
Abstract
Somatic gene copy number variation contributes to tumor progression. Using comparative genomic hybridization (CGH) array, the presence of genomic imbalances was evaluated in a series of 27 papillary thyroid carcinomas (PTCs). To detect only somatic imbalances, for each sample, the reference DNA was from normal thyroid tissue of the same patient. The presence of the BRAF V600E mutation was also evaluated. Both amplifications and deletions showed an uneven distribution along the entire PTC cohort; amplifications were more frequent than deletions (mean values of 17.5 and 7.2, respectively). Number of aberration events was not even among samples, the majority of them occurring only in a small fraction of PTCs. Most frequent amplifications were detected at regions 2q35, 4q26, and 4q34.1, containing FN1, PDE5A, and GALNTL6 genes, respectively. Most frequent deletions occurred at regions 6q25.2, containing OPMR1 and IPCEF1 genes and 7q14.2, containing AOAH and ELMO1 genes. Amplification of FN1 and PDE5A genomic regions was confirmed by quantitative PCR. Frequency of amplifications and deletions was in relationship with clinical features and BRAF mutation status of tumor. In fact, according to the American Joint Committee on Cancer stage and American Thyroid Association (ATA) risk classification, amplifications are more frequent in higher risk samples, while deletions tend to prevail in the lower risk tumors. Analysis of single aberrations according to the ATA risk grouping shows that amplifications containing PDE5A, GALNTL6, DHRS3, and DOCK9 genes are significantly more frequent in the intermediate/high risk group than in the low risk group. Thus, our data would indicate that analysis of somatic genome aberrations by CGH array can be useful to identify additional prognostic variables.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Identification of molecular mechanisms and markers of tumor progression and aggressiveness is a central theme in cancer research. Papillary thyroid carcinoma (PTC) represents the most frequent form (80–85 %) of thyroid malignancy [1]. Most PTCs are biologically indolent and have a very good prognosis (survival rates of 90–95 % at 5 years), thanks to their responsiveness to radioiodine treatment [2]. However, 5–10 % PTCs show a more aggressive behavior and a worse prognosis [3]. Knowledge of the molecular mechanisms that contribute to aggressiveness of PTCs is critical to propose tailored therapy approaches. Genetic and epigenetic alterations are both involved in the pathogenesis and progression of thyroid tumors [4–6]. Several studies indicate that the most common genetic alteration found in PTC is the V600E mutation of the BRAF gene [7]. Many studies indicate that the presence of this BRAF mutation in PTCs is associated to poor clinicopathological outcomes, including aggressive pathological features, increased recurrence and treatment failure [4, 8, 9]. Gene copy number variation may also contribute to PTC progression. In particular, gene amplification has been shown to be related to tumor aggressiveness [10, 11]. An efficient way to evaluate the presence of copy number variation at genomic levels is the comparative genomic hybridization (CGH) array [12]. In this microarray technology, gene dosage of a test genome is compared to that of a reference genome, and imbalance due to deletions or amplifications of genomic regions is easily detected. CGH array has been extensively used for the identification of genomic imbalances present in cancer [13]. Several data come from studies on thyroid tumors [14–16]. In most of them, however, the reference genome used in the CGH array was not that of the same patient. In this way, somatic copy number variations that occur in tumor cells (and that may have a role in tumor progression) cannot be distinguished from germline copy number variations existing in distinct human genomes [17].
The present study was conducted to identify copy number variations in PTCs by CGH array. In addition, we confirmed CGH array data by quantitative real-time PCR of selected genes and evaluated the presence of BRAF V600E mutation. Correlation analyses were performed with clinical/pathological characteristics according to the American Joint Committee on Cancer/Tumor Nodes Metastasis (AJCC/TNM) staging of tumors and the 2009 American Thyroid Association (ATA) initial risk stratification system, in order to find new genetic markers for prognostic predictions.
Materials and methods
Patients
For CGH array analysis, 27 subjects (23 females and 4 males) affected by PTC were examined. The mean age was 45 ± 10 (range 17–74) years. Thyroid tissues were obtained at thyroidectomy; for each subject, both tumor and non-tumor (normal) thyroid tissues were available. All samples were diagnosed by referral pathologists of the institutions and then reviewed by a single experienced pathologist, thus including only tumors with confirmed diagnosis. The study was approved by the medical ethics committee of the Azienda Ospedaliero-Universitaria S. Maria della Misericordia of Udine. Before surgery, each study participant provided written informed consent to the collection of thyroid tissue for genetic studies.
DNA extraction from thyroid tissues
For each patient, we used tumor and non-tumor tissue samples. From each tissue sample, formalin-fixed paraffin-embedded (FFPE) specimens were cut in 10-μm-thick sections on a microtome; one section treated with hematoxylin–eosin staining was used for histological confirmation of the presence of tumor cells, and the tumor area was highlighted by pathologist. Non-tumor tissue samples were controlled by the pathologist to completely exclude the presence of tumor cells. Genomic DNA was isolated from the FFPE specimens using NucleoSpin Tissue Kit (Macherey–Nagel, Düren, Germany) following manufacturer’s instructions. Extracted DNA was quantified using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA).
Evaluation of the BRAF V600E mutation
100 ng of isolated DNA was amplified by polymerase chain reaction (PCR) using 200 μM dNTPs, 1.5 mM MgCl2, 1 U of AmpliTaq Gold, and Buffer 1× AmpliTaq Gold DNA Polymerase (Applied Biosystems/Life technologies Italia, Monza, Italy). The PCR was carried out using 10 pmol of specific primers for exon 15 of the BRAF gene: (Forward): TCATAATGCTTGCTCTGATAGGA, (Reverse): GGCCAAAAATTTAATCAGTGG. The cycling condition for PCR program was 95 °C for 10 min, followed by 40 cycles of 95 °C for 1 min, 60 °C for 1 min, and 72 °C for 1 min. A final 7 min extension was included at the end of 40 cycles. The reactions were performed in a Veriti® 96-well Thermal Cycler (Applied Biosystems/Life technologies Italia). PCR products were subjected to 1.5 % agarose gel electrophoresis with ethidium bromide staining and purified with a commercial kit (NucleoSpin Gel and PCR Clean-up, from Macherey–Nagel).
All PCR products were sequenced using Big DyeTM Terminator version 3.1 Cycle sequencing kit (Applied Biosystems/Life technologies Italia), and one of the primers previously described was used for amplification. The sequencing reaction was carried out for 25 cycles (denaturation for 10 s at 96 °C, annealing for 5 s at 50 °C, and elongation for 4 min at 60 °C). The products of this reaction were purified using NucleoSeq Column (Macherey–Nagel) and subjected to capillary gel electrophoresis. Data collection and analysis were performed on an Applied Biosystems 3130xl automated sequencer (Applied Biosystems/Life technologies Italia). All PCR reactions and sequencing were repeated at least twice to confirm the presence of a mutation. This procedure allows to detect as low as 10 % mutated alleles, in agreement with most recent data [18].
CGH array
Because salt from Buffer B5 (NucleoSpin Tissue Kit, Macherey–Nagel) interferes with the labeling reaction, DNA was purified by precipitation with 1/10 vol of 0.3 M sodium acetate and 3 vol of ice-cold ethanol. The pellet obtained after centrifugation was washed with 1 vol of ice-cold 70 % ethanol and resuspended in nuclease free H2O.
CGH array experiments were performed using SurePrint G3 Human CGH Microarray 4 × 180 K cat n. G4449A (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions. Each array consists of 170,000 in situ synthesized 60 mer oligonucleotide probes that span coding and non coding sequences with an average spatial resolution of 13 kb.
To label DNA samples the one-step non-enzymatic Agilent Genomic DNA ULS labeling kit (Agilent Technologies) according to the ULS Labeling for Blood, Cells, Tissue, or FFPE (with a high throughput option) protocol v. 3.3 was utilized. As reference DNA for each sample, DNA extracted from non-tumor tissue of the same patient was used. 500 ng of sample DNA (tumor tissue) and 500 ng of reference DNA (non-tumor tissue) were labeled with ULS-Cy5 and ULS-Cy3, respectively. The degree of labeling was calculated using the Nanodrop ND 1000 Spectrophotometer (Thermo Scientific). The differentially labeled DNA samples were hybridized (65 °C) to the microarray for 40 h in a hybridization oven (Agilent Technologies). The slides, during hybridization, were rotated at 20 rpm. The array was scanned at 3 μm resolution using Agilent microarray scanner and analyzed using Feature Extraction v. 10.7.1.1 and FE Protocol CGH_107_Sept09 (Agilent Technologies).
CGH array data analysis
Copy number analysis was performed using the analytic module of Agilent Genomic Workbench Lite Edition 6.5.018 software (Agilent Technologies). The array CGH data are assessed with a series of quality control (QC) metrics then analyzed using the Aberration detection algorithm ADM2. The latter identifies all aberrant intervals in a given sample with consistently high or low log ratios, based on the statistical score derived from the average normalized log ratios of all probes in the genomic interval multiplied by the square root of the number of these probes. This score represents the deviation of the average of the normalized log ratios from its expected value of zero and is proportional to the height h (absolute average log ratio) of the genomic interval and to the square root of the number of probes in the interval. The ADM2 algorithm prompted by Genomic Workbench software was used to compute and assist the identification of aberrations for a given sample (threshold = 6.0), and detected regions were filtered for those spanning more than three consecutive probes with an average absolute log2 ratio >0.45 [19, 20]. The minimum average spatial resolution consists of 40 kb.
Statistical analysis
To identify genomic intervals that have statistically significant common aberrations was used the “context corrected” common aberration analysis (Agilent Genomic Workbench Lite Edition 6.5.018 software, p value threshold = 0.05, Overlap threshold = 0.1). This method adjusts the significance of a genomic interval according to the overall aberration state of the sample.
In order to evaluate correlation of genetic aberration detected by CGH array with AJCC staging, ATA risk, and BRAF mutational status, statistical analysis was performed by the t test using the Graphpad software. Values of p < 0.05 were considered statistical significant.
Quantitation of FN1 and PDE5A gene dosage by PCR
Five nano gram of DNA was amplified by real-time PCR (RT-PCR) reaction using buffer 1× Platinum SYBR Green qPCR SuperMix-UDG with ROX (Applied Biosystems/Life Technologies Italia). The RT-PCR was carried out using 3.75 pmol of the following specific primers: for FN1 gene (Forward): CCGAGGAGAGTGGAAGTGTGA, (Reverse): GAAAGATGGATTTGCGGAAATATT; for PDE5A gene (Forward): TTGGAGGTGGGTGAAGTTTAGG, (Reverse): TGAGTGATTATGAGGGAAAGGTAAAA; for ALB gene (used as a reference) (Forward): ATGCTGCACAGAATCCTTGGT, (Reverse): TCATCGACTTCCAGAGCTGAAA. The cycling condition for RT-PCR program was 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 1 min. The reactions were performed in a ABI Prism 7300 Sequence Detection System (Applied Biosystems/Life Technologies Italia). The 2−ΔΔCT (cycle threshold) method, by means of the SDS software (Applied Biosystems/Life Technologies Italia), was used to calculate the relative gene quantity [21]. The relative gene target quantity of each tumor sample was normalized to its healthy counterpart (calibrator), which has arbitrarily considered as 1.
Results
Clinicopathological features of investigated patients are shown in Table 1. Using CGH array, each tumor sample was compared to the normal genome of the same subject (from non-tumor thyroid tissue), thus only somatic mutations were identified. Examples of CGH array profiles are shown in supplemental Figs. 1 and 2. Multiple genomic imbalances were detected in all tumors. In terms of mean number of imbalances, though not reaching a statistical significance, a prevalence of amplifications over deletions was observed with mean values of 17.5 and 7.2 for amplifications and deletions, respectively (Fig. 1a). Panel b of Fig. 1 shows the percentage of imbalances of different length. As expected, short deletions/amplifications are prevailing: around 70 % of imbalances are below 200 kb.

Amplifications/deletions present in PTCs. a Each bar indicates the mean value ± SD of aberration found in PTCs. Amp amplifications, Del deletions. b Length ranges of aberrations found in PTCs. Percentage of amplifications is indicated by bars above the 0 line, percentage of deletions is indicated by bars below the 0 line

Distribution of genomic aberrations in PTCs. a Amplifications; b deletions. In either panel the dashed line indicates the theoretical even distribution of aberrations; each white dot indicates a single PTC. The black dots of a indicate 3 samples with no amplifications. The dark dots of b indicate 11 samples, with no deletions. In both panels, the gray areas indicate the top-ranking samples containing 75 % of amplifications (a) or deletions (b) of the entire cohort
The distribution of aberrations was extremely uneven among the entire cohort of PTCs. As shown in panel a of Fig. 2, the fraction of amplification events is not even, the majority of them occurring only in a fraction of PTCs. About 25 % of top-ranking samples (7 cases) contained the 75 % of amplifications. A similar scenario was observed for deletions: the 25 % of top-ranking samples (5 cases) contained the 75 % of deletions (Fig. 2b).
Patients’ tumors are grouped by AJCC stage, ATA risk, and BRAF mutational status (Table 1). We tested whether amplifications and deletions were differently distributed according to these criteria, and statistical significance was determined by the Student’s t test. As shown in Fig. 3, for the AJCC stage, amplifications were not significantly more prevalent in stage III-IV group than in stage I group (p: 0731), and the reverse phenomenon was detected for deletions (p: 0.249). Significant differences were detected between the ATA risk groups: amplifications were more prevalent in intermediate/high risk group than in low risk group (p: 0.046), while deletions were more prevalent in low risk group than in intermediate/high risk group (p: 0.049). No significant differences were detected between groups harboring or not the BRAF V600E mutation, even if amplifications were more prevalent in mutant group (p: 0.446) and deletions more prevalent in the wild-type group (p: 0.247).

Global analysis of amplifications/deletions in PTCs, grouped according to clinical and molecular features. a AJCC grouping; b ATA risk grouping; c BRAF mutational status grouping. In each panel, columns indicate the mean value ± SEM of total amplifications and deletions. p Values were obtained using the Student’s t test
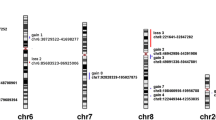
Then, our attention was focused on recurrent aberrations: we decided to define as recurrent aberrations those deletions or amplifications present in at least 5 samples of the whole cohort. Using this cut-off value, 30 aberrations were judged as recurrent, 23 aberrations were judged as amplifications, and 7 aberrations were judged as deletions (this difference is statistically significant according to the two-tailed binomial test; p = 0.0052). In Fig. 4, location of recurrent amplifications and deletions along chromosomes is depicted. Mean values of recurrent amplifications and deletions grouped according to the AJCC stage, ATA risk, and BRAF mutational status are shown in Fig. 5. Statistical significance was obtained for the ATA risk grouping, in which amplifications were more prevalent in intermediate/high risk than in low risk group (p: 0.033) and for the BRAF mutational status, in which amplification was more prevalent in the mutant group than in the wild-type group (p: 0.040). Table 2 shows locations, largeness as well as the number of samples containing recurrent aberrations. Most frequent amplifications occurred at regions 2q35 (in 15 samples), 4q26 (in 12 samples), and 4q34.1 (in 11 samples). Most frequent deletions occurred at regions 6q25.2 and 7q14.2 (both present in 7 samples). The deletion at 7q14.2 was the largest aberration (almost one megabase). The data of recurrent aberrations in the PTCs divided into subgroups according to AJCC stage, ATA risk, or BRAF mutational status are presented in Table 2. In the ATA risk grouping, amplifications at locations 1p36.22, 4q26, 4q34.1, and 13q32.3 were significantly more frequent in the intermediate/high risk group than in low risk group. According to the BRAF mutational status, deletions at locations 6q11.1 and 6q25.2 were more frequent in the wild-type group than in the samples bearing the V600E mutations. In order to confirm the CGH array data, dosage of two genes present in recurrent amplifications at chromosomes 2q35 and 4q26 (FN1 and PDE5A) was performed by quantitative PCR. These two genes were chosen for a potential functional role in thyroid tumorigenesis (see “Discussion” section). Thirteen samples with amplification of region containing FN1 gene and nine samples with amplification containing PDE5A gene have been evaluated. As shown in Fig. 6, tumor tissues in which amplification has been detected by array CGH showed significant higher relative quantity of both FN1 and PDE5A genes than corresponding normal tissues. Moreover, tumors in which no amplification has been detected by array CGH showed relative quantity similar to control tissues. These data confirm that genomic regions containing FN1 and PDE5A genes are amplified in several thyroid cancer tissues.

Recurrent amplifications/deletions in PTCs. Filled circles flanking chromosome ideograms indicate presence of amplifications or deletions. Circles on the right of each ideogram indicate amplifications, while symbols on the left indicate deletions

Global analysis of recurrent amplifications/deletions in PTCs, grouped according to clinical and molecular features. a AJCC grouping; b ATA risk grouping; c BRAF mutational status grouping. In each panel, columns indicate the mean value ± SEM of recurrent amplifications, and deletions p values were obtained using the Student’s t test

Gene dosage of FN1 and PDE5A in PTCs showing amplification of the regions containing the two genes (2q35 and 4q26, respectively). Thirteen PTCs containing the 2q35 amplification, 9 PTCs containing the 4q26 amplification, and 7 PTCs containing no amplification for both genes were analyzed. In the case of PTCs-containing amplifications, normal counterpart was analyzed. Relative quantity of FN1 and PDE5A was evaluated by quantitative PCR as described in the “materials and methods” section. The relative quantity of normal counterparts was considered as 1 (two gene copies). A relative quantity of 1.5 indicates the presence of three gene copies. NT indicates normal counterparts of amplified PTCs (light gray columns), T+ indicates amplified PTCs in CGH array analysis (dark gray columns), and T− indicates non-amplified PTCs in CGH array analysis (black columns)
Discussion
For several cancer types, genome-wide analysis by CGH array has provided an extensive high-resolution delineation of copy number changes present in tumor cells, adding important information on the genomic alterations occurring during cancer progression [22]. In this work, we have identified recurrent deletions and amplifications occurring in the genome of PTCs, also delineating differences among tumors divided according to the AJCC stage, ATA risk classification, and BRAF mutational status. Molecular karyotyping has been already used to study thyroid cancer. However, early CGH investigations were performed using metaphase chromosome spreads as detection system [23–28]; thus, the resolution power of this approach was extremely limited. More recently, microarray has been used as detection system. Most investigations, however, have been performed using BAC or cDNA microarrays [29–31], which still have a reduced power to detect genome imbalances, because arrays contain from few hundreds to few thousands immobilized probes. Oligonucleotide microarrays have been used to investigate thyroid cancer in one study only [32]. In that study, microarrays contained 33.000 probes (33 K) and had an average resolution power of 100 kb. Our study was performed by 180 K microarrays and, therefore, has the highest resolution power. A second major strong point of our investigation is the use of genomic DNA from the same patient as reference DNA. In this way, it has been possible to compare the tumor genome to the normal genome for each patient and, therefore, focus on somatic aberrations.
Using this approach, a major trend was overall observed, that is the prevalence, in our series of PTCs, of amplifications versus deletions. Prevalence of genomic gains with respect to losses has been previously observed in other neoplasms. For example, Sandgren and coworkers by investigating pheochromocytomas and paragangliomas have shown that genome gains were more frequent in malignant than benign tumors [33]. In contrast, in the case of PTCs, Unger et al. have shown higher frequency of deletions than amplifications [30], whereas Finn et al. have shown no significant predominance of one versus the other [29].
An integrated genomic characterization of PTC has been recently published [34]. In that study, to the identification of somatic copy number alterations, SNP array was informative in 495 PTCs. Only 135 of them (27.2 %) were positive for somatic aberrations. Thus, most tumors were considered negative for deletions/amplifications, four distinct classes were defined, and two of them were characterized for either 22q deletions or 1q amplifications. Instead, in our study, we have found copy number alterations in most PTCs. In addition, we have found a recurrent amplification in the 1q region, but not recurrent deletions were observed in the 22q region. Differences between our study and that of the TCGA Research Network likely arise from methodology. Besides technological differences, it is important to point out that the TCGA Research Network focused on chromosomal arm-level alterations, i.e., deletions or amplifications that comprise at least the 66 % of the chromosomal arm. Therefore, in that research authors focused on very large inbalances, while we analyzed also relatively small aberrations (40 kb is the minimal aberration length detection).
In our cohort of tumors, amplifications involving regions 2q35, 4q26, and 4q34.1 containing, respectively, FN1, PDE5A, and GALNTL6 genes resulted the most frequent (Table 2). FN1 encodes for fibronectin, an extracellular matrix glycoprotein that binds to integrins expressed in the cell membrane and plays a major role in migration, growth, cell adhesion, and differentiation [35]. Several studies have demonstrated that fibronectin degradation or its altered expression and organization are associated with a number of pathologies, including fibrosis and cancer [36]. Interestingly, a significant overexpression of FN1 mRNA has been reported in PTCs [37], as well as amplification of its genomic region [26]. Therefore, this gene has been proposed as a molecular marker of differentiated thyroid carcinomas, usable for discrimination between benign or not follicular thyroid tumors [38, 39]. On the other side, PDE5A belongs to the family of phosphodiesterase enzymes, which act hydrolyzing adenosine and guanosine 3′,5′-cyclic monophosphates (cAMP and cGMP) and therefore influencing the nucleotide signaling pathway [40, 41]. In particular, the PDE5A isoform is able to specifically hydrolyze cGMP, a second messenger acting as regulator of various physiological processes in many tissues, including thyroid [40, 41]. Expression of PDE5A has been demonstrated in normal thyroid tissue [42], where cGMP mediates inhibition of different steps of thyroid hormone biosynthesis [43], and very recent findings of our group have demonstrated an overexpression of both PDE5 mRNA and protein in a large series of PTCs [Sponziello et al. unpublished observations] GALNTL6 gene codes for N-acetylgalactosaminyltransferase-like 6 protein; no data are available on its expression in malignancies.
Difference in the distribution of amplifications and deletions was observed also when the tumors were analyzed according to the AJCC stage and ATA risk classification. In general, advanced TNM stages and intermediate/high risk status are considered predictors of a higher risk of disease mortality and disease persistence/recurrence, respectively, although these risk estimates may change overtime according to the clinical course of the disease and the response to therapy. In this series of PTCs, amplifications tend to be more frequent in higher risk samples, while deletions tend to be prevailing in the lower risk ones. A similar trend was observed also when only recurrent aberrations were considered. In our analysis, we considered recurrent mutations those present in at least 5 cases. Such a cut-off is roughly the same used in many other studies, in which recurrent mutations are defined those having a frequency equal or above 20 % [33, 44–46]. When the analysis was performed at the level of single aberrations, according to the ATA risk grouping, amplifications containing PDE5A, GALNTL6, DHRS3, and DOCK9 genes were significantly more frequent in the intermediate/high risk group than in the low risk group. DHRS3 (dehydrogenase/reductase (SDR family) member 3 or retSDR1) catalyzes the oxidation/reduction of a wide range of substrates, including retinoids and steroids [47], and is involved in a growth/tumor suppressive pathway [48]. DHRS3 is constitutively expressed in breast cancer cell lines, and alterations of DHRS3 have been described in metastatic PTCs, suggesting a role in PTC pathogenesis and progression, and, for this reason, is considered a possible molecular target for PTC therapy [49]. DOCK9 codes for a member of Dedicator of cytokinesis protein family involved in development [50]; however, no data are available for these proteins in thyroid tumorigenesis.
Finally, in our study, the results of genomic aberrations have been correlated with the presence or not of BRAF V600E mutation in the PTCs. In most studies, the BRAF V600E mutation is associated to PTC aggressiveness [51–53], owing to its association with several genetic and epigenetic damages observed in thyroid cancer cells [54, 55]. In general, amplifications in tumors with the BRAF V600E mutation are more frequent than in those without this point mutation, in agreement with the previous data in the more aggressive subgroups. However, the single aberrations detected with the highest difference between the two subgroups were the deletions in 6q chromosome corresponding to KHDRBS2 and OPMR1 genes, found in wild-type BRAF subgroup. For both genes, there are no evidences of a functional role in either normal or transformed thyrocytes [56–58].
In conclusion, our bona fide analysis of the somatic genomic aberrations occurring in PTCs revealed a number of amplifications/deletions, resulting presumably in altered gene expression (as demonstrated for those containing the FN1 and PDE5A genes), which characterize the tumor genotype and may contribute to the tumor development. Moreover, the finding of a higher frequency of particular aberrations in subgroups with higher AJCC stage and ATA risk level suggests that analysis of somatic genome aberrations by CGH array can be also useful to identify potential prognostic variables.
References
V.A. LiVolsi, Papillary thyroid carcinoma: an update. Mod. Pathol. 24, S1–S9 (2011)
M. Schlumberger, L. Lacroix, D. Russo, S. Filetti, J.M. Bidart, Defects in iodide metabolism in thyroid cancer and implications for the follow-up and treatment of patients. Nat. Clin. Pract. Endocrinol. Metab. 3, 260–269 (2007)
C.E. Silver, R.P. Owen, J.P. Rodrigo, A. Rinaldo, K.O. Devaney, A. Ferlito, Aggressive variants of papillary thyroid carcinoma. Head Neck 33, 1052–1059 (2011)
M. Xing, Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 13, 184–199 (2013)
D. Russo, G. Damante, E. Puxeddu, C. Durante, S. Filetti, Epigenetics of thyroid cancer and novel therapeutic targets. J. Mol. Endocrinol. 46, R73–R81 (2011)
C. Puppin, N. Passon, E. Lavarone, C. Di Loreto, F. Frasca, V. Vella, R. Vigneri, G. Damante, Levels of histone acetylation in thyroid tumors. Biochem. Biophys. Res. Commun. 411, 679–683 (2011)
R. Ciampi, Y.E. Nikiforov, Alterations of the BRAF gene in thyroid tumors. Endocr. Pathol. 16, 163–172 (2005)
E. Puxeddu, C. Durante, N. Avenia, S. Filetti, D. Russo, Clinical implication of BRAF mutation in thyroid carcinoma. Trends Endocrinol. Metab. 19, 138–145 (2008)
T.H. Kim, Y.J. Park, J.A. Lim, H.Y. Ahn, E.K. Lee, Y.J. Lee, K.W. Kim, S.K. Hahn, Y.K. Youn, K.H. Kim, B.Y. Cho, J. Park, The association of the BRAF(V600E) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: a meta-analysis. Cancer 118, 1764–1773 (2012)
J. Abubaker, Z. Jehan, P. Bavi, M. Sultana, S. Al-Harbi, M. Ibrahim, A. Al-Nuaim, M. Ahmed, T. Amin, M. Al-Fehaily, O. Al-Sanea, F. Al-Dayel, S. Uddin, K.S. Al-Kuraya, Clinicopathological analysis of papillary thyroid cancer with PIK3CA alterations in a Middle Eastern population. J. Clin. Endocrinol. Metab. 93, 611–618 (2008)
Z. Liu, P. Hou, M. Ji, H. Guan, K. Studeman, K. Jensen, V. Vasko, A.K. El-Naggar, M. Xing, Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 93, 3106–3116 (2008)
J.R. Pollack, C.M. Perou, A.A. Alizadeh, M.B. Eisen, A. Pergamenschikov, C.F. Williams, S.S. Jeffrey, D. Botstein, P.O. Brown, Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat. Genet. 23, 41–46 (1999)
D. Pinkel, D.G. Albertson, Comparative genomic hybridization. Annu. Rev. Genomics Hum. Genet. 6, 331–354 (2005)
M. Hermann, Standard and molecular cytogenetics of endocrine tumors. Am. J. Clin. Pathol. 119, S17–S38 (2003)
K. Unger, E. Malisch, G. Thomas, H. Braselmann, A. Walch, G. Jackl, P. Lewis, E. Lengfelder, T. Bogdanova, J. Wienberg, H. Zitzelsberger, Array CGH demonstrates characteristic aberration signatures in human papillary thyroid carcinomas governed by RET/PTC. Oncogene 27, 4592–4602 (2008)
H. Zitzelsberger, G. Thomas, K. Unger, Chromosomal aberrations in thyroid follicular-cell neoplasia: in the search of novel oncogenes and tumour suppressor genes. Mol. Cell. Endocrinol. 321, 57–66 (2010)
P.G. Buckley, K.K. Mantripragada, A. Piotrowski, T. Diaz de Ståhl, J.P. Dumanski, Copy-number polymorphisms: mining the tip of an iceberg. Trends Genet. 21, 315–317 (2005)
Ihle, M.A., Fassunke, J., Konig, K., Grunewald, I., Schlaak, M., Kreuzberg, N., Tietze, L., Schildhaus, H.U., Buttner, R., Merkelbach-Bruse, S.: Comparaison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer. 14 doi:10.1186/1471-2407-14-13. (2014)
M. Johansson, A. Oudin, K. Tiemann, A. Bernard, A. Golebiewska, O. Keunen, F. Fack, D. Stieber, B. Wang, H. Hedman, S.P. Niclou, The soluble form of the tumor suppressor Lrig1 potently inhibits in vivo glioma growth irrespective of EGF receptor status. Neuro-Oncol 15, 1200–1211 (2013)
S.H. Jang, J.W. Park, H.R. Kim, J.G. Seong, H.K. Kim, ADRM1 gene amplification is a candidate driver for gastric cancer. Clin. Exp. Metastasis 31, 727–733 (2014)
K.J. Livak, T.D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT. Methods 25(4), 402–408 (2001)
A. Kallioniemi, CGH microarrays and cancer. Curr. Opin. Biotechnol. 19, 36–40 (2008)
B. Singh, D. Lim, J.C. Cigudosa, R. Ghossein, A.R. Shaha, A. Poluri, V.B. Wreesmann, M. Tuttle, J.P. Shaha, P.H. Rao, Screening for genetic aberrations in papillary thyroid cancer by using comparative genomic hybridization. Surgery 128, 888–894 (2000)
V.B. Wreesman, R.A. Ghossein, S.G. Patel, C.P. Harris, E.A. Schnaser, A.R. Shaha, R.M. Tuttle, J.P. Shah, P.H. Rao, B. Singh, Genome-wide appraisal of thyroid cancer progression. Am. J. Pathol. 161, 1556–1559 (2002)
D. Miura, N. Wada, K. Chin, G.G. Magrane, M. Wong, Q.Y. Duh, O.H. Clark, Anaplastic thyroid cancer: cytogenetic patterns by comparative genomic hybridization. Thyroid 3, 283–290 (2003)
R.F. Rodrigues, L. Roque, J. Rosa-Santos, O. Cid, J. Soares, Chromosomal imbalances associated with anaplastic transformation of follicular thyroid carcinomas. Br. J. Cancer 90, 492–496 (2004)
R. Rodrigues, L. Roque, C. Espadinha, A. Pinto, R. Dominigues, J. Dinis, A. Catarino, T. Pereira, V. Leite, Comparative genomic hybridization, BRAF, RAS, RET, and oligo-array analysis in aneuploid papillary thyroid carcinomas. Oncol. Rep. 18, 917–926 (2007)
L. Roque, R. Rodrigues, A. Pinto, V. Moura-Nunes, J. Soares, Chromosomal imbalances in thyroid follicular neoplasms: a comparaison between follicular adenomas and carcinomas. Genes Chromosom. Cancer 36, 292–302 (2003)
S. Finn, P. Smyth, E. O’Regan, S. Cahill, M. Toner, C. Timon, R. Flavin, J. O’Leary, O. Sheils, Low-level genomic instability is a feature of papillary thyroid carcinoma: an array comparative genomic hybridization study of laser capture microdissected papillary thyroid carcinoma tumors and clonal cell lines. Arch. Pathol. Lab. Med. 1, 65–73 (2007)
K. Unger, E. Malisch, G. Thomas, H. Braselmann, A. Walch, G. Jackl, P. Lewis, E. Lengfelder, T. Bogdanova, J. Wienberg et al., Array CGH demonstrates characteristic aberration signatures in human papillary thyroid carcinomas governed by RET/PTC. Oncogene 27, 4592–4602 (2008)
L.Z. Fehér, G. Pocsay, L. Krenács, A. Zvara, E. Bagdi, R. Pocsay, G. Lukács, F. Gyory, A. Gazdag, E. Tarkó, L.G. Puskás, Amplification of thymosin beta 10 and AKAP13 genes in metastatic and aggressive papillary thyroid carcinomas. Pathol. Oncol. Res. 18, 449–458 (2012)
J.J. Lee, A.Y.M. Au, T. Foukakis, M. Barbaro, N. Kiss, R. Clifton-Bligh, J. Staaf, A. Borg, L. Delbridge, B.G. Robinson, G. Wallin, A. Hoog, C. Larsson, Array-CGH identifies cyclin D1and UBCH10 amplicons in anaplastic thyroid carcinoma. Endocr. Rel. Cancer 15, 801–815 (2008)
J. Sandgren, T. Diaz de Ståhl, R. Andersson, U. Menzel, A. Piotrowski, H. Nord, M. Bäckdahl, N.B. Kiss, M. Brauckhoff, J. Komorowski, H. Dralle, O. Hessman, C. Larsson, G. Akerström, C. Bruder, J.P. Dumanski, G. Westin, Recurrent genomic alterations in benign and malignant pheochromocytomas and paragangliomas revealed by whole-genome array comparative genomic hybridization analysis. Endocr Relat Cancer 17, 561–579 (2010)
The Cancer Genome Atlas Research Network, Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 (2014)
R. Pankov, K.M. Yamada, Fibronectin at a glance. J. Cell Sci. 115, 3861–3863 (2000)
C.M. Williams, A.J. Engler, R.D. Slone, L.L. Galant, J.E. Schwarzbauer, Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res. 68, 3185–3192 (2008)
C.A. da Silveira Mitteldorf, J.M. de Sousa-Canavez, K.R. Leite, C. Massumoto, L.H. Camara-Lopes, FN1, GALE, MET, and QPCT overexpression in papillary thyroid carcinoma: molecular analysis using frozen tissue and routine fine-needle aspiration biopsy samples. Diagn. Cytopathol. 39, 556–561 (2011)
E. Hesse, P.B. Musholt, E. Potter, T. Petrich, M. Wehmeier, R. von Wasielewski, R. Lichtinghagen, T.J. Musholt, Oncofoetal fibronectin—a tumour-specific marker in detecting minimal residual disease in differentiated thyroid carcinoma. Br. J. Cancer 93, 565–570 (2005)
M. Fryknäs, U. Wickenberg-Bolin, H. Göransson, M.G. Gustafsson, T. Foukakis, J.J. Lee, U. Landegren, A. Höög, C. Larsson, L. Grimelius, G. Wallin, U. Pettersson, A. Isaksson, Molecular markers for discrimination of benign and malignant follicular thyroid tumors. Tum. Biol. 27, 211–220 (2006)
H. Wang, Z. Yan, J. Cai, H. Robinson, H. Ke, Kinetic and structural studies of phosphodiesterase-8A and implication on the inhibitor selectivity. Biochemistry 47, 12760–12768 (2008)
L. Persani, A. Lania, L. Alberti, R. Romoli, G. Mantovani, S. Filetti, A. Spada, M. Conti, Induction of specific phosphodiesterase isoforms by constitutive activation of the cAMP pathway in autonomous thyroid adenomas. J. Clin. Endocrinol. Metab. 85, 2872–2878 (2000)
V. Lakics, E.H. Karran, F.G. Boess, Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 59, 367–374 (2010)
L.G. Bazzara, M.L. Vélez, M.E. Costamagna, A.M. Cabanillas, L. Fozzatti, A.M. Lucero, C.G. Pellizas, A.M. Masini-Repiso, Nitric oxide/cGMP signaling inhibits TSH-stimulated iodide uptake and expression of thyroid peroxidase and thyroglobulin mRNA in FRTL-5 thyroid cells. Thyroid 17, 717–727 (2007)
L. Sulaiman, I.L. Nilsson, C.C. Juhlin, F. Haglund, A. Höög, C. Larsson, J. Hashemi, Genetic characterization of large parathyroid adenomas. Endocr. Relat. Cancer 19, 389–407 (2012)
M. Raish, M. Khurshid, M.A. Ansari, P.K. Chaturvedi, S.M. Bae, J.H. Kim, E.K. Park, D.C. Park, W.S. Ahn, Analysis of molecular cytogenetic alterations in uterine leiomyosarcoma by array-based comparative genomic hybridization. J. Cancer Res. Clin. Oncol. 138, 1173–1186 (2012)
F. Persson, Y. Andrén, M. Winnes, B. Wedell, A. Nordkvist, G. Gudnadottir, R. Dahlenfors, H. Sjögren, J. Mark, G. Stenman, High-resolution genomic profiling of adenomas and carcinomas of the salivary glands reveals amplification, rearrangement, and fusion of HMGA2. Genes Chrom. Cancer 48, 69–82 (2009)
F. Haeseleer, K. Palczewski, Short-chain dehydrogenases/reductases in retina. Methods Enzymol. 316, 372–383 (2000)
F. Cerignoli, X. Guo, B. Cardinali, C. Rinaldi, J. Casaletto, L. Frati, I. Screpanti, L.J. Gudas, A. Gulino, C.J. Thiele, G. Giannini, retSDR1, a short-chain retinol dehydrogenase/reductase, is retinoic acid-inducible and frequently deleted in human neuroblastoma cell lines. Cancer Res. 62, 1196–1204 (2002)
G. Oler, C.P. Camacho, F.C. Hojaij, P. Michaluart Jr, G.J. Riggins, J.M. Cerutti, Gene expression profiling of papillary thyroid carcinoma identifies transcripts correlated with BRAF mutational status and lymph node metastasis. Clin. Cancer Res. 14, 4735–4742 (2008)
L. Shi, Dock protein family in brain development and neurological disease. Commun. Integr. Biol. 6, e26839 (2013)
L.M. Caronia, J.E. Phay, M.H. Shah, Role of BRAF in thyroid oncogenesis. Clin. Cancer Res. 17, 7511–7517 (2011)
R.P. Tufano, G.V. Teixeira, J. Bishop, K.A. Carson, M. Xing, BRAF mutation in papillary thyroid cancer and its value in tailoring initial treatment: a systematic review and meta-analysis. Medicine 91, 274–286 (2012)
E. Puxeddu, S. Filetti, BRAF mutation assessment in papillary thyroid cancer: are we ready to use it in clinical practice? Endocrine 45, 341–343 (2014)
M.S. Khan, A.A. Pandith, S.R. Masoodi, K.A. Wani, M.U.I. Hussain, S. Mudassar, Epigenetic silencing of TSHR gene in thyroid cancer patients in relation to their BRAF V600E mutation status. Endocrine 47, 449–455 (2014)
H. Shao, X. Yu, W. Cuifang, Q. Wang, H. Guan, Midkine expression is associated with clinicopathological features and BRAF mutation in papillary thyroid cancer. Endocrine 46, 285–291 (2014)
L. Wang, J. Xu, L. Zeng, X. Ye, Q. Wu, J. Dai, C. Ji, S. Gu, C. Zhao, Y. Xie, Y. Mao, Cloning and characterization of a novel human STAR domain containing cDNA KHDRBS2. Mol. Biol. Rep. 29, 369–375 (2002)
M.A. Attar, J.C. Salem, H.S. Pursel, L.C. Santy, CNK3 and IPCEF1 produce a single protein that is required for HGF dependent Arf6 activation and migration. Exp. Cell Res. 318, 228–237 (2012)
H.E. Speedy, M.C. Di Bernardo, G.P. Sava, M.J.S. Dyer, A. Holroyd, Y. Wang, N. Sunter, L. Mansouri, G. Juliusson, K.E. Smedby, G. Roos, S. Jayne, A. Majid, C. Dearden, A.G. Hall, T. Mainou-Fowler, G.H. Jackson, G. Summerfield, R.J. Harris, A.R. Pettitt, D.J. Allsup, J.R. Bailey, G. Pratt, C. Pepper, C. Fegan, R. Rosenquist, D. Catovsky, I.M. Allan, R.S. Houlston, A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat. Genet. 46, 56–60 (2014)
Acknowledgments
This work is funded by grants to GD from Associazione Italiana per la Ricerca sul Cancro (AIRC) (Project No IG 10296), to DR (MIUR: Grant 2010NFEB9L_003), and to MC (MIUR: Grant RBFR12FI27_003). Authors acknowledge Dr. W. Wilson for editorial work of English language.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Passon, N., Bregant, E., Sponziello, M. et al. Somatic amplifications and deletions in genome of papillary thyroid carcinomas. Endocrine 50, 453–464 (2015). https://doi.org/10.1007/s12020-015-0592-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-015-0592-z