
Abstract
Lipid peroxidation and mitochondrial damage impair insulin sensitivity in skeletal muscle. Sirtuin-1 (SIRT1) protects mitochondria and activates under energy restriction. Dapagliflozin (Dapa) is an antihyperglycaemic agent that belongs to the sodium-glucose cotransporter-2 (SGLT2) inhibitors. Evidence shows that Dapa can induce nutrient deprivation effects, providing additional metabolic benefits. This study investigates whether Dapa can trigger nutrient deprivation to activate SIRT1 and enhance insulin sensitivity in skeletal muscle. We treated diet-induced obese (DIO) mice with Dapa and measured metabolic parameters, lipid accumulation, oxidative stress, mitochondrial function, and glucose utilization in skeletal muscle. β-hydroxybutyric acid (β-HB) was intervened in C2C12 myotubes. The role of SIRT1 was verified by RNA interference. We found that Dapa treatment induced nutrient deprivation state and reduced lipid deposition and oxidative stress, improved mitochondrial function and glucose tolerance in skeletal muscle. The same positive effects were observed after β-HB intervening for C2C12 myotubes, and the promoting effects on glucose utilization were diminished by SIRT1 RNA interference. Thus, Dapa promotes a nutrient deprivation state and enhances skeletal muscle insulin sensitivity via SIRT1 activation. In this study, we identified a novel hypoglycemic mechanism of Dapa and the potential mechanistic targets.
Similar content being viewed by others
Introduction
Insulin resistance is defined as a reduced response of target tissues (such as the skeletal muscle, liver, and adipocytes) to insulin1; it is a key indicator for the risk of type 2 diabetes (T2DM) and plays a major role in the pathogenesis of multiple diseases, such as obesity, metabolic syndrome, polycystic ovary syndrome, non-alcoholic fatty liver disease, and cardiovascular disease2,3. Moreover, recent research found that insulin resistance can increase the risk of cancer and even the mortality4. Skeletal muscle is the main site for glucose disposition, accounting for approximately 80% of postprandial glucose uptake in humans5. Therefore, skeletal muscle insulin resistance is the initiating or primary defect that is evident decades before β-cell failure and overt hyperglycemia develops6,7.
Over the past two decades, the prevalence of T2DM has increased at an alarming speed8. The rapid increase in the prevalence of obesity in all age groups is one of the main causes of the exponential growth in the T2DM population9. In the obese state, accumulation of intramyocellular lipids (IMCL) is associated with the development of skeletal muscle insulin resistance, prediabetes and T2DM10. Increased IMCL storage results in the accumulation and dysregulation of detrimental lipid intermediates such as diacylglycerols (DAGs) and ceramides11. Lipid accumulation and peroxidation can damage mitochondrial biosynthesis and function. Thus, myocyte lipotoxicity and mitochondrial damage may be central to skeletal muscle insulin resistance and T2DM12.
Diet-induced obesity (DIO) in rodents has been shown to repress the expression of Proliferator-activated receptor-gamma co-activator-1α (PGC-1α), Nuclear Respiratory Factor-1 (NRF-1), and Transcription Factor A, Mitochondrial (TFAM), which are key molecules in mitochondrial biogenesis13. Additionally, because mtDNA is close to reactive oxygen species (ROS) production sites, it is more susceptible to lipid peroxidation damage, reducing mitochondrial content in muscle14. In obesity, skeletal muscle exhibits a smaller mitochondrial network, shorter mitochondrial length, and increased fragmentation, likely due to impaired mitochondrial dynamics15. Intramuscular lipid accumulation can also lead to mitochondrial dysfunction, disrupting multiple steps in the mitochondrial electron transport chain (ETC) and reducing ATP production16. Furthermore, the Silent information regulator 1 (SIRT1)-PGC-1α signaling pathway, which directly regulates mitochondrial biogenesis and function, is impaired in obesity17. Damaged mitochondrial synthesis and function greatly attenuate skeletal muscle glucose disposal ability, thereby disrupting glucose homeostasis18,19.
SIRT1 is a highly conserved NAD-dependent deacetylase and cellular regulator regarded as a sensor of cellular energy and metabolism20. As a key energy regulator, SIRT1 is activated under energy-restricted conditions such as hunger and fasting, whereas its expression is reduced in nutrient-overabundant conditions such as obesity and T2DM21. It is associated with many molecules related to glucose and lipid metabolism, including Adenosine 5′-monophosphate-activated protein kinase (AMPK), Forkhead box O (FOXO), Peroxisome proliferator-activated receptor γ (PPARγ), and PGC-1α22. The activation of SIRT1 can exert several actions. Firstly, SIRT1 activation inhibits energy storage and promotes lipid utilization. Specifically, SIRT1 inhibits the transcriptional activities of PPARγ and sterol regulatory element-binding protein 1c through deacetylation, suppressing adipocyte differentiation, reducing fat accumulation, and promoting fat mobilization22. Furthermore, activated SIRT1 can reduce phosphorylated-FOXO1 expression and activate FOXO1, thereby inhibiting ROS production and oxidative stress23. Importantly, SIRT1’s deacetylation of PGC-1α is extensively implicated in promoting mitochondrial biogenesis and function, as well as the destruction of damaged or aged mitochondria via mitophagy24. Therefore, promoting or restoring SIRT1 expression in skeletal muscles may be an effective strategy for improving skeletal muscle insulin resistance in obese individuals.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are antidiabetic drugs that lower blood glucose by increasing urinary glucose excretion through inhibiting glucose reabsorption in the renal proximal tubules25. Emerging evidence suggests that SGLT2 inhibitors can provide various metabolic benefits beyond glucose lowering, such as outperformed cardio and renal protective effect, weight and lipid reduction, and improved insulin resistance26, though the mechanisms remain unclear. Recent research suggests that the metabolic benefits of SGLT2 inhibitors may be related to their ability to induce a nutrient deprivation state. Notably, this state does not refer to a single metabolic pathway alteration but rather a series of metabolic shifts that mimic starvation. Specifically, SGLT2 inhibitors induce a state that mimics starvation, which is characterized at a systemic level by calorie loss in the urine, promotion of fat utilization over storage, activation of catabolic pathways, and enhanced hepatic ketogenesis27,28. At the cellular level, this starvation-mimicking state is sensed, triggering the upregulation of nutrient-deprivation signaling and downregulation of nutrient-surplus signaling27. SIRT1 represents a critical molecular response to caloric restriction and has been called the "energy sensor"29. However, current research on the benefits of nutrient deprivation primarily focuses on cardioprotection. Whether the SGLT2 inhibitor dapagliflozin (DAPA) can improve skeletal muscle insulin sensitivity by inducing nutrient deprivation and activating SIRT1 is still unclear.
Results
Dapa treatment induced a nutrient deprivation state and improved glucose tolerance in DIO mice
In our experiments, Dapa showed an evident effect on weight loss in DIO mice at a dosage of 1.5 mg/kg/day (Fig. 1A). In terms of body composition (Fig. 1B), compared with the high-fat diet (HFD) group, Dapa treatment significantly reduced body fat and moderately decreased body free fluid, but did not influence lean mass content. Dapa treatment also significantly reduced serum lipid levels, including triglycerides (TG), total cholesterol (TC), and low-density lipoprotein cholesterol (LDL-C) (Fig. 1C). Furthermore, the average respiratory quotient (RQ) of the natural control group (NC) group was approximately 0.85, consistent with the metabolism of glucose, fat, and protein. The RQ of HFD group was 0.75–0.85, which may be related to excessive fat accumulation leading to increased fat utilization. However, the mean RQ in the HFD + Dapa group was approximately 0.7, consistent with the conversion of metabolic substrates into fat (Fig. 1D). There were no significant differences in energy expenditure (EE) (Fig. 1E) and 24 h food uptake (Fig. 1F) between the HFD and HFD + Dapa groups. Due to the enhanced utilization of fat, our results also showed that Dapa intervention elevated the level of β-Hydroxybutyrate (β-HB) (Fig. 1G). Furthermore, Dapa treatment increased urine glucose concentration (Fig. 1H) but did not influence 24 h urine volume (Fig. 1I).

Dapa treatment induced nutrient deprivation state in diets induced obesity mice models. (A) Body weight changes in mice models. (B) Body composition analyzer measurements of body fat, free body fluid and lean mass. (C) Measurements of serum lipids included cholesterol (TC), triglyceride (TG), low density lipoprotein-cholesterol (LDL-C) and high-density lipoprotein-cholesterol (HDL-C). (D) Measurements of urine glucose concentration. (E) Metabolic cages collected 24 h’ urine from mice. (F–H) Monitor of metabolic parameters included respiratory quotient (RQ), energy expenditure (EE) and food uptake in 24 h period. N = 8 per groups. *p < 0.05, **p < 0.005, ***p < 0.001, §p < 0.0001 vs NC group. #p < 0.05, ##p < 0.005, ###p < 0.001, &p < 0.0001 vs HFD group. Statistical analyses were performed using one-way ANOVA with Tukey’s multiple comparison tests. Data are represented as means ± SEM values.
Dapa treatment alleviated ectopic lipid disposition and oxidative stress in skeletal muscle
Oil Red O staining showed that the marked lipid accumulation in the skeletal muscle tissue of the HFD group was alleviated in the HFD + Dapa group (Fig. 2A). As lipid peroxidation is the main source of ROS and oxidative stress, we evaluated the level of oxidative stress and found that malondialdehyde (MDA), an oxidative stress product, was overproduced in the HFD group, whereas the content of catalase (CAT), an antioxidant enzyme, decreased. These changes were completely reversed by Dapa treatment (Fig. 2B,C). We also investigated the RNA expression of oxidative stress-related factors in the skeletal muscle. We found that nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4), a major NADPH isoform that plays an important role in the redox process, increased significantly in the skeletal muscles of the HFD group, whereas the levels of antioxidant enzymes, including superoxide dismutase (SOD), CAT, and glutathione peroxidase (GSH-PX), decreased. Moreover, Dapa caused marked elevation in the mRNA expression of SOD and CAT, mild upregulation of GSH-PX, and inhibition of the gene expression of NOX4 (Fig. 2D).

Dapa treatment alleviated lipids ectopic disposition and inhibited oxidative stress in skeletal muscle of mice. (A) Oil red O staining detecting lipids accumulation in skeletal muscle sections. (B) Measurement of malondialdehyde (MDA) content in serum of mice. (C) Measurement of catalase (CAT) level in serum of mice. (D) Quantitative Real-Time PCR for Superoxide Dismutase (SOD), CAT and Glutathione-peroxidase (GSH-PX) and NADPH Oxidase 4 (NOX4) in skeletal muscle. Scale bar = 200 μm. N = 3–8 per groups. *p < 0.05, **p < 0.005, ***p < 0.001, §p < 0.0001 vs NC group. #p < 0.05, ##p < 0.005, ###p < 0.001, &p < 0.0001 vs HFD group. Statistical analyses were performed using one-way ANOVA with Tukey’s multiple comparison tests. Data are represented as means ± SEM values.
Dapa promoted mitochondrial biogenesis and protected mitochondrial function in skeletal muscle
Transmission electron microscopy (TEM) revealed a significant decrease in mitochondrial number in the gastrocnemius muscle of the HFD group. Furthermore, the mitochondria were morphologically fragmented, and there was increased deposition of lipid droplets. The extent of these pathological changes was reduced by Dapa (Fig. 3A). In addition, the gene expression of SIRT1, as well as downstream factors (PGC-1α, NRF-1, and TFAM) related to mitochondrial biogenesis, were impaired in HFD group, while Dapa treatment restored the expression levels to those of the NC group (Fig. 3B). In terms of protein expression, Dapa intervention restored SIRT1, PGC-1α, and NRF-1 expression to levels of the NC group, and TFAM expression was even higher than the NC group (Fig. 3C,D).

Dapa treatment prompt mitochondrial biosynthesis and function in skeletal muscle. (A) Transmission electron microscopy (TEM) observed the number of mitochondria in skeletal muscle sections. (B–D) Quantitative Real-Time PCR, Western-Blot analysis and densitometry quantification for mitochondrial biosynthesis-related factors Sirtuin 1(SIRT1), Peroxisome Proliferators-activated Receptor γ Coactivator-1α(PGC-1α), Nuclear Respiratory Factor-1(NRF-1) and Transcription Factor A, Mitochondrial (TFAM). (E–G) Quantitative Real-Time PCR, Western-Blot analysis and densitometry quantification for mitochondrial function related factors ATP Synthase 5Alpha Chain, Mitochondrial (ATP5A), Cytochrome-C (CytC), Optic Atrophy Protein-1 (OPA-1), Mitofusin-1(MFN-1), Mitofusin-2 (MFN-2) and Dynamin-Related Protein-1(DRP-1). Scale bar = 2-5 μm. N = 3–6 per groups. *p < 0.05, **p < 0.005, ***p < 0.001, §p < 0.0001 vs NC group. #p < 0.05, ##p < 0.005, ###p < 0.001, &p < 0.0001 vs HFD group. Statistical analyses were performed using one-way ANOVA with Tukey’s multiple comparison tests. Data are represented as means ± SEM values. Cropped gels and blots are shown, original gels and blots are presented in Supplementary Figs. 1–2.
Oxidative phosphorylation is the most important function of mitochondria; therefore, we examined the gene and protein expression of oxidative respiratory chain-related factors ATP Synthase 5Alpha Chain, Mitochondrial (ATP5A) and Cytochrome-C (CytC), the mitochondrial fission-related factor Dynamin-Related Protein-1 (DRP-1), and the pro-fusion factors Optic Atrophy Protein-1 (OPA-1), Mitofusin-1 (MFN-1), Mitofusin-2 (MFN-2). Our experiments found that the gene expression of these factors was inhibited in HFD group, whereas Dapa treatment significantly increased their expression (Fig. 3E). What’s more, the protein expression of mitochondrial function-related factors was significantly downregulated in the HFD group, Dapa intervention markedly improved this impairment, with no significant difference compared to the NC group (Fig. 3F,G).
Dapa improved glucose tolerance in the DIO mouse model and promoted the expression of glucose disposal-related factors in skeletal muscle
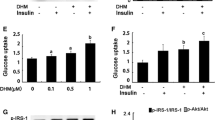
The HFD group showed increased fasting glucose and the 2 h glucose tolerance test was consistent with impaired glucose tolerance. Dapa intervention completely reversed these changes (Fig. 4A–C). The initial step in muscle glucose metabolism involves activation of the glucose transport system, leading to an influx of glucose into insulin target tissues30. Glucose Transporter 4 (GLUT4) is the most important functional molecule in the transport system and is regulated by the upstream factor Phosphoinositide 3 Kinase (PI3K)/Protein Kinase B (AKT)31. Our results showed that the gene expression of GLUT4 was significantly impaired in the HFD group. Dapa treatment significantly improved its expression, even surpassing the levels in the NC group (Fig. 4D). The protein expression of the PI3K/AKT/GLUT4 pathway was also significantly impaired in the HFD group. Dapa treatment enhanced the phosphorylation of PI3K(p-PI3K) and AKT (p-AKT) and upregulated the protein expression of GLUT4 to levels comparable to the NC group (Fig. 4E,F).

Dapa improved glucose tolerance in mice and the expression of glucose disposal-related factors in muscle. (A–C) Measurements of fasting glucose and area under curve (AUC) of 2 h OGTT. (D) PCR for detecting the expression of Glucose Transporter 4 (GLUT4). (E–G) Western-Blot analysis and densitometry quantification for Phosphoinositide 3 Kinase (PI3K), Phospho-PI3K (P-PI3K), Protein Kinase B (AKT), Phospho-AKT (P-AKT) and GLUT4. N = 4–8 per groups. *p < 0.05, **p < 0.005, ***p < 0.001, §p < 0.0001 vs NC group. #p < 0.05, ##p < 0.005, ###p < 0.001, &p < 0.0001 vs HFD group. Statistical analyses were performed using one-way ANOVA with Tukey’s multiple comparison tests. Data are represented as means ± SEM values. Cropped gels and blots are shown, original gels and blots are presented in Supplementary Fig. 3.
β-HB intervention inhibited lipid accumulation and peroxidation in C2C12 myotubes
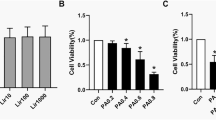
Given the multiple benefits observed from Dapa treatment, we explored whether these effects were related to the nutrient deprivation status. The ketogenesis effect is the most characteristic feature of the state, and β-HB is the most abundant type of ketone. Therefore, we verified the effects of β-HB in vivo. The Cell Counting Kit-8 (CCK-8) assay was used to assess the viability of cells treated with palmitic acid (PA) and β-HB; cell viability decreased significantly when the concentration of PA was over 0.3 mM (Fig. 5A). Cell viability was mildly increased after addition of 2 mM β-HB, however, it tended to decline when at higher concentrations (Fig. 5B). Therefore, we used 0.3 mM and 2 mM for the PA- and β-HB-intervention groups, respectively. In the PA group, lipids accumulated in C2C12 myotubes, while β-HB intervention significantly alleviated the effect (Fig. 5C). In addition, β-HB intervention downgraded the gene expression of NOX4 and promoted the gene expression of the antioxidant-related factors (Fig. 5D).

β-HB had the impact on decreasing lipids accumulation and oxidative stress in C2C12 myotubes. (A, B) Cell Counting Kit-8 (CCK-8) Assay measured the cell viability under PA or β-HB intervention in C2C12 myotubes. (C) Oil red O staining detecting the lipid accumulation situation in C2C12 myotubes. (D) Quantitative Real-Time PCR for Superoxide Dismutase (SOD), Catalase (CAT), Glutathione-peroxidase (GSH-PX) and NADPH Oxidase 4 (NOX4). Scale bar = 100 μm. N = 4–5 per groups. *p < 0.05, **p < 0.005, ***p < 0.001, §p < 0.0001 vs NC group. #p < 0.05, ##p < 0.005, ###p < 0.001, &p < 0.0001 vs PA group. Statistical analyses were performed using one-way ANOVA with Tukey’s multiple comparison tests. Data are represented as means ± SEM values.
β-HB increased mitochondrial numbers and improved mitochondrial function in C2C12 myotubes
Through MitoTracker fluorescence staining of C2C12 myotubes, we clearly observed the number and morphology of mitochondria under live-cell conditions. Our results showed that, compared with the NC group, mitochondrial numbers were significantly decreased after PA intervention, while β-HB intervention restored mitochondrial numbers to the level of NC group (Fig. 6A,B). Consistent with this, the results of western blotting demonstrated that β-HB promoted the expression of factors related to mitochondrial biogenesis in C2C12 myotubes (Fig. 6C,D). Furthermore, we observed a discrepancy in the expression of mitochondrial-function-related factors in the PA group. Interestingly, β-HB totally reversed the impacts of PA intervention on mitochondrial function (Fig. 6E,F).

β-HB intervention improved the mitochondrial biosynthesis and function in vivo. (A, B) Representative images and quantification of Mito-tracker fluorescent staining for detecting mitochondrial numbers in C2C12 myotubes. (C, D) Western-Blot analysis and densitometry quantification for mitochondrial biosynthesis-related factors Sirtuin-1 (SIRT1), Peroxisome Proliferators-activated Receptor γ Coactivator-1α(PGC-1α), Nuclear Respiratory Factor-1 (NRF-1) and Transcription Factor A, Mitochondrial (TFAM). (E, F) Western-Blot analysis and densitometry quantification for mitochondrial function related factors ATP Synthase 5Alpha Chain, Mitochondrial (ATP5A), Cytochrome-C (CytC), Optic Atrophy Protein-1 (OPA-1), Mitofusin-1 (MFN-1), Mitofusin-2 (MFN-2) and Dynamin-Related Protein-1 (DRP-1). Scale bar = 100 μm. N = 3–6 per groups. *p < 0.05, **p < 0.005, §p < 0.0001 vs NC group. #p < 0.05, ##p < 0.005, ###p < 0.001, &p < 0.0001 vs PA group. Statistical analyses were performed using one-way ANOVA with Tukey’s multiple comparison tests. Data are represented as means ± SEM values. Cropped gels and blots are shown, original gels and blots are presented in Supplementary Figs. 4–5.
β-HB activated the PI3K/AKT/GLUT4 pathway in vivo in a SIRT1-dependent manner
Considering the potentially protective effects of β-HB on PA-induced lipid accumulation in C2C12 myotubes, we sought to further examine its effect on glucose disposal. The expression of the PI3K/AKT/GLUT4 pathway was down-regulated in the PA group, which is consistent with the in vitro observations of a high-fat dietary intervention. Intriguingly, β-HB prominently elevated the gene expression of GLUT4 (Fig. 7A) and the protein expression levels of the pathway (Fig. 7B,C).

β-HB up-regulated the expression of glucose disposal-related factors in C2C12 myotubes in a SIRT1 dependent manner. (A) PCR for the expression of Glucose Transporter 4(GLUT4). (B, C) Western-Blot analysis and densitometry quantification for Phosphoinositide 3 Kinase (PI3K), Phospho-PI3K (P-PI3K), Protein Kinase B(AKT), Phospho-AKT (P-AKT) and GLUT4. (D–F) RNA interference technology inhibited the protein and gene expression of SIRT1 in C2C12 myotubes. (G, H) The protein expression of PI3K/AKT/GLUT4 pathway under SIRT1 knock out condition. N = 3–4 per groups. *p < 0.05, **p < 0.005, ***p < 0.001, §p < 0.0001 vs NC group. #p < 0.05, ##p < 0.005, ###p < 0.001, &p < 0.0001 vs PA group. $p < 0.0001 vs PA + β-HB group. Statistical comparison of two groups was performed by Student's t-test. Three or four conditions were conducted by one-way ANOVA followed by Tukey's multiple comparison correction. Data are represented as means ± SEM values. Cropped gels and blots are shown, original gels and blots are presented in Supplementary Figs. 6–8.
It has been shown that SIRT1 is the sensor of cellular energy status29. In the present study, we found that Dapa or β-HB increased the expression of SIRT1 at both mRNA and protein levels in vivo and in vitro. However, whether the effects of improving glucose disposal were associated with the activation of SIRT1 was unclear. We next used small interfering RNA (siRNA) for SIRT1 to determine whether the effect of β-HB on glucose disposal was dependent on SIRT1. The expression of SIRT1 was inhibited by a specific siRNA for SIRT1 but not by small interfering negative control (siNC) (Fig. 7D–F). The knockdown of SIRT1 largely blocked the effect of β-HB on the expression of the PI3K/AKT/GLUT4 pathway (Fig. 7G,H). Taken together, these results suggested that β-HB improved glucose disposal in a SIRT1-dependent manner.
Discussion
In our study, firstly, we observed that Dapa treatment induced a nutrient deprivation state in the DIO mouse model, which is characterized by loss of body weight, decreased body fat and serum lipids, conversion of metabolic substrates to fat, elevated ketone levels, and heat loss from the urine. In skeletal muscle, Dapa activated SIRT1, which been called an energy sensor. Meanwhile, Dapa alleviated ectopic lipid deposition and oxidative stress, promoted the expression of mitochondrial biosynthesis as well as function-related factors, improved glucose tolerance in mice, and upregulated the PI3K/Akt/GLUT4 pathway, which is the most important pathway for glucose disposal in the skeletal muscle. We next treated C2C12 myotubes with β-HB under the PA intervention condition, and similar benefits were detected. Finally, we verified whether the effect of β-HB in improving glucose disposal in myocytes was dependent on the activation of SIRT1. We found that the benefit was significantly attenuated after using siRNA to knock down SIRT1 expression.
There is compelling evidence from large-scale randomized controlled trials that SGLT2 inhibitors exert cardioprotective and renoprotective effects that are far greater than expected based on their effects on glycemia or glycosuria, as similar effects are not observed with antidiabetic drugs that have greater antihyperglycemic actions32,33. However, the underlying mechanisms are not well-understood. It has been hypothesized that SGLT2 inhibitors induce a “dormancy state” that mimics starvation34. In other words, the state is characterized by an increasing glucose extraction from the urine, a decrease in adipose tissue mass, and activation of catabolic pathways23, which is not observed with other antihyperglycemic drugs, with enhanced gluconeogenesis and ketogenesis35. Dapa treatment for five weeks lead to adaptive changes in skeletal muscle substrate metabolism, favoring metabolism of fatty acids and ketone bodies36, and Dapa treatment for 14 days in insulin-resistant individuals improved fat oxidation in skeletal muscle and mimicked the effects of calorie restriction37. In accordance with previous results, we observed the metabolic benefits of Dapa treatment, including decreases in body weight, body fat, and blood lipids, an increase in urine glucose concentration, an RQ value indicative of fat metabolism, and importantly, ketogenic effects; however, we did not observe a clear improvement in muscle mass, which has been controversial in previous research38,39.
When cells are stressed by starvation, they activate a transcriptional program that facilitates their adaptation to low-nutrient conditions. SIRT1 plays a critical role as a low-energy sensor31. Specifically, SIRT1 responds to nicotinamide adenine dinucleotide levels and serves as a redox rheostat. SIRT1 mutes oxidative stress by enhancing antioxidant activity40,41. In addition, SIRT1 inhibits energy storage while promoting energy utilization to facilitate adaption to a low-energy state. SIRT1 also can regulate mitochondrial quality control by promoting mitochondrial biogenesis and facilitating the disposal of dysfunctional organelles24. Recent experimental evidence suggests that SGLT2 inhibitors have activating effects on SIRT1; the mechanism may involve inhibiting endoplasmic reticulum stress and oxidative stress or maintaining mitochondrial homeostasis42,43. We measured the RNA and protein expression levels of SIRT1 in muscle, and the results showed that it was significantly inhibited under high-fat diet-induced obesity conditions, while Dapa treatment had a beneficial effect. Furthermore, we observed its effects on oxidative stress and mitochondrial biosynthesis and function to verify the probable benefits of SIRT1 activation. We found that Dapa can potentially display an antioxidant effect, and TEM showed increased mitochondrial numbers, and reduction of lipid accumulation in skeletal muscle. PGC-1α is part of the downstream signaling pathway of SIRT1, and the PGC-1α/NRF-1/TFAM pathway is critical for mitochondrial biosynthesis44, which was impaired in skeletal muscle of those with insulin resistance45. Our results demonstrate the protective effect of Dapa treatment on this pathway. In terms of mitochondrial function, we found that Dapa upregulated the expression of factors associated with oxidative phosphorylation as well as mitochondrial dynamics, both of which can be damaged by lipid peroxidation, which is consistent with the evidence18,46.
Ketogenesis induced by SGLT2 inhibitors represents a biomarker of nutrient deprivation status47. We examined whether nutrient deprivation status was associated with the range of metabolic benefits described above, by treating C2C12 myotubes with β-HB the most abundant type of ketone body. Intriguingly, β-HB boosted mitochondrial biosynthesis and function. Importantly, β-HB increased the expression of SIRT1 in vivo. Several previous studies also shown the activation of SIRT1 by β-HB, which exerts multiple metabolic benefits48,49. However, according to previous evidence, SGLT2 is expressed in many organs, including skeletal muscle50, and although its role remains unclear, it cannot be excluded that SGLT2 inhibitors may directly interact with SGLT2 on myocytes.
Finally, we evaluated whether Dapa or β-HB treatment could improve skeletal muscle insulin sensitivity in vitro and in vivo. After a high-fat diet, the mice showed a significant elevation in fasting blood glucose and impaired glucose tolerance, consistent with insulin resistance. Dapa treatment improved insulin sensitivity, as shown by decreased fasting glucose and normalization of the glucose tolerance test results. In skeletal muscle, insulin binds to the insulin receptor leading to phosphorylation of PI3K which then activates the downstream protein kinase B (also called Akt), eventually leading to the translocation of GLUT4 to the sarcolemma and subsequent entry of glucose into the cell30,31. We observed the impact of Dapa or β-HB on the PI3K/AKT/GLUT4 pathway to explore its effect on insulin-stimulated glucose processing in skeletal muscle. Our results demonstrate that both Dapa and β-HB have an active effect, consistent with a previous study51, which showed that Dapa activated the IRS-1/AKT/GLUT4 pathway in the soleus muscle and enhanced insulin sensitivity in rats with streptozotocin-induced T2DM. Clinical trials have also verified the benefits of Dapa on muscle insulin sensitivity among prediabetic insulin-resistant individuals37 and patients with T2DM52.
Pharmacological interventions targeting SGLT2 are widely used as therapeutic options for the treatment of diabetes. Increasing evidence has confirmed the excellent hypoglycemic effects of SGLT2 inhibitors53. However, the mechanism through which SGLT2 inhibitors exert this protective effect has been poorly understood. The conventional opinion is that SGLT2 inhibitors reduce renal glucose reabsorption in the proximal tubules and increase urinary glucose excretion54, less research has focused on the effects on other organs related to glucose metabolism. Skeletal muscle is one of the most important organs for maintaining glucose homeostasis3. In the early stages of T2DM, impaired glycogen synthesis in muscles is the primary cause of insulin resistance55. Our study suggests that SGLT2 inhibitors have the potential to improve skeletal muscle insulin sensitivity, which may broaden the horizons of the hypoglycemic mechanisms of SGLT2 inhibitors and the potential benefits of nutrient deprivation, as well as fill the treatment gap in obesity-induced insulin resistance. However, there were also some limitations in our study. Firstly, as mentioned before, there may be mechanisms other than ketogenic effects for Dapa to act on myotubes, but the present study focused on exploring the benefits of nutrient deprivation state induced by Dapa. In addition, while animal research has contributed significantly to our understanding of human biology and physiology, its limitations must be considered that they cannot achieve the accurate representation of human.
In conclusion, our results suggest that Dapa treatment may have the potential to induce nutrient deprivation and activate the energy sensor SIRT1, thereby alleviating ectopic lipid deposition and oxidative stress, improving mitochondrial biosynthesis and function, and eventually ameliorating the loss of insulin sensitivity of skeletal muscle in the face of high-fat diets.
Materials and methods
Animals and diets
C57BL/6 male mice aged 5–6 weeks were purchased from HFK Bioscience Co. Ltd., Beijing, China. The mice were housed under a 12/12 h light/dark cycle circumstances at 23 ± 1 °C and given standard chow diets with free access to water. Mice were maintained in cages with a maximum of five mice per cage. After one-week adaptive feeding, the animals were randomly divided into two groups, the NC group fed a chow-diet, and the DIO group fed with a high-fat diet (D12492, Research Diets, New Brunswick, USA). After 12 weeks, the mice in the DIO group were randomly divided into a high-fat diet fed dapagliflozin (Selleck Chemicals, Houston, USA) treatment group (HFD + Dapa) and the group only fed with high-fat diet. Dapa was administered via oral gavage at 1.5 mg/kg/day for eight weeks. This dosage was calculated based on the therapeutic dose used in humans and is consistent with other studies56. An equal volume of vehicle was administered to the NC and HFD groups.
The animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Chu Hsien-I Memorial Hospital of Tianjin Medical University, approval numbers DXBYY-IACUC-2021038. All experiments were performed in accordance with relevant named guidelines and regulations, and all authors complied with the ARRIVE guidelines.
Body weight and body composition
The animals were weighed every weekend during the 20 weeks period. Body composition, including body fat, free body fluid and lean mass, were analyzed by Bruker Mini spec Whole Body Composition Analyzer (Bruker, MA, USA).
Metabolic parameters
The metabolic parameters of the mice were monitored using a Comprehensive Lab Animal Monitoring System (Columbus Instruments, Columbus, USA). Mice were acclimated for 24 h before measurements were taken (from 7 to 7 am at the next day). The RQ, EE, and food uptake of each mouse were determined every 24 h.
Measurement of serum lipids
Fasting serum samples were separated and stored at − 80 °C. Serum levels of TC, TG, lLDL-C, and high-density lipoprotein cholesterol (HDL-C) were measured using commercial kits (Biosino Bio-Technology Inc, Beijing, China).
Two-hour glucose tolerance test
Mice were intraperitoneally injected with filter-sterilized glucose (1.5 g/kg body weight) after fasting for 8 h. Blood glucose from tail tip was measured at 0, 15, 30, 60, 90, and 120 min after glucose administration using a glucometer (Roche).
Urine volume and urinary glucose concentration
The 24 h urine output of the mice was collected using Metabolic Cages (Tongan Medical Experimental Equipment, Suzhou, China) and the volume was recorded. The glucose concentration in urine was measured using ELISA kits (Gelatins, Shanghai, China).
Measurement of serum β-HB
Blood was collected through the angular vein. To avoid interference from fasting, we chose to measure β-HB levels in a fed state. To ensure the mice were in a fed state, we provided ample food the night before the experiment. We kept them in a quiet, comfortable environment to minimize stress and avoid disruption of feeding behavior. Moreover, we checked the weight of the added and remaining food to ensure that the mice had eaten. The level of β-HB was measured with an ELISA kit (Meimian Industrial Co. Ltd., Jiangsu, China).
Measurement of serum MDA and CAT
MDA, a marker of lipid peroxidation, was determined using an MDA Detection Kit (Jiancheng Biotech Inc., Nanjing, China). Catalase, an indicator of antioxidant enzymes, was measured using a CAT Assay Kit (Jiancheng Biotech Inc.). Serum MDA and CAT levels were measured after an 8-h fasting period.
TEM
Skeletal tissue was immediately rinsed in phosphate buffered saline (PBS) after removal from the mice. The tissue was diced into 1 mm3 cubes and fixed in 2% glutaraldehyde in 0.1 mM phosphate buffer (pH 7.3) for 12 h at 4 °C. The samples were washed with PBS and post-fixed in 1% osmium tetroxide for 2 h. Samples were then dehydrated through a graded series of acetone and embedded in SPI-Pon 812 resin (SPICo., West Chester, PA, USA) for 24 h at 36 °C, 48 °C, and 60 °C, respectively. The embedded tissue was cut into 70 nm sections and stained with uranyl acetate (30 min), followed by 0.2% lead citrate (30 min). Images were captured using an electron microscope (Philips, Amsterdam, Netherlands).
Cell culture and interventions
C2C12 mouse myoblasts (Procell Life Science and Technology, Wuhan, China) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco Life Technologies, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco Life Technologies) and 2% penicillin and streptomycin (Solarbio Science & Technology Co., Ltd., Beijing, China). Differentiation medium (DM) was composed of DMEM containing 2% horse serum (Gibco Life Technologies). When cell confluence reached 80%, the medium was exchanged for DM to induce differentiation. Cells were cultured at 37 °C in 5% CO2. The differentiated mature C2C12 myotubes were divided into three groups for interventions: the NC, the PA intervention group, and the palmitic acid and β-HB intervention group (PA + β-HB). The intervention concentrations of PA and β-HB were determined based on the cell counting kit-8 (CCK-8) test, and are 0.3 mM and 2 mM, respectively. All myotubes were cultured in experimental media for 24 h before harvesting for subsequent experiments.
Cell viability measurement
C2C12 myotubes were cultured in 96-well plates at a density of 1 × 104 cells/well and treated with PA (0, 0.2, 0.3, 0.4, 0.5, 0.75 and 1 mM) and β-HB (0, 0.5, 1, 2, 3, 4 and 5 mM) for 24 h. The Cell Counting Kit-8 (CCK-8) reagent (NCM Biotech, Suzhou, China) was used to measure cell viability.
Mito tracker green staining
Myotubes differentiated from C2C12 myoblasts for 5 days were washed with PBS and incubated at 37 °C for 45 min with 100 nM MitoTracker Green FM (Beyotime, Shanghai, China). Fluorescence signals were detected using a confocal fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
RNA interference
C2C12 myotubes were seeded in 6-well plates. When the cell density reached 70–80%, 10 μL Lipofectamine 3000 (Invitrogen, USA), 250 μL Opti-MEM (Gibco Life Technologies) and 10 μL of siSIRT1 or siNC (Sigma-Aldrich, St. Louis, MO, USA) was added to each well. Cells were grown for 24 h and then incubated with PA or β-HB for 24 h.
Oil Red O staining
A portion of the freshly extracted skeletal muscle tissue was embedded with optimal cutting temperature compound followed immediately by freezing at − 80 °C. The frozen tissues were sliced into 8 µm sections. C2C12 myotubes were inoculated into six-well plates for staining. Tissues and cells were stained with Oil Red O stain solution (Solarbio Science & Technology Co.) according to the manufacturer's protocol, and images were acquired using a Leica microscope (Leica Microsystems). The magnification used for Oil Red O staining is 20 ×.
RNA isolation and quantitative real-time PCR
Total RNA was isolated from the skeletal muscle and C2C12 myotubes using TRIzol Reagent (Transgene Biotech, Beijing, China), according to the manufacturer's instructions. A total of 1 μg RNA was reverse-transcribed into cDNA, and then the cDNA was amplified and quantified with an Applied Biosystems real-time PCR system using SYBR Green PCR master mix and the ΔΔCt threshold cycle method. Gene expression levels were normalized to β-actin mRNA and expressed relative to the control. The primer sequences are listed in Table 1.
Protein extraction and western blot analysis
Skeletal muscle tissue samples and C2C12 myotubes were lysed in RIPA buffer containing a mixture of protease and phosphatase inhibitors, centrifuged at 15,000×g for 15 min, and protein concentrations were measured. Equal amounts of protein were resolved by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (10–12%) and transferred onto a nitrocellulose membrane (Millipore, MA, USA). After incubation with 5% non-fat milk for 1 h at room temperature, the following primary antibodies were used: anti-SIRT1 (1:1000, Cat. no. 9475s; Cell Signaling Technology, Danvers, MA, USA), anti- PGC-1α (1:5000, Cat. No. 66369-1-2g; Proteintech, Chicago, USA) and anti-NRF-1 (1:1000, Cat. No. 12482-1-AP; Proteintech), anti-TFAM (1:2000, Cat. No. 22586-1-AP; Proteintech), anti- ATP5A (1:1000, Cat. No. ab14748; Abcam, Cambridge, UK), anti-CytC (1:1000, Cat. No. T55734F; Abmart, Shanghai, China), anti-OPA-1 (1:2000, Cat. No. ab157457; Abcam), anti-MFN-1 (1:3000, Cat. No. ab126575; Abcam), anti-MFN-2 (1:1000, Cat. no. 9482s; Cell Signaling Technology), anti-DRP-1 (1:1000, Cat. No. ab140494; Abcam), anti-PI3K (1:1000, Cat. No. 4249s; Cell Signaling Technology), anti- P-PI3K (1:1000, Cat. No. ab86714; Abcam), anti-AKT (1:1000, Cat. no. 4685s; Cell Signaling Technology), anti-P-AKT (1:2000, Cat. no. 4060s; Cell Signaling Technology), anti-GLUT4 (1:1000, Cat. No. ab654; Abcam) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:5000, Cat. No. M20006F; Abmart). The membranes were then incubated with the corresponding secondary HRP-conjugated antibodies. GAPDH was used as a loading control. The proteins were quantified using the ImageJ software (National Institutes of Health, MD, USA). Protein bands were visualized in ECL (NCM Biotech, SuZhou, China) and the targeted proteins were scanned with the Molecular Imager ChemiDoc XRS Imaging System (Bio-Rad, Hercules, CA, USA). Due to the blots being cut prior to antibody hybridization, original gels and blots with complete membrane edges are presented in the Supplementary figure.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9.4.0 software (GraphPad Software, San Diego, CA, USA). Results are expressed as the mean ± SEM. Statistical comparison of two groups was performed by Student's t-test, and three or more conditions were compared by one-way ANOVA followed by Tukey's multiple comparison correction; P < 0.05 was considered statistically significant.
Data availability
The data supporting the findings of this study are not publicly accessible due to sensitivity concerns. Access to these data can be granted by the corresponding author upon a reasonable request, subject to certain restrictions.
References
Samuel, V. T. & Shulman, G. I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Invest. 126(1), 12–22. https://doi.org/10.1172/JCI77812 (2016).
DeFronzo, R. A. et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers. 1, 15019. https://doi.org/10.1038/nrdp.2015.19 (2015).
Petersen, M. C. & Shulman, G. I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 98(4), 2133–2223. https://doi.org/10.1152/physrev.00063.2017 (2018).
Szablewski, L. Insulin resistance: The increased risk of cancers. Curr. Oncol. 31(2), 998–1027. https://doi.org/10.3390/curroncol31020075 (2024).
Thiebaud, D. et al. The effect of graded doses of insulin on total glucose uptake, glucose oxidation, and glucose storage in man. Diabetes. 31(11), 957–963. https://doi.org/10.2337/diacare.31.11.957 (1982).
Lillioja, S. et al. Impaired glucose tolerance as a disorder of insulin action: Longitudinal and cross-sectional studies in Pima Indians. N. Engl. J. Med. 318(19), 1217–1225. https://doi.org/10.1056/NEJM198805123181901 (1988).
Sylow, L., Tokarz, V. L., Richter, E. A. & Klip, A. The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell. Metab. 33(4), 758–780. https://doi.org/10.1016/j.cmet.2021.03.020 (2021).
Sun, H. et al. IDF diabetes atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119. https://doi.org/10.1016/j.diabres.2021.109119 (2022).
Ruze, R. et al. Obesity and type 2 diabetes mellitus: Connections in epidemiology, pathogenesis, and treatments. Front. Endocrinol. 14, 1161521. https://doi.org/10.3389/fendo.2023.1161521 (2023).
Badin, P. M., Langin, D. & Moro, C. Dynamics of skeletal muscle lipid pools. Trends Endocrinol. Metab. 24(12), 607–615. https://doi.org/10.1016/j.tem.2013.08.001 (2013).
Meex, R. C. R., Blaak, E. E. & van Loon, L. J. C. Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes. Obes. Rev. 20(9), 1205–1217. https://doi.org/10.1111/obr.12862 (2019).
Schrauwen, P. High-fat diet, muscular lipotoxicity and insulin resistance. Proc. Nutr. Soc. 66(1), 33–41. https://doi.org/10.1017/S0029665107005277 (2007).
Sparks, L. M. et al. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 54(7), 1926–1933. https://doi.org/10.2337/diabetes.54.7.1926 (2005).
Yakes, F. M. & Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA. 94(2), 514–519. https://doi.org/10.1073/pnas.94.2.514 (1997).
Jheng, H. F. et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell Biol. 32(2), 309–319. https://doi.org/10.1128/MCB.05603-11 (2012).
Pileggi, C. A., Parmar, G. & Harper, M. E. The lifecycle of skeletal muscle mitochondria in obesity. Obes. Rev. 22(5), e13164. https://doi.org/10.1111/obr.13164 (2021).
Chen, X. et al. Mitochondrial dysfunction: Roles in skeletal muscle atrophy. J. Transl. Med. 21(1), 503. https://doi.org/10.1186/s12967-023-04369-z (2023).
Kim, J. A., Wei, Y. & Sowers, J. R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 102(4), 401–414. https://doi.org/10.1161/CIRCRESAHA.107.165472 (2008).
Di Meo, S., Iossa, S. & Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 233(1), R15–R42. https://doi.org/10.1530/JOE-16-0598 (2017).
Gurd, B. J. Deacetylation of PGC-1α by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 36(5), 589–597. https://doi.org/10.1139/h11-070 (2011).
Knutti, D. & Kralli, A. PGC-1, a versatile coactivator. Trends Endocrinol. Metab. 12(8), 360–365. https://doi.org/10.1016/s1043-2760(01)00457-x (2001).
Lu, C. et al. Novel role of the SIRT1 in endocrine and metabolic diseases. Int. J. Biol. Sci. 19(2), 484–501. https://doi.org/10.7150/ijbs.78654 (2023).
Thirupathi, A. & de Souza, C. T. Multi-regulatory network of ROS: The interconnection of ROS, PGC-1 alpha, and AMPK-SIRT1 during exercise. J. Physiol. Biochem. 73(4), 487–494. https://doi.org/10.1007/s13105-017-0576-y (2017).
Tang, B. L. Sirt1 and the mitochondria. Mol. Cells. 39(2), 87–95. https://doi.org/10.14348/molcells.2016.2318 (2016).
Xu, B., Li, S., Kang, B. & Zhou, J. The current role of sodium-glucose cotransporter 2 inhibitors in type 2 diabetes mellitus management. Cardiovasc. Diabetol. 21(1), 83. https://doi.org/10.1186/s12933-022-01512-w (2022).
Preda, A. et al. SGLT2 inhibitors: From glucose-lowering to cardiovascular benefits. Cardiovasc. Res. 120(5), 443–460. https://doi.org/10.1093/cvr/cvae047 (2024).
Packer, M. SGLT2 inhibitors: Role in protective reprogramming of cardiac nutrient transport and metabolism. Nat. Rev. Cardiol. 20(7), 443–462. https://doi.org/10.1038/s41569-022-00824-4 (2023).
Packer, M. Critical reanalysis of the mechanisms underlying the cardiorenal benefits of SGLT2 inhibitors and reaffirmation of the nutrient deprivation signaling/autophagy hypothesis. Circulation. 146(18), 1383–1405. https://doi.org/10.1161/CIRCULATIONAHA.122.061732 (2022).
Rodgers, J. T. et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 434, 113118. https://doi.org/10.1038/nature03354 (2005).
Taniguchi, C. M., Emanuelli, B. & Kahn, C. R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 7(2), 85–96. https://doi.org/10.1038/nrm1837 (2006).
Krook, A. et al. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes. 49(2), 284–292. https://doi.org/10.2337/diabetes.49.2.284 (2000).
Heerspink, H. J. L. et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 383(15), 1436–1446. https://doi.org/10.1056/NEJMoa2024816 (2020).
McMurray, J. J. V. et al. DAPA-HF Trial Committees and Investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 381(21), 1995–2008. https://doi.org/10.1056/NEJMoa1911303 (2019).
Avogaro, A., Fadini, G. P. & Del Prato, S. Reinterpreting cardiorenal protection of renal sodium-glucose cotransporter 2 inhibitors via cellular life history programming. Diabetes Care. 43(3), 501–507. https://doi.org/10.2337/dc19-1410 (2020).
Swe, M. T. et al. Dapagliflozin not only improves hepatic injury and pancreatic endoplasmic reticulum stress, but also induces hepatic gluconeogenic enzymes expression in obese rats. Clin. Sci. 133(23), 2415–2430. https://doi.org/10.1042/CS20190863 (2019).
Op den Kamp, Y. J. M. et al. Effects of SGLT2 inhibitor dapagliflozin in patients with type 2 diabetes on skeletal muscle cellular metabolism. Mol. Metab. 66, 101620. https://doi.org/10.1016/j.molmet.2022.101620 (2022).
Veelen, A. et al. Effects of the sodium-glucose cotransporter 2 inhibitor dapagliflozin on substrate metabolism in prediabetic insulin resistant individuals: A randomized, double-blind crossover trial. Metabolism. 140, 155396. https://doi.org/10.1016/j.metabol.2022.155396 (2023).
Sugiyama, S. et al. Dapagliflozin reduces fat mass without affecting muscle mass in type 2 diabetes. J. Atheroscler. Thromb. 25(6), 467–476. https://doi.org/10.5551/jat.40873 (2018).
Zhang, S., Qi, Z., Wang, Y., Song, D. & Zhu, D. Effect of sodium-glucose transporter 2 inhibitors on sarcopenia in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Front. Endocrinol. 14, 1203666. https://doi.org/10.3389/fendo.2023.1203666 (2023).
Gillum, M. P., Erion, D. M. & Shulman, G. I. Sirtuin-1 regulation of mammalian metabolism. Trends Mol. Med. 17(1), 8–13. https://doi.org/10.1016/j.molmed.2010.09.005 (2011).
Alcendor, R. R. et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100(10), 1512–1521. https://doi.org/10.1161/01.RES.0000267723.65696.4a (2007).
He, L. et al. Dapagliflozin improves endothelial cell dysfunction by regulating mitochondrial production via the SIRT1/PGC-1α pathway in obese mice. Biochem. Biophys. Res. Commun. 615, 123–130. https://doi.org/10.1016/j.bbrc.2022.05.022 (2022).
Ren, F. F. et al. Dapagliflozin attenuates pressure overload-induced myocardial remodeling in mice via activating SIRT1 and inhibiting endoplasmic reticulum stress. Acta Pharmacol. Sin. 43(7), 1721–1732. https://doi.org/10.1038/s41401-021-00805-2 (2022).
Pagel-Langenickel, I., Bao, J., Pang, L. & Sack, M. N. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr. Rev. 31(1), 25–51. https://doi.org/10.1210/er.2009-0003 (2010).
Patti, M. E. et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA. 100(14), 8466–8471. https://doi.org/10.1073/pnas.1032913100 (2003).
Shao, Q. et al. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 18(1), 165. https://doi.org/10.1186/s12933-019-0964-4 (2019).
Packer, M. Role of deranged energy deprivation signaling in the pathogenesis of cardiac and renal disease in states of perceived nutrient overabundance. Circulation. 141(25), 2095–2105. https://doi.org/10.1161/CIRCULATIONAHA.119.045561 (2020).
Abduraman, M. A., Azizan, N. A., Teoh, S. H. & Tan, M. L. Ketogenesis and SIRT1 as a tool in managing obesity. Obes. Res. Clin. Pract. 15(1), 10–18. https://doi.org/10.1016/j.orcp.2020.12.001 (2021).
McCarty, M. F., DiNicolantonio, J. J. & O’Keefe, J. H. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses. 85(5), 631–639. https://doi.org/10.1016/j.mehy.2015.08.002 (2015).
Wright, E. M., Loo, D. D. & Hirayama, B. A. Biology of human sodium glucose transporters. Physiol. Rev. 91(2), 733–794. https://doi.org/10.1152/physrev.00055.2009 (2011).
Fleifel, A. M., Soubh, A. A., Abdallah, D. M., Ahmed, K. A. & El-Abhar, H. S. Preferential effect of montelukast on dapagliflozin: Modulation of IRS-1/AKT/GLUT4 and ER stress response elements improves insulin sensitivity in soleus muscle of a type-2 diabetic rat model. Life Sci. 307, 120865. https://doi.org/10.1016/j.lfs.2022.120865 (2022).
Goto, Y. et al. Improvement of skeletal muscle insulin sensitivity by 1 week of SGLT2 inhibitor use. Endocr. Connect. 9(7), 599–606. https://doi.org/10.1530/EC-20-008 (2020).
Scheen, A. J. Sodium-glucose cotransporter type 2 inhibitors for the treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 16(10), 556–577. https://doi.org/10.1038/s41574-020-0392- (2020).
Lee, P. C., Ganguly, S. & Goh, S. Y. Weight loss associated with sodium-glucose cotransporter-2 inhibition: A review of evidence and underlying mechanisms. Obes. Rev. 19(12), 1630–1641. https://doi.org/10.1111/obr.12755 (2018).
DeFronzo, R. A. & Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 32(Suppl 2), S157–S163. https://doi.org/10.2337/dc09-S302 (2009).
Wu, Q. et al. Dapagliflozin protects against chronic heart failure in mice by inhibiting macrophage-mediated inflammation, independent of SGLT2. Cell Rep. Med. 4(12), 101334. https://doi.org/10.1016/j.xcrm.2023.101334 (2023).
Funding
This research was funded by the Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-032A), Scientific Research Funding of Tianjin Medical University Chu Hsien-I Memorial Hospital (ZXY-ZDSYS2023-3, ZXY-ZDSYSZD2023-01) and Tianjin Natural Science Foundation Projects (21JCQNJC00460).
Author information
Authors and Affiliations
Contributions
Qi Gao designed the research study, conducted experiments, acquired and analyzed the data, and wrote the manuscript. Yingying Jiang co-designed and conducted the experiments, acquired and analyzed the data. Zhenqiang Song, Huizhu Ren and Yan Kong acquired the data and revised the manuscript, Cong Wang and Miaoyan Zheng analyzed the data and discussed the study. Yanhui Yang and Chunyan Shan co-designed the study, discussed analyses and data interpretation, revised and approved the manuscript, and collaborated with all other authors. All authors have reviewed the final version of the manuscript and approved it for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, Q., Jiang, Y., Song, Z. et al. Dapagliflozin improves skeletal muscle insulin sensitivity through SIRT1 activation induced by nutrient deprivation state. Sci Rep 14, 16878 (2024). https://doi.org/10.1038/s41598-024-67755-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67755-7
- Springer Nature Limited