
Abstract
Osteoporosis is characterized by the reduced mass as well as quality of bone, and increased risk of fragility fracture. Skeletal homeostasis is maintained through the balanced activities of osteoclasts and osteoblasts, and osteoporosis is considered to be a metabolic disorder caused by an imbalance between bone resorption and formation. Whereas osteoclasts and osteoblasts have been the primary targets for elucidating the cell-based mechanisms underlying the pathophysiology of osteoporosis, much less attention has been paid to the role of osteocytes. This review focuses on the physiologic function of osteocytes in the regulation of bone and mineral metabolism, summarizing the findings from human disease and mouse genetics, and then extending the discussion to the pathogenetic roles of osteocytes in skeletal aging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Differentiation of Osteocytes from Osteoblasts: The Gene Expression Signature
The osteocyte is a form of the terminally differentiated osteoblast [1]. The bone-forming osteoblasts synthesize collagen matrix and then mineralize it. A sub-population of osteoblasts, which has not been defined yet, takes on the cell fate of embedding in the matrix and becoming osteocytes. Osteocytes are postmitotic and quiescent cells, and unlike osteoblasts and osteoclasts, which are clustered on the bone surface, they are separated from each other physically while being connected functionally through gap junctions at the ends of their cell processes [2–4].
The osteocyte differentiation process has been extensively characterized by gene expression studies [5–7]. The typical genes expressed in osteoblasts include type I collagen, alkaline phosphatase, osteocalcin and RANKL, which are involved in bone formation and also the regulation of osteoclastic bone resorption. On the other hand, dentin matrix protein (DMP)-1, sclerostin, E11/podoplanin and FGF23 are among the products of osteocytes [8–12]. The differential expression of these genes in osteoblasts vis-a-vis osteocytes has been analyzed by expression profiling techniques [5, 6, 13]. Using transgenic mice expressing fluorescent proteins in either osteocytes or osteoblasts driven by the Dmp1 or Col1a1 promoter, respectively, Paic et al. showed that Bmp4, Bmp8a, Enpp1 and Ank, as well as Dmp1 and Phex, which are known to be osteocytic genes, are expressed in osteocytes at higher levels than in osteoblasts [6]. By comparing the established cell lines osteoblastic 2T3 versus osteocytic MLO-Y4, it has been shown that certain transcription factors, such as Vdr, Tcf7 and Irx5, are predominantly expressed in osteocytes [13].
Another transcription factor, Dlx3, a gene responsible for Tricho-dento-osseous syndrome, has also been reported to be expressed in osteocytes [14]. Sclerostin, an antagonist of Wnt signaling, is known as a secretory product of osteocytes, and the expression of Sost, the gene encoding sclerostin, is regulated by a MEF2 family transcription factor [15]. It has recently been reported that the expression of Osterix (Osx), encoded by Sp7, a gene essential for osteoblast differentiation, is also highly expressed in osteocytes and that conditional deletion in postnatal mice results in down-regulation of Dmp1, Phex, and Sost [16]. In the conditional Osx KO mice, impaired mineralization and disorganization of collagen fibers were observed, while the number of proliferating osteoblasts and the expression of Runx2 were increased, suggesting that Osx may play a role in osteocyte maturation and function [16]. Tcf7 and Dlx3 are downstream regulators of the Wnt/β-catenin signaling pathway [17, 18], and Osx has been reported to inhibit its signaling [19]. It is conceivable that TCF7, Dlx3, MEF2 and Osx, which are not osteocyte-specific transcription factors, function co-operatively in the differentiation and maturation of osteocytes. Further studies are required to better address the function in osteocyte physiology in vivo of the transcription network involving these factors.
As osteoblasts terminally differentiate into osteocytes, the cells become deeply embedded in the bone matrix, where nutrient and oxygen supplies are considered to be quite limited. Hirao et al. demonstrated that differentiation of cultured osteoblasts is accelerated under hypoxic (5% O2) conditions and that the expression of osteocytic genes, such as Dmp1, Mepe, and Fgf23, is elevated compared with that in osteoblasts cultured under normoxic (20% O2) condition [20]. Intriguingly, osteocytic gene expression was reported to be upregulated in a three dimensional culture of osteoblasts. Boukhechba et al. reported that osteoblasts cultured densely on hydroxyapatite/tricalcium phosphate biphasic calcium phosphate ceramic particles exhibit accelerated differentiation, and exhibited an osteocytic gene expression pattern within a week of culture [21]. Thus, in addition to the intrinsic transcriptional network within osteocytes themselves, environmental cues, such as the oxygen tension and 3D architecture, are likely to play critical roles in osteocytogenesis and cell fate.
The Dendritic Morphology of Osteocytes: An Application of Mouse Genetics
Osteocytes possess a unique morphology with numerous dendritic processes, which has been implicated in their function of sensing the mechanical stress and matrix properties around them [4, 22]. The development and establishment of the lacuno-canalicular system of osteocytes has been shown to be correlated with bone matrix maturation [4, 22, 23]. The lacuno-canalicular system can be visualized by silver staining [23–26], and using this technique, Hirose et al. reported that in the hindlimb of very young mice, the alignment of round-shaped osteocytes and the orientation of cell processes are random and that along with maturation of the skeleton, the cells become flattened and regularly aligned at certain intervals [24]. Thus, at 12 weeks of age, most osteocytes exhibit a flattened shape, and their cellular processes are extended both in parallel and perpendicularly to the longitudinal axis of the hindlimb toward the cortical bone surface [24]. Osteocytic processes have also been shown to be stained with phalloidin [27], suggesting that the processes consist of actin-based cytoskeletons similar to microvilli, which sense both nutrients and the extracellular fluid flow.
Studies on genetically modified mice have revealed morphological alterations in osteocytes and the lacuno-canalicular network. Matrix metalloproteinases (MMPs) play critical roles in the remodeling and maintenance of extracellular matrix, and mice lacking MT1-MMP, encoded by Mmp14, exhibit a massive loss of osteocytic dendritic processes in the cortical bone, concomitantly with diminished collagen cleavage in the lacuno-canalicular system, suggesting that cleavage of collagen is required for the development and maintenance of these osteocytic processes [25].
An autosomal recessive form of multicentric osteolysis with severe osteoporosis is caused by loss-of-function mutations in the MMP2 gene [28]. In the calvaria of Mmp2 knockout mice, the proportion of empty lacunae was significantly increased, while the number of osteocytic processes and their connections were decreased, indicating that the formation of osteocytic network is impaired in Mmp2 KO mice [29]. Since the impairment of the osteocytic network evidently preceded osteocytic death, it is inferred that such network formation is crucial for osteocyte survival.
A disorganized lacuno-canalicular system is also a feature of Dmp1 knockout mice [30]. Osteocytic lacunae in Dmp1KO mice were much larger than those of wild-type mice, with a decreased number and chaotic orientation [30]. The cell surface of the mutant osteocytes was frizzy, and the osteocytic processes were abnormal [30]. An altered osteocyte morphology was also noted in Osx KO mice [16], and like the case with the Dmp1KO mice, the number of dendritic processes was decreased. Taken together with the finding that Dmp1 expression is significantly reduced in Osx-deficient osteocytes, it is suggested that the Osx-DMP1 axis is required for the normal morphology and function of osteocytes.
Another extracellular matrix component, perlecan, a large heparan sulfate proteoglycan, has been implicated in the physiology of the lacuno-canalicular system [31, 32]. The pericellular matrix of osteocytes is thought to consist of polysaccharide moieties, forming the so-called glycocalyx [33–35]. The heparan sulfate proteoglycan perlecan possesses a large amount of sulfated glycosaminoglycans, the proposed functions of which include acting as a reservoir for growth factors and their regulators, cell adhesion and ionic filtering in the basement membrane [36, 37]. Mice lacking Hspg2, encoding perlecan, died in utero or perinatally due to defects in the integrity of the basement membrane during the course of heart and brain development [38]. Mice carrying a hypomorphic mutation in Hspg2 gene exhibited a less severe phenotype, exhibiting dwarfism and skeletal defects [39] which resembled the phenotype of type 1 Schwartz-Jampel syndrome, in which mutations in the HSPG2 gene have been found [39, 40]. Thompson et al. have recently reported that perlecan is localized in the pericellular space of the lacuno-canalicular system and that Hspg2 hypomorphic mice displayed significant decreases in the area and size of the osteocytic canaliculi [41]. How the alterations in the canalicular system affect osteocyte function in terms of mineral metabolism and mechanotransduction has not been described in detail as yet, and further information on the morphology-function relationship is much anticipated.
Osteocytes in Mineral Homeostasis
Osteocytic genes, such as Phex, Fgf23, and Dmp1, have been implicated in phosphate metabolism [42, 43]. PHEX was identified as a candidate gene for X-linked hypophosphatemia (XLH) through positional cloning approach [44]. Hyp mice with spontaneous hypophosphatemia and osteomalacia carry a loss-of-function mutation in the Phex gene. Phex encodes a type II membrane protein which is homologous to a Zn-dependent protease [44]. In Phex mutants, the osteocytic expression of FGF23 is elevated, which is suggested to be responsible for the hypophosphatemic phenotype [12]. Yuan et al. [45] demonstrated that conditional knockout mice for Phex in the osteoblast lineage driven by the osteocalcin promoter exhibited a hypophosphatemia phenotype and that the phenotype in hyp mice was rescued by osteoblast-specific expression of wild-type Phex. These results are consistent with the notion that the Phex expressed in the osteoblast-osteocyte lineage plays a key role in phosphate homeostasis.
FGF23 is a member of the FGF19 subfamily of the FGF superfamily that exerts systemic and endocrine modes of activity [46, 47]. Mutations of the FGF23 gene have been identified in patients with autosomal dominant hypophosphatemic rickets (ADHR), who exhibit enhanced FGF23 activity in a gain-of-function manner due to resistance to the proteolytic cleavage of the molecule [48]. Although the PHEX protein possesses proteolytic activity, the cleavage of FGF23 has not been demonstrated [49]. FGF23 acts on target cells through the conventional FGF receptors (FGFRs), and a co-receptor, α-klotho, that has been identified as a gene mutated in a premature-aging mouse, klotho [50, 51]. Both klotho mice and Fgf23 KO mice exhibit hyperphosphatemia, with a high serum concentration of vitamin D, and a premature-aging phenotype [52].
DMP1 has been identified as one of the genes responsible for autosomal recessive hypophosphatemic rickets (ARHR) [30, 53]. ARHR, characterized by renal phosphate wasting, altered regulation of 1α-hydroxylase activity and rickets/osteomalacia, resembles XLH and ADHR [54]. In Dmp1 KO mice, which exhibit hypophosphatemic rickets [30], both the serum FGF23 levels and osteocytic Fgf23 expression are elevated, similar to Phex-deficient mice. Aberrant expression of Col1a1 and elevated Podoplanin/E11, a marker of early osteocytes, were observed in the osteocytes of the Dmp1 KO mice, suggesting that DMP1 is involved in osteocyte maturation [30].
Matrix extracellular phosphoglycoprotein (MEPE), also known as OF45, was identified as a gene upregulated in the tumors of oncogenic osteomalacia patients [55, 56], although its in vivo function in phosphate metabolism is not known [57]. MEPE is highly expressed in osteocytes and, together with DMP1 and osteopontin, which is encoded by Spp1, belongs to the family of small integrin-binding ligand, N-linked glycoproteins (SIBLINGs) [58]. These SIBLING genes are clustered in a region of chromosome 4q21 and are thought to have derived from an ancestor gene by duplications [59, 60]. It is reported that proteolytically cleaved peptides derived from MEPE inhibit matrix mineralization [55, 61].
Loss-of-function mutations of the ENPP1 gene, which is reportedly expressed in osteocytes, have been found in patients with ARHR [62, 63]. ENPP1 encodes ecto-nucleotide pyrophosphatase/phosphodiesterase, which generates inorganic pyrophosphate, an inhibitor of mineralization [64]. Mutations of the gene in humans and mice cause ectopic calcification, such as generalized arterial calcification in infancy and ossification of the posterior longitudinal ligament of the spine [65, 66]. ARHR patients with an ENPP1 mutation presented with normal levels of FGF23, PTH and vitamin D metabolites [62, 63], suggesting that ENPP1 may function locally in matrix mineralization. How the loss of Enpp1 activity specifically causes under-mineralization in bone, vis-a-vis the ectopic calcification phenotype in extra-skeletal tissues, remains to be elucidated.
In summary, although the kidney is considered to be the main player in phosphate homeostasis, osteocytes nevertheless do play a key role. Osteocytes produce two proteins, Phex and FGF23, with opposite actions in phosphate metabolism, as evidenced by the hypo- or hyper-phosphatemia caused by loss of the respective gene [45, 67]. The causative role of FGF23 in the Phex mutant has been demonstrated by showing that hyp phenotype was reversible by crossing with Fgf23 KO mice [68] or by the administration of FGF23 blocking antibodies [69]. Thus, osteocytes deploy two methods of coupling and balancing the processes of phosphate metabolism and matrix mineralization. First, they produce proteins with opposite biological activities, FGF23 and Phex; second, they utilize a local as well as systemic (i.e. communicating with the kidney via FGF23) mode of regulation. It is worth noting that these methods are very well adapted to maintaining these cells, allowing a fine-tuning of the matrix stiffness and/or quality surrounding them.
PTH Activity in Osteocytes
The calcitrophic hormone PTH is a central regulator of bone metabolism. Intermittent administration of PTH increases bone mass through robust stimulation of bone formation and currently is the only anabolic therapy for osteoporotic patients [70, 71]. The molecular and cellular basis underling PTH-mediated activation of osteoblastic function has not been fully elucidated. Recently, osteocytes have received attention as one of the primary targets of PTH action in bone, as the receptor for PTH, PTHR1, is highly expressed in them. O’Brien et al. generated a transgenic mouse model expressing constitutively active PTHR1 specifically in osteocytes and have presented evidence that the mice exhibit accelerated bone turnover and increased bone mass [72].
It has been reported that osteocytic expression of two Wnt antagonists, sclerostin and Dkk1, is significantly reduced by PTH treatment [73–75], suggesting that PTH stimulates bone formation via down-regulation of the Wnt antagonists and therefore, facilitated Wnt signaling. Furthermore, PTH induces stabilization of β-catenin, which mimics the canonical pathway of Wnt signaling through the activation of PTHR1 and PKA [76, 77]. Since the Wnt canonical pathway is known to activate cell proliferation, e.g. through the induction of cyclin D1 [78], it was expected at first to stimulate bone formation. In fact, ectopic expression of constitutively stabilized β-catenin in osteoblasts did result in a high bone mass phenotype. However, this turned out to be caused not by stimulation of bone formation but by reduced bone resorption through up-regulation of OPG, an antagonist of RANKL [79]. Holmen et al. [80] analyzed conditional KO mice for β-catenin or Apc, a negative regulator of β-catenin, in osteoblasts using an osteocalcin-Cre deletor, and demonstrated that the Wnt canonical signaling increases Opg expression, while decreasing Rankl expression. Although osteoblasts from Apc-conditional KO mice exhibited normal differentiation ex vivo, those from β-catenin conditional KO mice exhibited retarded differentiation [80], suggesting that β-catenin also has certain cell autonomous functions in osteoblasts.
Recently, Kramer et al. [81] reported the development of osteocyte-specific β-catenin knockout mice driven by Dmp1 promoter. The mice exhibited mild postnatal growth retardation and premature lethality with early-onset and progressive bone loss along with accelerated osteoclastic bone resorption, whereas the parameters for osteoblasts and osteocytes were normal [81]. The increased bone resorption was ascribed to a reduction in Opg expression, which is consistent with the notion that β-catenin signaling in osteocytes plays a role in the control of bone resorption via the regulation of Opg expression [81]. In contrast, it is widely recognized that the PKA pathway activated by PTH-PTHR1 signaling up-regulates RANKL expression in osteoblasts [82–84] and suppresses the osteocytic expression of Sost via MEF2 [15], thereby leading to the stimulation of both the resorption and formation of bone. Further studies are required to clarify the relevance of the signaling pathway involving PTH-R, PKA, β-catenin and RANKL/OPG in osteocyte function to better understand the molecular basis of the anabolic signaling of PTH in osteocytes.
Osteocytic Osteolysis
While bone resorption by osteoclasts is believed to be a major force driving the mobilization of minerals from bone, there has been a long-standing hypothesis that osteocytes themselves elute minerals from the peri-lacunar matrix, a phenomenon called “osteocytic osteolysis” [85, 86]. This concept is supported by both the ontology of osteocytes in bone and a number of experimental observations. Osteocytes are the most abundant cell type in bone and, as mentioned previously, extend numerous and long cell processes through canaliculi. As a result, the cell surface area facing the bone matrix far exceeds that of osteoblasts and osteoclasts. Thus, osteocytes, forming an extensive network throughout the skeleton, are best suited to monitor and maintain bone matrix quality. Conceivably, the ontology of osteocytes enables an efficient mobilization of calcium, with a minimum amount of mineral dissolution on an individual cell basis, and without provoking a resorption cavity and concomitant alteration in bone structure accompanying bona fide bone resorption by osteoclasts.
Experimentally, the phenomenon of osteocytic osteolysis has been reported in relation to PTH action on calcium metabolism. Talmage et al., based on the observations that PTH administration to thyroparathyroidectomized rats induced a rapid release of calcium into the plasma, proposed that the acute change they observed in the plasma calcium level resulted from an increase in calcium transport through the osteocyte-lining cell complex, rather than osteoclastic bone resorption [87]. Krempien et al. observed changes in the morphology of osteocytes as well as the distribution of intracellular organelles, with osteolysis of their lacunar walls, following chronic administration of PTH in rats [88]. Tazawa et al. have shown that continuous administration of PTH into 8-month-old rats caused enlargement of osteocytic lacunae and up-regulation of acid phosphatase activity in osteocytes [89]. Thus, although the concept remains to be proven experimentally, it is tempting to speculate that osteocytes have the potential to mobilize minerals through an as yet undefined mechanism and serve important physiologic functions, especially in the case of the need for calcium.
Osteocytes in Osteoporosis
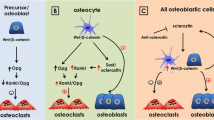
Osteocytes produce sclerostin and Dkk1, both of which are antagonists of Wnt signaling, and major regulators of bone accrual and osteoblast differentiation. Thus, a negative feedback loop is envisioned in which osteoblasts differentiate into osteocytes, which in turn suppress the differentiation of osteoblasts by antagonizing Wnt signaling. Osteocytes are also supposed to generate a repellent signal against osteoclasts in light of the observations that osteoclastic bone resorption is targeted toward damaged or empty lacunae in which osteocytes are dying [90, 91]. These lines of evidence are compatible with the working hypothesis that osteocytes keep both osteoblastic and osteoclastic activities in check, thereby regulating the net balance of bone remodeling (Fig. 1). According to this model, it would follow that a loss of osteocytic control alone could manifest as an imbalance between bone formation and resorption, a metabolic hallmark of osteoporosis.

Osteocytic control of BMU activity: the ‘metabolostat’ concept. 1 In the resting state of the basic multicelluar unit (BMU), osteocytes keep the activities of osteoclasts and osteoblasts in check by sending them ‘cytostatic’ signals. 2 When osteocytes are damaged or lost, the ‘cytostatic’ signals to both osteoblasts and osteoclasts are eliminated. 3 As a net result, osteoclastic activation and bone resorption prevail, according to the observations in the osteocyte ablation model [92]. 4 Following a reversal phase, osteoblasts start to be recruited to the resorbed cavity. 5 As osteoblasts synthesize bone matrix, some of them become embedded in the newly formed matrix and undergo terminal differentiation into osteocytes. During this process, osteoid osteocytes may be active in matrix mineralization. 6 Upon completion of BMU renewal, fully differentiated mature osteocytes regain the ‘cytostatic’ control over osteoblasts and osteoclasts
Tatsumi et al. [92] developed a transgenic mouse model expressing the receptor for diphtheria toxin (DT), a human-specific toxin, specifically in osteocytes, using the Dmp1-promoter. Whereas the transgenic mouse itself displayed apparently normal bone development and metabolism, a single injection of DT induced a massive loss of osteocytes within 2–3 days [92]. The ablation of osteocytes in these mice caused an acute increase in bone resorption along with an upregulation of RANKL expression [92], which findings are consistent with the concept that osteocytes normally function to keep osteoclasts from aberrant activation (Fig. 1).
The osteocyte-less model provides a suitable experimental system for addressing the role of osteocytes in mechanotransduction in an intact animal [92]. Unloading on hindlimbs by tail suspension usually induces acute and profound bone loss due to the activation of osteoclastic bone resorption, while osteocyte-ablated mice were resistant to the unloading-induced bone loss as well as the activation of osteoclasts. The osteocyte-less mice responded normally, however, to re-loading (following tail suspension), and bone mass lost during unloading was restored just as in control mice with intact osteocytes. Collectively, these data were interpreted to suggest that osteocytes play an essential role in sensing and/or transducing mechano-signals, at least during the catabolic phase of unloading, whereas the intact osteocytic network is not an absolute requirement for the robust anabolic response following the catabolic phase.
Another intriguing finding of the osteocyte ablation model was the occurrence, even in young animals, of microfractures in the cortical bone reminiscent of the microcracks observed in human subjects with aging [92], implying that osteocytes may be active in the detection and/or repair of microdamage. Inversely, microcracks have been reported to trigger osteocytic death by disrupting the lacuno-canalicular system [93, 94]. Thus, a loss of osteocytes could be either a cause or a result of microdamage propagation, depending on the extent of the damage.
Estrogen is an essential hormone for the growth and maintenance of the skeleton, and postmenopausal estrogen deficiency causes bone loss. Tomkinson et al. reported that in cases of estrogen withdrawal following the treatment of women with a gonadotropin-releasing hormone analogue, osteocyte viability was reduced due to increased apoptosis [95]. Regarding the relationship between osteocytes and aging, Vashishth et al. reported that a reduced density of osteocyte lacunae in human cortical bone is associated with an accumulation of microcracks in aging [96]. Qiu et al. reported the osteocyte density in human cancellous bone is reduced in osteoporotic patients with vertebral fracture, compared with non-fracture healthy controls [97]. They also noted that the osteocyte density in iliac bone biopsies declines with aging [98]. Taken together with the findings in the osteocyte ablation model [92], although an acutely massive cell loss does not occur physiologically, or even in the usual pathological cases, these clinical observations imply that osteocytes serve an important physiologic function in the maintenance of skeletal health. Furthermore, a decline in their viability and/or activity could be a substantial risk, predisposing to bone fragility and fracture.
Glucocorticoid excess is recognized as a secondary aspect of osteoporosis and a major cause of fracture. The glucocorticoid receptor is expressed in human osteocytes and osteoblasts [99]. Weinstein et al. reported that osteocyte apoptosis is increased in patients with glucocorticoid-induced osteonecrosis of the hip [100]. They demonstrated experimentally that glucocorticoid administration causes apoptosis of osteocytes as well as osteoblasts [101]. Thus, glucocorticoid activity in the osteoblast-osteocyte lineage may play an etiologically causal role in the development of bone fragility.
Currently, osteoporosis is most often treated with bisphosphonates (BPs), which are known as anti-resorptive drugs. It has been reported that BPs promote survival of osteocytes [102] and that labeled BP was found to be present in osteocytic lacunae [103], raising the intriguing possibility that BPs protect bone by modulating osteocytic function.
Conclusion: The ‘metabolostat’ Hypothesis
Accumulating evidence points to important roles for osteocytes in bone and mineral homeostasis. In view of the findings that osteocytes produce the Wnt antagonists, sclerostin and Dkk1, to suppress bone formation and that loss of osteocytes leads to accelerated bone resorption, it is suggested that osteocytes play primarily a “regulatory” role in bone remodeling by acting as a ‘metabolostat’ (Fig. 1). According to this model, osteocytes send ‘cytostatic’ signals to both osteoblasts and osteoclasts in order to keep their functions in proper check (Fig. 1).
Mechanical and hormonal signals are sensed by osteocytes and modulate the ‘metabolostat’ function (Fig. 2). Aging, mechanical overload or glucocorticoid excess as well as decreased blood supply causes osteocytic loss and/or dysfunction, ultimately leading to bone fragility. Osteocyte function depends on the integrity of the lacuno-canalicular network, which senses mechanical stress, regulates surrounding matrix properties, and under certain circumstances sends information to osteoblasts and bone-lining cells at the bone surface. Osteocytes have often been introduced as ‘the third cell’ of bone, but in fact they represent the principal cell type controlling bone remodeling, coordinately regulating the activities of two effector cells, osteoblasts and osteoclasts, in response to mechanical and hormonal stimuli (Figs. 1, 2).

Triangular regulation by the osteocyte, osteoblast and osteoclast. Osteocytes sense mechanical stress and receive PTH and other hormonal stimuli, such as estrogen and glucocorticoid, and integrate and translate them into anabolic or catabolic signals to osteoblasts or osteoclasts (the direct actions of these hormonal and mechanical signals on osteoblasts and/or osteoclasts are assumed, but omitted in this illustration). Candidates for the ‘cytostatic’ signals from osteocytes to osteoblasts include sclerostin and Dkk1, which are downregulated by mechanical loading. As a ‘cytostatic’ signal from osteocytes to osteoclasts, OPG is conceivable, but remains to be proven. The black and white arrows indicate catabolic and anabolic signals, respectively
References
Nijweide PJ, Burger EH, Klein-Nulend J. The osteocyte. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of bone biology. 2nd ed. San Diego: Academic Press; 2002. p. 93–107.
Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42:606–15.
Noble BS. The osteocyte lineage. Arch Biochem Biophys. 2008;473:106–11.
Burger EH, Klein-Nulend J. Mechanotransduction in bone–role of the lacuno-canalicular network. FASEB J. 1999;13(Suppl):S101–12.
Dean AK, Harris SE, Kalajzic I, Ruan J. A systems biology approach to the identification and analysis of transcriptional regulatory networks in osteocytes. BMC Bioinformatics. 2009;10(Suppl 9):S5.
Paic F, Igwe JC, Nori R, Kronenberg MS, Franceschetti T, Harrington P, Kuo L, Shin DG, Rowe DW, Harris SE, Kalajzic I. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone. 2009;45:682–92.
Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Dev Dyn. 2006;235:176–90.
Kalajzic I, Braut A, Guo D, Jiang X, Kronenberg MS, Mina M, Harris MA, Harris SE, Rowe DW. Dentin matrix protein 1 expression during osteoblastic differentiation, generation of an osteocyte GFP-transgene. Bone. 2004;35:74–82.
Wetterwald A, Hoffstetter W, Cecchini MG, Lanske B, Wagner C, Fleisch H, Atkinson M. Characterization and cloning of the E11 antigen, a marker expressed by rat osteoblasts and osteocytes. Bone. 1996;18:125–32.
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–76.
Kobayashi K, Imanishi Y, Koshiyama H, Miyauchi A, Wakasa K, Kawata T, Goto H, Ohashi H, Koyano HM, Mochizuki R, Miki T, Inaba M, Nishizawa Y. Expression of FGF23 is correlated with serum phosphate level in isolated fibrous dysplasia. Life Sci. 2006;78:2295–301.
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291:E38–49.
Yang W, Harris MA, Heinrich JG, Guo D, Bonewald LF, Harris SE. Gene expression signatures of a fibroblastoid preosteoblast and cuboidal osteoblast cell model compared to the MLO-Y4 osteocyte cell model. Bone. 2009;44:32–45.
Li H, Marijanovic I, Kronenberg MS, Erceg I, Stover ML, Velonis D, Mina M, Heinrich JG, Harris SE, Upholt WB, Kalajzic I, Lichtler AC. Expression and function of Dlx genes in the osteoblast lineage. Dev Biol. 2008;316:458–70.
Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, Keller H. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res. 2007;22:1957–67.
Zhou X, Zhang Z, Feng JQ, Dusevich VM, Sinha K, Zhang H, Darnay BG, de Crombrugghe B. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc Natl Acad Sci USA. 2010;107:12919–24.
Hwang J, Mehrani T, Millar SE, Morasso MI. Dlx3 is a crucial regulator of hair follicle differentiation and cycling. Development. 2008;135:3149–59.
Willert K, Jones KA. Wnt signaling: is the party in the nucleus? Genes Dev. 2006;20:1394–404.
Zhang C, Cho K, Huang Y, Lyons JP, Zhou X, Sinha K, McCrea PD, de Crombrugghe B. Inhibition of Wnt signaling by the osteoblast-specific transcription factor Osterix. Proc Natl Acad Sci USA. 2008;105:6936–41.
Hirao M, Hashimoto J, Yamasaki N, Ando W, Tsuboi H, Myoui A, Yoshikawa H. Oxygen tension is an important mediator of the transformation of osteoblasts to osteocytes. J Bone Miner Metab. 2007;25:266–76.
Boukhechba F, Balaguer T, Michiels JF, Ackermann K, Quincey D, Bouler JM, Pyerin W, Carle GF, Rochet N. Human primary osteocyte differentiation in a 3D culture system. J Bone Miner Res. 2009;24:1927–35.
Turner CH, Robling AG, Duncan RL, Burr DB. Do bone cells behave like a neuronal network? Calcif Tissue Int. 2002;70:435–42.
Kusuzaki K, Kageyama N, Shinjo H, Takeshita H, Murata H, Hashiguchi S, Ashihara T, Hirasawa Y. Development of bone canaliculi during bone repair. Bone. 2000;27:655–9.
Hirose S, Li M, Kojima T, de Freitas PH, Ubaidus S, Oda K, Saito C, Amizuka N. A histological assessment on the distribution of the osteocytic lacunar canalicular system using silver staining. J Bone Miner Metab. 2007;25:374–82.
Holmbeck K, Bianco P, Pidoux I, Inoue S, Billinghurst RC, Wu W, Chrysovergis K, Yamada S, Birkedal-Hansen H, Poole AR. The metalloproteinase MT1-MMP is required for normal development and maintenance of osteocyte processes in bone. J Cell Sci. 2005;118:147–56.
Schoen FA. A method to stain decalcified bone without loss of structural detail. Biotech Histochem. 1991;66:216–9.
Kamioka H, Honjo T, Takano-Yamamoto T. A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone. 2001;28:145–9.
Martignetti JA, Aqeel AA, Sewairi WA, Boumah CE, Kambouris M, Mayouf SA, Sheth KV, Eid WA, Dowling O, Harris J, Glucksman MJ, Bahabri S, Meyer BF, Desnick RJ. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 2001;28:261–5.
Inoue K, Mikuni-Takagaki Y, Oikawa K, Itoh T, Inada M, Noguchi T, Park JS, Onodera T, Krane SM, Noda M, Itohara S. A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J Biol Chem. 2006;281:33814–24.
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–5.
You LD, Weinbaum S, Cowin SC, Schaffler MB. Ultrastructure of the osteocyte process and its pericellular matrix. Anat Rec A Discov Mol Cell Evol Biol. 2004;278:505–13.
Sauren YM, Mieremet RH, Groot CG, Scherft JP. An electron microscopic study on the presence of proteoglycans in the mineralized matrix of rat and human compact lamellar bone. Anat Rec. 1992;232:36–44.
Burra S, Nicolella DP, Francis WL, Freitas CJ, Mueschke NJ, Poole K, Jiang JX. Dendritic processes of osteocytes are mechanotransducers that induce the opening of hemichannels. Proc Natl Acad Sci USA. 2010;107:13648–53.
Wang L, Wang Y, Han Y, Henderson SC, Majeska RJ, Weinbaum S, Schaffler MB. In situ measurement of solute transport in the bone lacunar-canalicular system. Proc Natl Acad Sci USA. 2005;102:11911–6.
Cowin SC, Weinbaum S, Zeng Y. A case for bone canaliculi as the anatomical site of strain generated potentials. J Biomech. 1995;28:1281–97.
Forsberg E, Kjellen L. Heparan sulfate: lessons from knockout mice. J Clin Invest. 2001;108:175–80.
Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–7.
Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–8.
Rodgers KD, Sasaki T, Aszodi A, Jacenko O. Reduced perlecan in mice results in chondrodysplasia resembling Schwartz-Jampel syndrome. Hum Mol Genet. 2007;16:515–28.
Nicole S, Davoine CS, Topaloglu H, Cattolico L, Barral D, Beighton P, Hamida CB, Hammouda H, Cruaud C, White PS, Samson D, Urtizberea JA, Lehmann-Horn F, Weissenbach J, Hentati F, Fontaine B. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat Genet. 2000;26:480–3.
Thompson WR, Modla S, Grindel BJ, Czymmek KJ, Kirn-Safran CB, Wang L, Duncan RL, Farach-Carson MC. Perlecan/Hspg2 deficiency alters the pericellular space of the lacuno-canalicular system surrounding osteocytic processes in cortical bone. J Bone Miner Res. 2010.
Fukumoto S. The role of bone in phosphate metabolism. Mol Cell Endocrinol. 2009;310:63–70.
Bonewald LF. Osteocytes as dynamic multifunctional cells. Ann NY Acad Sci. 2007;1116:281–90.
Francis F, Hennig S, Korn B, Reinhardt R, de Jong P, Poustka A, Lehrach H, Rowe PSN, Goulding JN, Summerfield T, Mountford R, Read AP, Popowska E, Pronicka E, Davies KE, O’Riordan JLH, Econs MJ, Nesbitt T, Drezner MK, Oudet CL, Pannetier S, Hanauer A, Strom TM, Meindl A, Lorenz B, Cagnoli B, Mohnike KL, Murken J, Meitinger T. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11:130–6.
Yuan B, Takaiwa M, Clemens TL, Feng JQ, Kumar R, Rowe PS, Xie Y, Drezner MK. Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest. 2008;118:722–34.
Fukumoto S. Actions and mode of actions of FGF19 subfamily members. Endocr J. 2008;55:23–31.
Kharitonenkov A. FGFs and metabolism. Curr Opin Pharmacol. 2009;9:805–10.
ADHR-Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–8.
Rowe PS. The wrickkened pathways of FGF23, MEPE and PHEX. Crit Rev Oral Biol Med. 2004;15:264–81.
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51.
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–4.
Razzaque MS, Lanske B. Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med. 2006;12:298–305.
Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38:1248–50.
Miller WL, Portale AA. Genetic causes of rickets. Curr Opin Pediatr. 1999;11:333–9.
Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000;67:54–68.
Petersen DN, Tkalcevic GT, Mansolf AL, Rivera-Gonzalez R, Brown TA. Identification of osteoblast/osteocyte factor 45 (OF45), a bone-specific cDNA encoding an RGD-containing protein that is highly expressed in osteoblasts and osteocytes. J Biol Chem. 2000;275:36172–80.
Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, Simmons HA, Crawford DT, Chidsey-Frink KL, Ke HZ, McNeish JD, Brown TA. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003;278:1998–2007.
Fisher LW, Fedarko NS. Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res. 2003;44(Suppl 1):33–40.
Fisher LW, Torchia DA, Fohr B, Young MF, Fedarko NS. Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochem Biophys Res Commun. 2001;280:460–5.
Bellahcene A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer. 2008;8:212–26.
Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone. 2004;34:303–19.
Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86:267–72.
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86:273–8.
Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millan JL. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164:1199–209.
Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nurnberg P. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–81.
Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet. 1998;19:271–3.
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–8.
Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Juppner H, Lanske B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–32.
Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S, Shimada T. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009;24:1879–88.
Tashjian AH Jr, Gagel RF. Teriparatide [human PTH(1–34)]: 2.5 years of experience on the use and safety of the drug for the treatment of osteoporosis. J Bone Miner Res. 2006;21:354–65.
Canalis E. Update in new anabolic therapies for osteoporosis. J Clin Endocrinol Metab. 2010;95:1496–504.
O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One. 2008;3:e2942.
Guo J, Liu M, Yang D, Bouxsein ML, Saito H, Galvin RJ, Kuhstoss SA, Thomas CC, Schipani E, Baron R, Bringhurst FR, Kronenberg HM. Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab. 2010;11:161–71.
Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–83.
Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–58.
Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008;22:2968–79.
Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJ, Martin TJ, Gillespie MT, Onyia JE. Effects of parathyroid hormone on Wnt signaling pathway in bone. J Cell Biochem. 2005;95:1178–90.
Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–7.
Glass DA II, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64.
Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, Williams BO. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280:21162–8.
Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–85.
Fu Q, Jilka RL, Manolagas SC, O’Brien CA. Parathyroid hormone stimulates receptor activator of NFkappa B ligand and inhibits osteoprotegerin expression via protein kinase A activation of cAMP-response element-binding protein. J Biol Chem. 2002;277:48868–75.
Kondo H, Guo J, Bringhurst FR. Cyclic adenosine monophosphate/protein kinase A mediates parathyroid hormone/parathyroid hormone-related protein receptor regulation of osteoclastogenesis and expression of RANKL and osteoprotegerin mRNAs by marrow stromal cells. J Bone Miner Res. 2002;17:1667–79.
Lee SK, Lorenzo JA. Regulation of receptor activator of nuclear factor-kappa B ligand and osteoprotegerin mRNA expression by parathyroid hormone is predominantly mediated by the protein kinase a pathway in murine bone marrow cultures. Bone. 2002;31:252–9.
Qing H, Bonewald LF. Osteocyte remodeling of the perilacunar and pericanalicular matrix. Int J Oral Sci. 2009;1:59–65.
Teti A, Zallone A. Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone. 2009;44:11–6.
Talmage RV, Grubb SA. A laboratory model demonstrating osteocyte-osteoblast control of plasma calcium concentrations. Table model for plasma calcium control. Clin Orthop Relat Res. 1977;122:299–306.
Krempien B, Friedrich E, Ritz E. Effect of PTH on osteocyte ultrastructure. Adv Exp Med Biol. 1978;103:437–50.
Tazawa K, Hoshi K, Kawamoto S, Tanaka M, Ejiri S, Ozawa H. Osteocytic osteolysis observed in rats to which parathyroid hormone was continuously administered. J Bone Miner Metab. 2004;22:524–9.
Noble BS, Peet N, Stevens HY, Brabbs A, Mosley JR, Reilly GC, Reeve J, Skerry TM, Lanyon LE. Mechanical loading: biphasic osteocyte survival and targeting of osteoclasts for bone destruction in rat cortical bone. Am J Physiol Cell Physiol. 2003;284:C934–43.
Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res. 2000;15:60–7.
Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–75.
Hazenberg JG, Freeley M, Foran E, Lee TC, Taylor D. Microdamage: a cell transducing mechanism based on ruptured osteocyte processes. J Biomech. 2006;39:2096–103.
Noble B. Bone microdamage and cell apoptosis. Eur Cell Mater. 2003;6:46–55. discussion 55.
Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS. The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res. 1998;13:1243–50.
Vashishth D, Verborgt O, Divine G, Schaffler MB, Fyhrie DP. Decline in osteocyte lacunar density in human cortical bone is associated with accumulation of microcracks with age. Bone. 2000;26:375–80.
Qiu S, Rao DS, Palnitkar S, Parfitt AM. Reduced iliac cancellous osteocyte density in patients with osteoporotic vertebral fracture. J Bone Miner Res. 2003;18:1657–63.
Qiu S, Rao DS, Palnitkar S, Parfitt AM. Age and distance from the surface but not menopause reduce osteocyte density in human cancellous bone. Bone. 2002;31:313–8.
Abu EO, Horner A, Kusec V, Triffitt JT, Compston JE. The localization of the functional glucocorticoid receptor alpha in human bone. J Clin Endocrinol Metab. 2000;85:883–9.
Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab. 2000;85:2907–12.
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–82.
Plotkin LI, Aguirre JI, Kousteni S, Manolagas SC, Bellido T. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of extracellular signal-regulated kinase activation. J Biol Chem. 2005;280:7317–25.
Roelofs AJ, Coxon FP, Ebetino FH, Lundy MW, Henneman ZJ, Nancollas GH, Sun S, Blazewska KM, Bala JL, Kashemirov BA, Khalid AB, McKenna CE, Rogers MJ. Fluorescent risedronate analogues reveal bisphosphonate uptake by bone marrow monocytes and localization around osteocytes in vivo. J Bone Miner Res. 2010;25:606–16.
Acknowledgments
This study was supported in part by grants for the Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO) of Japan (to K.I.) and from Takeda Science Foundation (to K.I.). Pacific Edit reviewed the manuscript before submission.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Watanabe, K., Ikeda, K. Osteocytes in Normal Physiology and Osteoporosis. Clinic Rev Bone Miner Metab 8, 224–232 (2010). https://doi.org/10.1007/s12018-010-9076-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-010-9076-0