
Abstract
Chronic kidney disease-mineral and bone disorder (CKD-MBD) is a complex interplay of risk factors, diseases and outcomes. In the last two decades, there has been a steady drift towards definitions depending ever more closely on laboratory abnormalities, which only represent at best possible risk factors for downstream pathology. In the early years of nephrology, especially when the therapeutic agents we had available to combat CKD–MBD were modest, bone and muscle problems were often advanced, and symptomatic, with bone and muscle pain, fractures and myopathy. This ‘old style’ hyperparathyroidism paradigm has become substantially less common now, and we can forget how symptomatic, and how draining of life-quality, CKD-MBD can be. In this chapter, we explore in the main the most feared complication of all, that of a fracture, which not only are dramatically more common in CKD patients, but also have a much heightened mortality over similar fractures but not in a CKD setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The very first descriptions of chronic kidney disease-mineral and bone disorder (CKD-MBD) (or renal rickets, or renal osteodystrophy) were nearly 100 years ago now, well before the modern phenomenon of mass dialysis therapy was remotely envisaged.
In 1921, ten cases of stunted development associated with bone deformities caused by chronic nephritis were described [1]. It took several more decades to appreciate that vitamin D deficiency in children led to rickets (the lack of the ‘anti-rachitic factor’) but there was another contribution to bone pathology beyond vitamin D—renal rickets—in which the cartilaginous growth plate in uraemic children was not widened, but devoid of cartilage cells, with advanced fibrosis in severe cases.
At the beginning of the era of dialysis, in the early to mid-1960s, with so many people surviving to some degree with advanced CKD, a wealth of new signs, symptoms and syndromes were described by the pioneers in our field. In 1966, Stanbury and Lumb correlated plasma calcium and phosphate values with bone disease in 134 uraemic individuals [2]. In patients with osteitis fibrosa, plasma calcium, phosphate and product were all significantly higher than seen with osteomalacia or rickets. In this era, severe bone pain, resorption of phalanges, myalgia, myopathy, bone fractures, tendon snapping and avulsion, large bony deformities due to Brown tumours, tumoural calcification, calcinosis, vascular calcification and calciphylaxis were all well recognized as signs of advanced renal bone diseases.

Calciphylaxis (calcific uraemic arteriolopathy): lower panel is distal necrotic gangrenous variant, upper panel is central fat necrotic variant. Permission from Manson Publishers, London. Clinical Handbook of Renal Medicine Ed Pattison, Goldsmith, Fervenza, Hartley and el Gramde. London, 2004
Parathyroidectomy, or if fortunate, transplantation, were the only safe solutions to such advanced high-turnover bone disease, as it would not be until the mid-1970s that vitamin D compounds became available for clinical use.
Pathological Basis of CKD-MBD
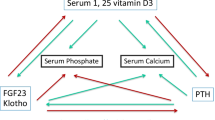
The major factors responsible for continued stimulation of parathyroid gland activity, function and eventual hyperplasia in renal failure are hypocalcaemia, diminished 1,25-dihydroxyvitamin D levels and hyperphosphatemia. CKD profoundly disrupts systemic calcium and phosphate homoeostasis and affects the bone, gut and parathyroid glands. This occurs because of decreased renal excretion of phosphate and diminished renal hydroxylation of 25-hydroxyvitamin D to calcitriol (1,25-dihydroxyvitamin D).
Circulating calcitriol levels begin to fall when the glomerular filtration rate is less than 60 ml/min [3] and are typically markedly reduced in subjects with end-stage renal failure. The loss of functioning renal tissue, physiologic suppression by hyperphosphatemia (a relatively late phenomenon in CKD) and rise in serum FGF-23 levels all participate in the marked and progressive decline in calcitriol synthesis [4].
The Calcium Sending Receptor (CaSR), which is highly expressed in the parathyroid glands, permits variations in the serum calcium concentration to be instantaneously sensed by the parathyroid gland, leading to appropriate changes in PTH secretion. The fall in serum calcium concentration with renal failure, as sensed by the CaSR, is a potent stimulus to the release of PTH. Extracellular calcium acting through the CaSR plays the predominant role in regulating parathyroid function, resulting in this receptor being the major therapeutic target for suppressing parathyroid gland function [5].
Since vitamin D stimulates intestinal phosphate absorption, the decrease in active vitamin D production may be viewed as an adaptive response to minimize hyperphosphatemia in the setting of reduced renal phosphate excretion in patients with kidney failure. Calcitriol deficiency also affects a variety of factors regulating calcium homoeostasis, thereby altering PTH levels. There are reductions in calcitriol-regulated calcium absorption in the gut and calcium release from bone, both of which promote the development of hypocalcaemia; this is a potent additional stimulus to the release of parathyroid hormone [4, 6].
A decrease in calcitriol levels also lowers the number of vitamin D receptors in the parathyroid cells. The lack of calcitriol and the decreased number of receptors may both directly promote parathyroid chief cell hyperplasia and nodule formation through non-genomic effects.
Hyperphosphatemia is also an important factor that promotes and accelerates hyperparathyroidism. Although the identity of the extracellular phosphate sensor is unknown, a novel phosphaturic factor, FGF23, regulated by phosphate and vitamin D [7] has a potentially important role in regulating parathyroid gland function in end-stage renal disease (ESRD). Hyperphosphatemia also lowers the levels of ionized calcium and interferes with the production of 1,25-dihydroxyvitamin D, thereby resulting in increased PTH levels [6].
Symptoms and Signs of CKD-MBD
The first manifestations of CKD-MBD are biochemical, and these derangements happen, as we have discussed, early in CKD and a long time before any symptomatology ensues [8]. Indeed, symptomatic CKD-MBD is much rarer now than was the case two or more decades ago, as better detection, better prevention and better treatment options all now exist for the majority of CKD patients. However, occasional patients do present late with advanced CKD with symptomatic bone pathologies, and also, there are non-compliant patients whose bone disease can run out of control, despite the opportunities for treatment. Thus, it is important not to forget the symptoms and signs of CKD-MBD.
However, if the physiological biochemical abnormalities are not corrected, renal bone disease, referred to as renal osteodystrophy, will inevitably develop over time. Although frequently asymptomatic, this disorder can result in weakness, fractures, bone and muscle pain and avascular necrosis. These symptoms and signs do not generally occur until the patient is undergoing maintenance dialysis. Severe bone pain and myopathy more likely represents osteomalacia than osteitis fibrosa cystica.
There are several forms of renal osteodystrophy, including osteitis fibrosa cystica, adynamic bone disease and osteomalacia. In some patients, there is evidence of more than one type, which is called mixed osteodystrophy. Osteitis fibrosa cystica and mixed osteodystrophy are largely the direct result of increased PTH levels, while adynamic bone disease is a consequence of excessive suppression of the parathyroid gland with current therapies [4, 6].
Bone biopsies of affected patients with osteitis fibrosa cystica demonstrate increased bone turnover activity and defective mineralization. This disorder is generally asymptomatic, but is associated with bone pain in a minority of patients. There is an increased risk of fractures. Severe symptomatic disease is currently uncommon with modern therapy. Adynamic bone disease, which is characterized by low osteoclastic and osteoblastic activity and bone formation rates, is due in part at least to excess suppression of the parathyroid gland with current therapies, particularly calcium-containing phosphate binders and vitamin D analogues. These patients typically maintain a low serum intact PTH concentration, which is frequently accompanied by an elevated serum calcium level. Adynamic bone disease increases the risk for fractures and metastatic calcification [9].
Features of Some of the Clinical Manifestations of CKD-MBD
Fractures
Fracture rates are elevated in patients with predialysis chronic kidney disease [10, 11]. Cross-sectional data suggest fracture risk increases as kidney function declines and by CKD stage 4 approximates that for ESRD [12]. Furthermore, fracture-related mortality for patients with predialysis CKD is approximately twofold higher than the general population. These data are alarming because CKD and osteoporosis frequently co-localize, and the incidence and prevalence of predialysis CKD, osteoporosis and fragility fracture are expected to increase exponentially as the population ages. Reliable methods to identify patients with CKD at risk for fracture are currently lacking. Thus, there is a critical need to develop diagnostic tests and assess their utility to detect increased fracture risk in the burgeoning CKD population. The vexed issue of bisphosphonates in CKD is dealt with in this journal issue [13].
Fracture rates in patients with CKD-5D are reported to be similar to or greater than that of people 10 or 20 years older in the general population (see [14]). In a study using data from the U.S. Renal Data System (USRDS) Registry, hip fractures were increased fourfold in Caucasian dialysis patients compared with a matched sample from the general population. Although the fracture risk was higher for all age groups of CKD-5D in this study, for those less than 65 years old the relative risk (RR) ranged between 10- and 100-fold higher, most likely due to the low incidence of hip fracture in the general population in this age range [15]. The incidence of hip fracture was 7.5 per 1,000 patient-years for men and 13.6 per 1,000 patient-years for women and the RR increased with time on dialysis [15].
In the second phase of the Dialysis Outcomes and Practice Patterns Study (DOPPS), which recorded information on 12,782 haemodialysis (HD) patients from 320 dialysis facilities across 12 countries (2002–2004), the incidence of hip fracture was 8.9 per 1,000 patient-years and all fractures 25.6 per 1,000 patient-years [16]. There were no major differences in fracture rates between countries after adjustment for demographics and co-morbidities. The incidence of long-bone fractures in over 7,000 patients studied over a 5-year period in the Dialysis Morbidity and Mortality Study (DMMS) was 16.9 per 1,000 patient-years, with the femoral neck being the most common site (59.8%) [17]. One single-centre study of Japanese HD patients reported the prevalence of fractures was 15% in men and 30% in women and another Japanese study showed the prevalence of vertebral fractures in diabetic HD patients to be greater compared with non-diabetic dialysis patients (32 vs. 13%).
Few studies have addressed the risk of fracture and its consequences at earlier stages of CKD, but there is increasing evidence to suggest that patients with CKD-3 to 5 are at greater fracture risk than the general population. One recent study of 13,177 people aged 75 years and over from the United Kingdom reported that levels of estimated glomerular filtration rate (eGFR) less than 45 ml/min/1.73 m2 were associated with an almost twofold increase in mortality after hip fracture [18]. In a cross-sectional analysis of the National Health and Nutrition Examination Survey (NHANES) III study, moderate to severe CKD was independently associated with a more than twofold increase in hip fracture and this association was stronger than several traditional risk factors including age and gender [19]. In another retrospective cohort from Veterans Affairs medical centres in the United States, the RR of hip fracture for men with levels of eGFR between 30–59 and 15–29 ml/min/1.73 m2 was 1.28 and 3.98, respectively. These studies suggest that the risk of hip fracture in patients with advanced renal impairment is similar to that of dialysis patients [20].
Bone strength is determined by the amount of bone present and the quality of that bone. In the clinic, bone mass is usually assessed by dual energy X-ray absorptiometry (DXA), which provides an estimate of bone mineral density (BMD, g/cm2). Bone quality is related to other material properties of the skeleton, including bone microarchitecture and remodelling activity. Microarchitecture and remodelling are linked; low and high remodelling lead to loss of bone structural integrity and increased fracture risk.
An important very recent study examined the possible diagnosis of future fracture in CKD using biomarkers of bone quality and turnover [10]. The main findings demonstrating that certain Bone Turnover Markers were able to distinguish between patients with and without fracture. The highest tertile of formation (osteocalcin, P1NP) and resorption (Trap-5b) markers were independently associated with increased odds of prior fracture. Combining the highest tertile level of osteocalcin, P1NP, or Trap-5b with FN T-score improved discrimination of fracture history over measurement of FN T-score or BTMs alone. Fracture prevalence was highest in CKD patients with a FN T-score of -2.0 or less and either osteocalcin, P1NP, or Trap-5b in the highest two tertiles. Indeed, for any combination of elevated BTM and low FN BMD, specificity and negative predictive value for fracture were around 80%.
The authors suggested that predialysis CKD patients can be risk stratified for fragility by combining BTMs with BMD. Interestingly, whether BTMs were renally excreted or not did not affect fracture discrimination. In terms of pathogenesis, high iPTH and BTM levels were associated with low bone mass, smaller bone size, and a disrupted trabecular network, abnormalities which have found to be associated with fracture.
In prospective studies, high BSAP levels predicted abnormalities associated with fracture, such as bone loss, turnover and mineralization defects, but have not been shown to predict fracture per se until very recently, with a large study reported from Japan in which BMD was measured annually and serum biochemistry monthly for 485 haemodialysed patients from April 2003 to March 2008, and all fractures were recorded. Forty-six new episodes of any type of fracture and 29 cases of prevalent spine fracture were recorded. Serum bone-specific alkaline phosphatase (b-AP) was a very useful surrogate marker for any type of incident fracture risk [area under curve (AUC) = 0.766, P < 0.0001]. A significantly greater risk of any type of incident fracture was associated with parathyroid hormone (PTH) levels either less than 150 pg/ml [hazard ratio (HR) = 3.47, P < 0.01] or greater than 300 pg/ml (HR = 5.88, P < 0.0001) compared with 150–300 pg/ml. Receiver-operating characteristic analysis demonstrated a significant predictive power for incident of any type of fracture by BMD at the total hip (AUC = 0.760, P < 0.0001) and other hip regions in women in the lower PTH group (PTH < 204 pg/ml). BMDs at every site but whole body or lumbar spine had significant power to discriminate prevalent spine fracture regardless of gender or PTH [11].
Brown Tumours
Osteoclastomas or Brown tumours are caused by localized replacement of bone by vascularized fibrous tissue (osteitis fibrosa cystica) resulting from PTH-stimulated osteoelastic activity. They are often painful, and in addition, can cause significant symptoms by local compression.

Parathyroid Brown tumour: upper panel is isotopic bone scan showing avidity, lower panel is X-ray of tibia, showing bone cystic changes. Permission from Manson Publishers, London. Clinical Handbook of Renal Medicine Ed Pattison, Goldsmith, Fervenza, Hartley and el Gramde. London, 2004
The fibrous tissue contains giant cells and the lesions may become cystic following necrosis and liquefaction. Radiographically, Brown tumours are well-defined lytic lesions, often eccentric or cortical, that may cause endosteal scalloping and osseous expansion. These lesions occur more frequently in primary hyperparathyroidism, and their manifestation in secondary hyperparathyroidism was not described until 1963. The prevalence of osteoclastoma in secondary hyperparathyroidism has been cited by Katz et al. and Griffiths et al. as 1.5 and 1.7%, respectively [21]. Brown tumours are frequently solitary, although multiple lesions have been reported. Ribs, pelvis, especially facial bones and femurs are common locations, and the axial skeleton may be involved with neurobiological sequelae. After successful treatment of hyperparathyroidism, typically after parathyroidectomy, Brown tumours may heal with calcification, sclerosis and lesion disappearance or the lytic area may persist.

Phalangeal erosions and scalloping (above) and vertebral striping (‘rugger jersey’) in advanced secondary hyperparathyroidism. Permission from Manson Publishers, London. Clinical Handbook of Renal Medicine Ed Pattison, Goldsmith, Fervenza, Hartley and el Gramde. London, 2004
Calciphylaxis (Calcific Uraemic Arteriolopathy)
Calciphylaxis is a rare and often lethal disorder characterized by systemic medial calcification of arterioles, which leads to ischaemia and subcutaneous necrosis. Histological examination reveals small vessel mural calcification with or without endovascular fibrosis, extravascular calcification (amorphous luminal calcium) and thrombotic vaso-occlusion (leading to ischaemic skin necrosis) [22].
Calciphylaxis is one of several types of extra-osseous calcification (which also include intimal, medial and valvular calcifications) that may occur in patients with end-stage renal disease (ESRD) [22–24]. Traditionally, it has been classified as metastatic calcification indicating passive mineralization of serum calcium and phosphate crystals.
Calciphylaxis most commonly occurs in patients with ESRD who are on HD or who have recently received a renal transplant, but it can occur with primary hyperparathyroidism without significant CKD.
Clinical calciphylaxis was first described very early in the dialysis era (1950s to 1960s). Although previously rare, the incidence of this disorder has appeared to be increasing, due in part to the awareness and the recognition of clinical signs and risk factors associated with calciphylaxis. Multiple studies have identified major clinical factors and co-morbid conditions that may help recognize patients at risk for the development of calciphylaxis in both ESRD and non-ESRD patients [22, 24].
Women undergoing dialysis have higher risk of developing calciphylaxis. Among patients with ESRD, obesity (BMI > 30) may lead to excess stress on dermal and hypodermal arterioles, resulting in focally dystrophic calcification of arterioles. Increasing increments in phosphorus levels among those with ESRD is a risk factor [24] but so are a great number of other parameters and clinical characteristics. The administration of different medications is associated with an increased risk of calciphylaxis in patients with ESRD. These include warfarin, calcium-based binders and vitamin D analogues and systemic corticosteroids [24].
Calciphylaxis is typically characterized by areas of excruciatingly painful ischaemic necrosis that usually develop on areas with greatest adiposity including abdomen, buttock and thigh. There are violaceous, painful, plaque-like subcutaneous nodules. The initial purpuric plaques and nodules subsequently progress to ischaemic/necrotic ulcers with eschars that often become superinfected [22, 24].
Ischaemic myopathy is a less frequent complication that can occur without skin necrosis. Digital ischaemia has a somewhat better prognosis than proximal skin necrosis, but these patients are still at appreciable risk. Novel therapies are being investigated [24–26]. More information and therapeutic insights into this devastating condition can be gleaned from [27].
Conclusions
Although the current diagnostic and interventional paradigms for CKD-MBD are almost exclusively biochemical in nature, well before patients have any significant symptomatology, it should be remembered that CKD-MBD can be a significant cause of morbidity, symptoms, loss of quality of life, and in the case of fractures and calciphylaxis, can definitely shorten life expectancy significantly. Being aware of these symptoms is important for those patients who either present acutely with severe and advanced CKD, or, who refuse treatment and whose bone and mineral disorders relentlessly progress to become more and more clinically evident.
References
Feldenfeld AJ, Torres A. Bone histomorphometry in chronic kidney disease, chapter 10. In: Olgaard PK, Silver J, Salusky IB, editors. The spectrum of mineral and bone disorder in chronic kidney disease. Oxford: Oxford University Press; 2010.
Stanbury SW, Lumb GA. Parathyroid function in chronic renal failure. A statistical survey of the plasma biochemistry in azotaemic renal osteodystrophy. Q J Med. 1966;35(137):1–23.
Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, Andress DL. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71(1):31–8.
Sprague SM. Renal bone disease. Curr Opin Endocrinol Diabetes Obes. 2010;17(6):535–9.
Geibel JP. The calcium-sensing receptor. J Nephrol. 2010;23(Suppl 16):S130–5.
Cunningham J, Locatelli F, Rodriguez M. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin J Am Soc Nephrol. 2011;6(4):913–21.
John GB, Cheng CY, Kuro OM. Role of Klotho in aging, phosphate metabolism, and CKD. Am J Kidney Dis. 2011;58(1):127–34.
Goldsmith DJ, Cunningham J. Mineral metabolism and vitamin D in chronic kidney disease—more questions than answers. Nat Rev Nephrol. 2011;7(6):341–6.
Moore C, Yee J, Malluche H, Rao DS, Monier-Faugere MC, Adams E, Daramola-Ogunwuyi O, Fehmi H, Bhat S, Osman-Malik Y. Relationship between bone histology and markers of bone and mineral metabolism in African-American hemodialysis patients. Clin J Am Soc Nephrol. 2009;4(9):1484–93.
Nickolas TL, Cremers S, Zhang A, Thomas V, Stein E, Cohen A, Chauncey R, Nikkel L, Yin MT, Liu XS, Boutroy S, Staron RB, Leonard MB, McMahon DJ, Dworakowski E, Shane E. Discriminants of prevalent fractures in chronic kidney disease. J Am Soc Nephrol. 2011;22(8):1560–72.
Iimori S, Mori Y, Akita W, Kuyama T, Takada S, Asai T, Kuwahara M, Sasaki S, Tsukamoto Y. Diagnostic usefulness of bone mineral density and biochemical markers of bone turnover in predicting fracture in CKD stage 5D patients—a single-center cohort study. Nephrol Dial Transplant. 2011. doi:10.1093/ndt/gfr317.
Toussaint ND, Elder GJ, Kerr PG. A rational guide to reducing fracture risk in dialysis patients. Semin Dial. 2010;23(1):43–54.
Miller J, Stoller ML. Urological aspects of management. Clinic Rev Bone Miner Metab. 2011. doi:10.1007/s12018-011-9109-3.
Nickolas TL, McMahon DJ, Shane E. Relationship between moderate to severe kidney disease and hip fracture in the United States. J Am Soc Nephrol. 2006;17:3223–32.
Collins AJ, Kasiske B, Herzog C, Chen SC, Everson S, Constantini E, Grimm R, McBean M, Xue J, Chavers B, Matas A, Manning W, Louis T, Pan W, Liu J, Li S, Roberts T, Dalleska F, Snyder J, Ebben J, Frazier E, Sheets D, Johnson R, Li S, Dunning S, Berrini D, Guo H, Solid C, Arko C, Daniels F, Wang X, Forrest B, Gilbertson D, St Peter W, Frederick P, Eggers P, Agodoa L. Excerpts from the United States renal data system 2003 annual data report: atlas of end-stage renal disease in the United States. Am J Kidney Dis. 2003;42(6 Suppl 5):A5–7, S1–230.
Blayney MJ, Tentori F. Trends and consequences of mineral bone disorder in haemodialysis patients: lessons from The Dialysis Outcomes and Practice Patterns Study (DOPPS). J Ren Care. 2009;35(Suppl 1):7–13.
Dooley AC, Weiss NS, Kestenbaum B. Increased risk of hip fracture among men with CKD. Am J Kidney Dis. 2008;51:38–44.
Fried LF, Biggs ML, Shlipak MG, Seliger S, Kestenbaum B, Stehman-Breen C, Sarnak M, Siscovick D, Harris T, Cauley J, Newman AB, Robbins J. Association of kidney function with incident hip fracture in older adults. J Am Soc Nephrol. 2007;18:282–6.
Danese MD, Kim J, Doan QV, Dylan M, Griffiths R, Chertow GM. PTH and the risks for hip, vertebral, and pelvic fractures among patients on dialysis. Am J Kidney Dis. 2006;47(1):149–56.
Nitsch D, Mylne A, Roderick PJ, Smeeth L, Hubbard R, Fletcher A. Chronic kidney disease and hip fracture-related mortality in older people in the UK. Nephrol Dial Transplant. 2009;24:1539–44.
Klawansky S, Komaroff E, Cavanaugh PF Jr, Mitchell DY, Gordon MJ, Connelly JE, Ross SD. Relationship between age, renal function and bone mineral density in the US population. Osteoporos Int. 2003;14:570–6.
Rix M, Andreassen H, Eskildsen P, Langdahl B, Olgaard K. Bone mineral density and biochemical markers of bone turnover in patients with predialysis chronic renal failure. Kidney Int. 1999;56:1084–93.
Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15(1):48–54.
Brandenburg VM, Cozzolino M, Ketteler M. Calciphylaxis: a still unmet challenge. J Nephrol. 2011;24(2):142–8.
Covic A, Kanbay M, Voroneanu L, Turgut F, Serban DN, Serban IL, Goldsmith DJ. Vascular calcification in chronic kidney disease. Clin Sci (Lond). 2010;119(3):111–21.
Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23(3):258–62.
Morrow B, Qunibi W. Specific bone and mineral disorders in patients with chronic kidney disease. Clinic Rev Bone Miner Metab. 2011. doi:10.1007/s12018-011-9114-6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Heymann, E.P., Jenkins, M. & Goldsmith, D. Clinical Features and Manifestations of CKD-MBD. Clinic Rev Bone Miner Metab 10, 142–148 (2012). https://doi.org/10.1007/s12018-011-9115-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-011-9115-5