and oxidative metabolism, as well as oxidative stress and cellular injury. [28, 29] Recently, a study showed that expression of Sirt3 decreases in a diabetic mouse model. And KD of Sirt3 in the C2C12 cell line results in impaired insulin signaling [30]. In the present study, we found that DHM increased Sirt3 expression in skeletal muscle in vitro and in vivo. In addition, Sirt3 siRNA and the Sirt3−/− genotype markedly abrogated DHM-induced autophagy, thereby down-regulating insulin signaling and glucose uptake induced by DHM. These results indicated that Sirt3 plays a critical role in DHM-induced autophagy in skeletal muscle.
In addition to Sirt3, the mechanism of DHM-induced Sirt3 activation was also investigated. Studies have demonstrated that Sirt3 functions as a downstream target of PGC-1α, which is directly regulated by AMPK. AMPK is a key player in the regulation of energy metabolism and is considered to be a therapeutic target for T2D because of its reported anti-insulin resistance properties. Fasting or nutrient excess may trigger the activation or inhibition of AMPK, respectively, which leads to alteration of PGC-1α activity. A previous report suggested that PGC-1α stimulated mouse Sirt3 activity in both muscle cells and hepatocytes, indicating that PGC-1α acts as an endogenous regulator of Sirt3 [31]. Our group has found that DHM improved the physical performance under acute hypoxic conditions by partially activating AMPK in skeletal muscle [11]. We therefore postulated that DHM regulated Sirt3 expression through activation of the AMPK-PGC-1α axis. As expected, our results showed that DHM triggered AMPK phosphorylation, which is required for PGC-1α expression in skeletal muscle in vitro and in vivo. And PGC-1α or AMPK siRNA significantly decreased the expression of Sirt3 induced by DHM. These findings suggested that the AMPK-PGC-1α signaling pathway could be a key route in DHM-induced Sirt3 regulation.
In conclusion, the results of the present study indicated that DHM improves skeletal muscle insulin sensitivity by partially inducing autophagy via regulating the AMPK-PGC1α-Sirt3 signaling pathway (Fig. 7). Our findings suggested a potential role for DHM in the prevention and treatment of T2D as well as other insulin resistance-related metabolic diseases such as nonalcoholic fatty liver disease.

Proposed mechanisms of DHM-induced autophagy in skeletal muscle myotubes. DHM improves insulin sensitivity by inducing autophagy via the proposed pathways: DHM activates AMPK, thereby increasing PGC-1α and Sirt3 expressions, and ultimately inducing autophagy
Abbreviations
- 3-MA:
-
3-Methyladenine
- Akt:
-
Protein kinase B
- AMPK:
-
AMP-activated protein kinase
- Atg:
-
Autophagy-related gene
- BafA1:
-
Bafilomycin A1
- DHM:
-
Dihydromyricetin
- IRS-1:
-
Insulin receptor substrate-1
- LC3:
-
Light chain 3
- PGC-1α:
-
Peroxisome proliferator-activated receptor coactivator-1α
- siRNA:
-
Small interfering RNA
- T2D:
-
Type 2 diabetes
- Sirt3:
-
Silent mating-type information regulation 2 homolog 3
References
Y.S. Zhang, Z.X. Ning, S.Z. Yang, H. Wu, Yao Xue Xue Bao 38(4), 241–244 (2003)
J.M. Harnly, R.F. Doherty, G.R. Beecher, J.M. Holden, D.B. Haytowitz, S. Bhagwat, S. Gebhardt, J. Agric. Food Chem. 54(26), 9966–9977 (2006)
P. Pu, D.M. Gao, S. Mohamed, J. Chen, J. Zhang, X.Y. Zhou, N.J. Zhou, J. Xie, H. Jiang, Arch. Biochem. Biophys. 518(1), 61–70 (2012)
X. Dai, Y. Ding, Z. Zhang, X. Cai, L. Bao, Y. Li, Biol. Pharm. Bull. 36(5), 788–795 (2013)
Y. Kondo, S. Kondo, Autophagy 2(2), 85–90 (2006)
H.S. Jung, K.W. Chung, K.J. Won, J. Kim, M. Komatsu, K. Tanaka, Y.H. Nguyen, T.M. Kang, K.H. Yoon, J.W. Kim, Y.T. Jeong, M.S. Han, M.K. Lee, K.W. Kim, J. Shin, M.S. Lee, Cell Metab. 8(4), 318–324 (2008)
C. He, M.C. Bassik, V. Moresi, K. Sun, Y. Wei, Z. Zou, Z. An, J. Loh, J. Fisher, Q. Sun, S. Korsmeyer, M. Packer, H.I. May, J.A. Hill, H.W. Virgin, C. Gilpin, G. Xiao, R. Bassel-Duby, P.E. Scherer, B. Levine, Nature 481(7382), 511–515 (2012)
Y. Zhou, X. Liang, H. Chang, F. Shu, Y. Wu, T. Zhang, Y. Fu, Q. Zhang, J.D. Zhu, M. Mi, Cancer Sci. 105(10), 1279–1287 (2014)
E. Jing, B. Emanuelli, M.D. Hirschey, J. Boucher, K.Y. Lee, D. Lombard, E.M. Verdin, C.R. Kahn, Proc Natl Acad Sci USA 108(35), 14608–14613 (2011)
V.K. Yechoor, M.E. Patti, K. Ueki, P.G. Laustsen, R. Saccone, R. Rauniyar, C.R. Kahn, Proc Natl Acad Sci USA 101(47), 16525–16530 (2004)
D. Zou, K. Chen, P. Liu, H. Chang, J. Zhu, M. Mi. Med. Sci. Sports Exerc. (2014)
M.L. Chen, L. Yi, X. Jin, X.Y. Liang, Y. Zhou, T. Zhang, Q. Xie, X. Zhou, H. Chang, Y.J. Fu, J.D. Zhu, Q.Y. Zhang, M.T. Mi, Autophagy 9(12), 2033–2045 (2013)
K. Sawada, K. Kawabata, T. Yamashita, K. Kawasaki, N. Yamamoto, H. Ashida, Lipids Health Dis 11, 36 (2012)
D.J. Powell, S. Turban, A. Gray, E. Hajduch, H.S. Hundal, Biochem. J. 382(Pt 2), 619–629 (2004)
J.J. Senn, J. Biol. Chem. 281(37), 26865–26875 (2006)
C. Schmitz-Peiffer, D.L. Craig, T.J. Biden, J. Biol. Chem. 274(34), 24202–24210 (1999)
B. Levine, G. Kroemer, Cell 132(1), 27–42 (2008)
H.S. Jung, K.W. Chung, K.J. Won, J. Kim, M. Komatsu, K. Tanaka, Y.H. Nguyen, T.M. Kang, K.H. Yoon, J.W. Kim, Y.T. Jeong, M.S. Han, M.K. Lee, K.W. Kim, J. Shin, M.S. Lee, Cell Metab. 8(4), 318–324 (2008)
C. Ebato, T. Uchida, M. Arakawa, M. Komatsu, T. Ueno, K. Komiya, K. Azuma, T. Hirose, K. Tanaka, E. Kominami, R. Kawamori, Y. Fujitani, H. Watada, Cell Metab. 8(4), 325–332 (2008)
H.Y. Liu, J. Han, S.Y. Cao, T. Hong, D. Zhuo, J. Shi, Z. Liu, W. Cao, J. Biol. Chem. 284(45), 31484–31492 (2009)
R. Singh, S. Kaushik, Y. Wang, Y. Xiang, I. Novak, M. Komatsu, K. Tanaka, A.M. Cuervo, M.J. Czaja, Nature 458(7242), 1131–1135 (2009)
T. Yoshizaki, C. Kusunoki, M. Kondo, M. Yasuda, S. Kume, K. Morino, O. Sekine, S. Ugi, T. Uzu, Y. Nishio, A. Kashiwagi, H. Maegawa, Biochem. Biophys. Res. Commun. 417(1), 352–357 (2012)
C. He, M.C. Bassik, V. Moresi, K. Sun, Y. Wei, Z. Zou, Z. An, J. Loh, J. Fisher, Q. Sun, S. Korsmeyer, M. Packer, H.I. May, J.A. Hill, H.W. Virgin, C. Gilpin, G. Xiao, R. Bassel-Duby, P.E. Scherer, B. Levine, Nature 481(7382), 511–515 (2012)
Q. Meng, D. Cai, J. Biol. Chem. 286(37), 32324–32332 (2011)
S. Kume, M.C. Thomas, D. Koya, Diabetes 61(1), 23–29 (2012)
K.H. Kim, Y.T. Jeong, H. Oh, S.H. Kim, J.M. Cho, Y.N. Kim, S.S. Kim, D.H. Kim, K.Y. Hur, H.K. Kim, T. Ko, J. Han, H.L. Kim, J. Kim, S.H. Back, M. Komatsu, H. Chen, D.C. Chan, M. Konishi, N. Itoh, C.S. Choi, M.S. Lee, Nat. Med. 19(1), 83–92 (2013)
S. Imai, F.B. Johnson, R.A. Marciniak, M. McVey, P.U. Park, L. Guarente, Cold Spring Harb. Symp. Quant. Biol. 65, 297–302 (2000)
P. Onyango, I. Celic, J.M. McCaffery, J.D. Boeke, A.P. Feinberg, Proc Natl Acad Sci USA 99(21), 13653–13658 (2002)
B. Schwer, B.J. North, R.A. Frye, M. Ott, E. Verdin, J. Cell Biol. 158(4), 647–657 (2002)
E. Jing, B. Emanuelli, M.D. Hirschey, J. Boucher, K.Y. Lee, D. Lombard, E.M. Verdin, C.R. Kahn, Proc Natl Acad Sci USA 108(35), 14608–14613 (2011)
S.J. Park, F. Ahmad, A. Philp, K. Baar, T. Williams, H. Luo, H. Ke, H. Rehmann, R. Taussig, A.L. Brown, M.K. Kim, M.A. Beaven, A.B. Burgin, V. Manganiello, J.H. Chung, Cell 148(3), 421–433 (2012)
Acknowledgments
This work was supported by the research Grant from National Natural Science Foundation of China (81372975) and the special fund of Chongqing key laboratory (CSTC) for Nutrition and Food Safety.
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Shi, L., Zhang, T., Zhou, Y. et al. Dihydromyricetin improves skeletal muscle insulin sensitivity by inducing autophagy via the AMPK-PGC-1α-Sirt3 signaling pathway. Endocrine 50, 378–389 (2015). https://doi.org/10.1007/s12020-015-0599-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-015-0599-5