
Abstract
The model of asthma as a single entity has now been replaced by a much more complex biological network of distinct and interrelating inflammatory pathways. The term asthma is now considered an umbrella diagnosis for several diseases with distinct mechanistic pathways (endotypes) and variable clinical presentations (phenotypes). The precise definition of these endotypes is central to asthma management due to inherent therapeutic and prognostic implications. This review presents the molecular mechanisms behind the heterogeneity of airway inflammation in asthmatic patients. Asthma endotypes may be broadly regarded as type 2 (T2) high or T2-low. Several biologic agents have been approved for T2-high asthma, with numerous other therapeutics that are incipient and similarly targeted at specific molecular mechanisms. Collectively, these advances have shifted existing paradigms in the approach to asthma to tailor novel therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The model of asthma as a single entity is now obsolete due to an increased understanding of its underlying heterogeneity. The traditional dogma of asthma is that of excessive T-helper cell type 2 (Th2) cell responses and specific IgE driving airway hyperresponsiveness. While this accurately conveys the dominant mechanisms of allergic asthma, the term “asthma” is now considered an umbrella diagnosis for a collection of several other distinct diseases (endotypes) and varying phenotypes (young atopic, obese middle aged, and elderly), all of which manifest with symptoms of wheezing and shortness of breath to cough and chest tightness, and are accompanied by variable airflow obstruction. Until a few years ago, treatments have been applied universally to all patients with asthma. However, the diverseness of this disease results in varying responses to therapies. The exact prevalence of severe asthma is unclear but thought to be between 5 and 10% of all-comers with asthma. Patients with severe disease have refractory symptoms despite high-intensity therapy. This population accounts for much of the morbidity and mortality related to asthma and have a clear unmet need. Therefore, recent attempts have been made to deconstruct asthma into its pathological components to further understand the heterogeneity of asthma profiles.
The recent identification of fundamental inflammatory endotypes have presented a more granular approach to the study of asthma (Table 1). The focus has shifted to the precise delineation of molecular pathways (endotypes) driving disease. The theoretical basis of endotyping corresponds with the current interest in personalized medicine. With the advent of an ever-expanding repertoire of biologic agents, an appropriate classification system, with meaningful biomarkers, is needed to leverage molecular data and tailor treatment decisions. Therefore, stratification according to inflammatory endotype is now deemed a central component in the algorithm for management of severe asthma.
In this review, we will provide an overview of molecular endotypes, asthma phenotypes, and existing biomarkers, with a focus on Th2 (also referred to as type 2 or T2) and non-type 2 pathways.
Asthma Classification: Phenotype Versus Endotype
Asthma is heterogeneous in terms of severity, natural history, and treatment responsiveness, and this heterogeneity reflects the underlying mechanisms. A longstanding approach to asthma has been to group patients based on observable combinations of clinical, biological, and physiological characteristics into so-called phenotypes. Simply put, phenotypes are defined as “observable characteristics that result from a combination of hereditary and environmental influences.” However, the strategy is now evolving to associate molecular mechanisms to phenotype. Asthma endotypes describe these distinct pathophysiologic mechanisms at a cellular and molecular level. Despite similar clinical symptoms, patients may respond very differently to the same therapeutic interventions. Precision medicine is used to describe treatment targeted at patient endotype. For instance, the delineation of the complex type 2 inflammatory network in severe asthma has inspired the design of several incipient targeted biologic therapies.
Immunopathogenesis of Asthma
One of the initial mechanistic insights into asthma endotyping was based on an understanding of the dominant CD4+ T-cell response. CD4+ T-cell responses in asthma are heterogeneous, comprising multiple subsets that promote the underlying inflammatory pathways of a particular asthma subtype. Since the discovery of classical CD4+ T-cell subsets (Th1 and Th2 subpopulations)—over 30 years ago, it was swiftly recognized that Th2 cells are the principal driver of eosinophilic airway inflammation by generating abundant quantities of IL-4, IL-5, and IL-13. Nearly two decades ago, Wenzel et al. [1] stratified corticosteroid-dependent asthma into two different subtypes based on the presence of airway eosinophilia. This subsequently led to polarization into two major asthma endotypes: Th2-high (eosinophilic) and Th2-low (non-eosinophilic) which is the most well-established classification of severe asthma endotypes.
Recently, a substantial body of evidence has proven that group 2 innate lymphoid cells (ILC2s) also play an equally critical role in type 2 immune responses. ILCs constitute a family of lineage-negative innate lymphocytes that are distinct from T and B cells, and do not participate in classical allergen-specific activation. Yet, ILCs are potent producers of prototypic type 2 cytokines. While they have widespread tissue distribution, ILC2s are particularly high in airway tissues and produce large quantities of IL-5 and IL-13 in response to alarmins, mediators released from epithelial cells in response to stressors such as infection or inflammation. In asthma, ILC2s appear to play an early and key role in augmenting the type 2 responses in the airway. Thus, it is increasingly recognized that innate immunity has a leading role in the pathophysiology of asthma, with intricate interconnections between innate and adaptive immunity. Together, Th2 cells and ILC2s are the primary regulators of type 2 immunity, and express the master transcription factor GATA3, which governs the production of type 2 cytokines. Therefore, Th2-high inflammation is alternatively labeled as type 2 (or T2) inflammation, so as to account for the role of other immune cells involved with Th2 airway inflammation.
Asthma Phenotypes
Asthma was long thought to manifest as two major phenotypes, non-atopic or “intrinsic” asthma, and atopic or “extrinsic” asthma. Early-onset atopic asthma is most prevalent during childhood and into young adulthood, after which the non-atopic form predominates among older age groups. Additional asthma phenotypes were defined using a hypothesis-based approach, which classified patients into broad categories based on a single variable, including disease severity, symptom triggers, age at onset, inflammatory patterns, exacerbations, and airflow obstruction [1, 2]. A major limitation with this approach arose because the categories could not distinguish the groups, and many overlapped.
In contrast, the newer approaches have used a systems biology methodology that mitigates the effect of preconceived biases. These cluster analyses have applied algorithms that integrate the effect of multiple interacting components in large cohorts to describe and predict clinical phenotypes as well as molecular mechanisms of asthma. These include the Severe Asthma Research Program (SARP) [3], the Unbiased Biomarkers for the Prediction of Respiratory Disease Outcome (U-BIOPRED) [4], and Airways Disease Endotyping for Personalized Therapeutics (ADEPT) [5]. Although differences in clusters were found, a consensus on specific subsets have emerged. They include two major groups: T2-high and non-T2-high groups.
Type 2 (T2)-High Endotype
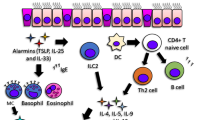
The type 2 immune response-driven endotype consists of an intricate interplay of several individual pathways (Fig. 1). The T2-high asthma endotype encompasses several related subtypes in both children and adults. Since most emerging biologic treatments are targeted toward T2 cytokines, we will attempt to delineate the pathophysiology of T2 asthma in the context of relevant cytokines and other molecular targets.

T2-high inflammatory pathways in asthma. A dysregulated epithelial barrier facilitates translocation of allergens, air pollution, and viruses, leading to release of alarmins such as thymic stromal lymphopoietin (TSLP), IL-25, and IL-33. TSLP primes dendritic cells to induce the differentiation of naïve T cells into Th2 cells. Th2 cells activate B cells via IL-4 to differentiate into plasma cells that generate IgE required for mast cell responses to allergens. The alarmins IL-25 and IL-33 can activate group 2 innate lymphoid cells (ILC2s), mast cells, eosinophils, and basophils. Activated ILC2s, like Th2 cells, produce IL-5 and IL-13. IL-5 promotes eosinophil differentiation and survival. IL-13, IL-4, and inflammatory mediators from mast cells, basophils, and eosinophils have effects on airway hyperresponsiveness, smooth muscle hypertrophy, and airway remodeling. CysLT, cysteinyl leukotrienes; ECP, eosinophil cationic protein; EDN, eosinophil-derived neurotoxin; EPx, eosinophil peroxidase; MBP, major basic protein; PGD2, prostaglandin D2
Pathophysiology
Alarmins (TSLP, IL-25, IL-33)
The innate immune pathway originates in the airway epithelium, which is now known to be a dynamic orchestrator of immune responses in T2 high asthma. Most asthmatics exhibit a dysregulated epithelial barrier, with marked loss of E-cadherin [6] and claudin-18 [7] that mediate tight junctions (TJs). Reduced barrier integrity caused by damage to the airway epithelium plays an integral role in asthma pathogenesis, facilitating access to the stromal tissue by allergens and microbes. Further, protease-containing allergens such as house dust mite (HDM) are able to directly cleave epithelial TJs and disrupt barrier structures. Interestingly, airway epithelial cells (AECs) can act as sentinel cells that enable detection of microbial and non-microbial agents including allergens through pattern recognition receptors (PRRs). The AECs respond rapidly to stimuli with release of cytokines termed alarmins, which then propagate ensuing adaptive type 2 immune pathways.
Alarmins are epithelial-derived mediators, including thymic stromal lymphopoietin (TSLP), IL-25, and IL-33. These are upstream cytokine mediators that initiate multiple type 2 signaling pathways in response to infection and allergen-driven inflammation. While IL-33 and IL-25 mainly activate ILC2s, TSLP also primes antigen-presenting cells (APCs), typified by dendritic cells (DCs), to promote type 2 immunity by activating T cells and B cells. IL-33 is a member of the IL-1 family of cytokines that has been highlighted in type 2 immunity due to the association of genetic variants of both IL-33 and its receptor, ST2, with atopic asthma. Although IL-25 and IL-33 have overlapping immunological effects, IL-33-ST2 appears to be the crucial amplifier of T2 asthma [8]. In other studies, the IL-33 gene has been closely linked with severe exacerbations in early-onset asthma. IL-33 also causes airway remodeling in severe refractory disease [9]. Studies have consistently demonstrated expanded expression of these markers in airways of asthmatics compared with healthy individuals, and this expression correlated with disease severity [10,11,12,13]. To show the direct role in asthma pathogenesis, increased transcription of these cytokines has been demonstrated in airway tissue in lung biopsies from asthmatic patients after exposure to allergen and are involved in delayed asthmatic responses [13].
ILC2
Alarmins then serve to activate ILC2s that are lineage-negative cells and lack lymphocyte surface markers and antigen-specific receptors. Following stimulation, they are potent producers of IL-5 and IL-13 and thus propagate early type 2 immune responses [14]. Of note, ILC2s produce 10-fold more IL-5 and IL-13 compared with activated Th2 cells and thus, ILC2s are a critical source of these cytokines to augment the T2 response [15]. Their discovery led to the change in nomenclature from Th2-high asthma, which implied that these cytokines are exclusive products of Th2 cells, to type 2. Recent evidence also supports a role of ILC2s in the remodeling and repair of damaged tissue of asthma, disclosing new potential therapeutic target acting on the remodeling process [16].
Th2 Cytokines (IL-4, IL-5, IL-13)
Th2-high asthma generally goes hand in hand with eosinophilic inflammation. Eosinophils are key elements in long-term perpetuation of type 2 inflammation in asthmatics and will be described further in more detail.
Activated dendritic cells (DCs) induce the expression of a Th2 pathogenic signature in the presence of the master transcription factor GATA-3. Th2 cells stimulate type 2 immunity through the secretion of the cytokines IL-4, IL-5, and IL-13, which is manifested as high IgE antibody titers and eosinophilia. It is now known that Th2 memory cells can still be a source of these prototypical cytokines even in the absence of CD4+–DC interactions in non-allergic T2-high asthma [17].
Both IL-4 as well as IL-13 utilize a common IL-4Rɑ chain (which forms a heterodimer with IL-2Rɣc and IL-13Rɑ, respectively) to initiate signaling through the phosphorylation and activation of the transcription factor signal transducer and activator of transcription-6 (STAT6) [18]. IL-13 as well as IL-4 promote goblet cell overexpression, increased mucus secretion, as well as airway hyperresponsiveness. However, only IL-4 is able to drive the induction of Th2 cells since T cells do not bind IL-13. IL-4 is the predominant cytokine that drives Th2 cell differentiation and production of downstream cytokines, including IL-5 and IL-13, as well as B-cell activation of IgE isotype switching. Furthermore, Th2 cytokines possess fibrogenic functions and stimulates additional airway remodeling through activation of fibroblasts.
IL-5 plays a pivotal role in the promoting the differentiation and maturation of IL-5Rα + eosinophil progenitors in the bone marrow, as well as their subsequent mobilization and survival. IL-5 also supports the development of other type 2 cells including MCs and basophils [19].
Eosinophils
Eosinophils are the cardinal cell type associated with type 2 asthma and have pleiotropic effects on various inflammatory cells. Large numbers of eosinophils are recruited to the site of inflammation through eotaxins, which are chemokines that respond to triggers via CCR3 receptors expressed on eosinophils. They have the capacity to synthesize and store cytotoxic proteins within intracellular granules. Upon stimulation, they release a myriad of inflammatory mediators including cytokines (including IL-13 and IL-5), chemokines (such as eotaxins), granule mediators, as well as cysteinyl leukotrienes (cysLT) such as LTC4, LTD4, and LTE4. Eosinophil cytotoxic proteins include major basic protein (MBP), eosinophil peroxidase (EPX), eosinophil cationic protein (ECP), and eosinophil-derived neurotoxin (EDN). More recent studies suggest that eosinophil degranulation products might be a better indicator of eosinophil activation status as opposed to absolute numbers [20].
Eosinophils also activate bronchial fibroblasts through the production of profibrotic factors and are thus associated with features of remodeling, in particular, the thickening of the basement membrane [21]. Their inflammatory factors support airway smooth muscle contractility and inhibit their relaxation [22]. Cysteinyl leukotrienes (CysLTs) are potent bronchoconstrictors that contribute to ILC2 production of type 2 cytokines by acting in synergy with IL-33 and further drive the self-amplifying loop that characterizes Th2 inflammation. The relationship of eosinophilia with inflammation in asthma will be discussed later in greater detail.
Mast Cells and Basophils
Mast cells and basophils play a pathogenic role in allergy. While the role of MCs is well established, the functional significance of basophils has recently gained attention. Both mast cells and basophils express high-affinity IgE receptor (FcεR1) and ST2 on their surfaces and are activated by IgE cross-linking as well as IL-33 [23]. Interestingly, mast cells are resident tissue based, but basophils are blood-borne and often recruited to tissues where they attain final activation status. Thus, basophils have been shown to be increased in bronchial walls of T2 high asthma likely due to increased inflammation. Upon activation, both mast cells and basophils secrete histamine and lipid mediators—prostaglandin D2 (PGD2) and CysLTs—while tryptase and proteases are unique to mast cells [24]. More interestingly, basophils have been shown to secrete IL-4 that directly modulates ILC2 function [25, 26] along with other roles in B-cell differentiation. Tissue basophils have been strongly associated with patients with allergic disease [27, 28] and the mediators secreted by mast cells and basophils have been shown to correlate with disease severity in asthma.
Serum IgE
Serum IgE is used as a surrogate marker of atopy. Specific IgE that develops in response to allergen exposure binds to its high-affinity receptor FcepsilonRI (FcεRI), expressed most prominently on mast cells (MCs), and the low affinity receptor FcεRII or CD23, on APCs. In addition to mediating the immediate hypersensitivity response in allergic asthma through MC activation, allergen-specific IgE also induces a delayed phase reaction characterized by the massive influx of eosinophils and other inflammatory cells. It also facilitates antigen presentation through enhanced antigen uptake by B cells and DCs. Moreover, recent data demonstrating expression of FcεRI receptors on airway smooth muscle cells implicates IgE may have a direct role in the pathogenesis of airway remodeling [29]. Thus, IgE may be indirectly associated with airway remodeling in severe asthma. Finally, ongoing IgE-dependent activation of mast cells may cause an increase in vascular damage and infiltration by inflammatory cells that may also contribute to airway remodeling [30].
Prostaglandin D2 (PGD2)
Expanded MC populations have been observed in the airways of T2-high asthma, and MCs produce PGD2 which induces vasodilation and increased vascular permeability. Recent data strongly suggest that an altered functional subtype of MC may have greater potential to generate PGD2. These PGD2-high MCs strongly predict poorly controlled T2 high asthma and are associated with more severe disease [31, 32]. A significantly higher concentration of PGD2 has been reported in bronchoalveolar lavage (BAL) fluid of patients with severe asthma as compared with mild-to-moderate asthmatics, and it is an important predictor of severe asthma [31]. Similarly, PGD2 receptors, such as the D-Prostanoid (DP) receptor and the homologous receptor mutant molecule expressed on Th2 cells (CRTH2), are expressed on Th2 cells, eosinophils, ILC2s, and epithelium with highest expression in bronchial biopsy samples of patients with severe asthmatics [32].
Classification of T2-High Phenotypes
T2-high phenotypes have been classified into three groups: early-onset allergic asthma, late-onset eosinophilic asthma, and aspirin-exacerbated respiratory disease (AERD). Clinical characteristics of these groups are described along with growing evidence of clinical correlates that may result in potential etiologies of T2-high groups.
Early-Onset Allergic Asthma
Early onset or “extrinsic” allergic asthma is the archetypal asthma phenotype. The presentation ranges from mild to severe, and it has not been elucidated whether severe asthma is the result of evolution from a milder form or instead arises de novo as a severe type during childhood [2]. This phenotype is distinguished from T2-high non-atopic asthma by positive allergy skin tests and increased serum-specific IgE. It is important to note that the presence alone of elevated total or specific IgE are specific biomarkers for allergic asthma, as allergy testing may be positive in up to 50% of the general population. Otherwise, little is known regarding precise roles of innate and adaptive immune cells specifically in early-onset allergic asthma as compared to other forms of Th2-high non-atopic asthma.
Late-Onset Eosinophilic Asthma
A subset of T2-high asthmatics with adult-onset disease have a distinct steroid-resistant eosinophilic phenotype of unknown molecular mechanism [1]. Airway T2 inflammation is not ameliorated by ICS therapy in approximately half of asthmatics, and these patients are older and have more severe asthma [33] with fixed airflow obstruction. The great majority of these patients have comorbid chronic rhinosinusitis with nasal polyps (CRSwNP) which generally precedes asthma development. This phenotype is generally characterized by prominent blood and sputum eosinophilia refractory to inhaled/oral corticosteroid treatment. There is generally no evidence of atopy, but is characterized by an intense, ILC2-driven production of IL-5 and IL-13. A recent cluster analysis identified an endotype of asthma with CRSwNP that highly expresses Staphylococcus aureus enterotoxin (SE) specific IgE and high levels of IL-5 and IgE [34]. Some of these patients have sputum neutrophilia in addition to eosinophilia, implicating Th2/Th17 interactions [35]. These patients generally have also high FeNO and normal or elevated serum total IgE but probably with a lower etiopathogenetic importance. The recognition of this phenotype may be an indication to escalate therapy earlier.
Aspirin-Exacerbated Respiratory Disease (AERD)
A subset of the above-described late-onset phenotype is AERD, characterized by asthma, CRSwNP, and COX-1 inhibitor-induced respiratory reactions. Although the mechanisms underlying AERD are not fully elucidated, its development appears to be contingent upon dysregulated arachidonic acid (AA) metabolism and cysLT production. Baseline levels of prostaglandin E2 (PGE2) levels are markedly deficient along with its receptor EP2. PGE2 is critical in inhibiting the activation of ILC2s, mast cells, and eosinophils. The loss of homeostatic PGE2 expression removes negative feedback on the 5-lipoxygenase (5-LOX) pathway and thus upregulates constitutive cysLT synthesis.
Aspirin is a potent COX-1 and COX-2 inhibitor. COX inhibition shifts AA metabolism shifted from the COX to the 5-LOX pathway. The upshot of this inflammatory cascade is the suppression of residual homeostatic PGE2 and culminates in CysLT overproduction from mast cells, eosinophils, and macrophages. This aberrant cysLT production is mediated by leukotriene C4 synthase (LTC4S). CysLTs including leukotriene C4 (LTC4), LTD4, and LTE4 are potent bronchoconstrictors that are responsible for most of the symptoms in AERD.
These lipid mediators also regulate the alarmin/ILC2/IL-5/IL-13 pathway, which drives the profound tissue and blood eosinophilia characteristic of AERD [36, 37]. This effect of innate type 2 mediators is further amplified by PGD2 and cysLTs. The ultimate result is severe persistent upper as well as lower airway disease with refractory NPs and asthma.
Clinical Correlates of T2-High Asthma
An array of host factors (such as genetics and comorbid disease) and environmental exposures (including viral infections, cigarette smoking, and air pollution) contribute to steroid-refractory T2-high disease.
Host Genetic Factors
Genome-wide association studies (GWAS) have evidenced a genetic bias specifically linked to early-onset asthma, and the genes associated with this phenotype are epithelial-related rather than allergy-related [8]. Variations at chromosome 17q21 have been consistently highlighted as the most striking and consistently recapitulated locus in asthma. Two genes that activate fibrogenic pathways, ORM1-like 3 (ORMDL3) and gasdermin B (GSDMB), have specifically been recognized as likely candidates at this locus [38].
Viral Etiologies
The role of viruses in asthma exacerbations further contributes to pathologic mechanisms in severe allergic asthma [39]. While respiratory viruses (especially rhinovirus) are the most common trigger of asthma exacerbations, the T2-high subgroup appears to be particularly exacerbation prone with viral infections. Recent studies show deficient local innate antiviral immune responses, as well as upregulated pro-T2 response with increased production of Th2 cytokines, in the setting of respiratory viral infections. In addition, recent evidence suggests that this susceptibility to viruses in asthmatics may be secondary to a defect in innate immunity (interferon signaling pathways) [40]. AECs in these patients demonstrate deficient type I and type III interferon (IFN) production in response to viral infections. Molecular mechanisms include IgE/FcεR1 cross-linking inhibits virus-induced IFN-α responses of plasmacytoid dendritic cells (pDCs), which could explain increased susceptibility in allergic asthma.
In addition, the cadherin-related family member 3 (CDHR3) gene mediates tight junctions in the airway epithelium and polymorphisms in this gene are associated with severe asthma exacerbations. Rhinovirus-C is dependent on host epithelial CDHR3 as a receptor for cell entry thereby decreasing tight junctions in an already tenuous epithelial barrier. These observations, taken together, imply a mechanistic link between this host genomic variation and asthma risk [40].
Air Pollution
Another contributory factor to worsening allergic asthma is air pollution. Environmental pollutants, such as ozone and particulate matter, are not only associated with increased asthma morbidity but also contribute to development of disease. Diesel exhaust particles (DEPs) augment primary IgE sensitization to allergens, as well as immediate and late-phase allergic responses [41].
Neurogenic Inflammation
The role of neurogenic inflammation in allergic airway inflammation has been recently highlighted [42]. Transient receptor potential vanilloid 1 (TRPV1) is the hub of almost all neuronal inflammatory signaling pathways. This receptor regulates the activation and inflammatory properties of CD4 T cells [43, 44] and modulates inflammatory and structural changes in chronic asthma. There is accumulating data to support that symptoms of chronic allergic asthma might be linked to the effects of allergen-induced neuromodulation. Nerve growth factor (NGF) is a neurotrophin that mediates neuroplasticity. Additionally, high expression levels of NGF have been shown to enhance the activity of eosinophils, allergen-mediated eosinophil inflammation, and consequently airway hyperreactivity [45].
T2-High Biomarkers
The ultimate clinical goal in severe asthma is the measurement of disease severity and response to treatment. The identification of biomarkers to recognize endotypes and guide therapy has been a recent priority in asthma. A biomarker is defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes or pharmacologic responses to a therapeutic intervention” [46]. Currently available biomarkers provide a platform to dichotomize individuals to either T2-high or non-T2-high groups. This approach is meant to establish candidacy for T2 targeted therapies and has been incorporated into standard practice. Furthermore, uncomplicated point-of-care testing, such as blood and exhaled breath tests, facilitates biomarker use.
There is a need for additional validation of biomarkers to optimize definitions of asthma phenotypes. Some biomarkers may offer highest yields when applied in the background of certain clinical characteristics (e.g., more reversible airway disease, age at disease onset). A combination of different biomarkers, or a biomarker panel, may be more suitable than a single one to refine selection of biological therapies.
The canonical marker of T2-high asthma is elevated airway eosinophil counts, but elevated peripheral eosinophil counts, blood periostin level, FeNO, and allergen-specific IgE levels have been used as substitute markers. While all of these values would be expected to coincide, they may be elevated independent of each other due to the selective action of individual mechanistic pathways. It is important to note that while T2-high asthma is typically associated with eosinophils, yet the presence of eosinophils in itself does not automatically mean that they are the most influential pathogenic cell type and does not equate with response to eosinophil ablation.
Sputum Eosinophils
Eosinophilic airway inflammation is one of the most influential traits in asthma, and accounts for approximately 40–60% of patients with severe asthma [46]. The analysis of eosinophils in induced sputum is a common technique to characterize T2 asthma. Sputum eosinophils are considered the “gold standard” type 2 biomarker (with cutoff points of > 3%). However, the procedure may be suboptimal for routine testing due to its slightly invasive and cumbersome nature.
Peripheral Blood Eosinophils
Peripheral blood eosinophils are phenotypically distinct from those in the lung, and the relationship between peripheral eosinophilia and airway inflammation has been inconsistent across studies. This discrepancy may reflect the patient populations studied, the experimental setting, and baseline medications. Thus, blood eosinophils in some studies did not fully represent airway eosinophil activity [47] further complicating its utility as a reliable biomarker. Although the blood AEC has been readily adopted in practice, the absolute eosinophil count (AEC) may not be sensitive at identifying the most severe T2-high asthmatics. This is in part due to the inverse correlation between sputum/blood eosinophilia ratio and increasing disease severity, especially in steroid-dependent asthmatics [47]. There is also data to suggest a discrepancy between blood eosinophil counts and eosinophilic airway inflammation in morbidly obese patients, and the blood AEC might be a poor indicator of airway type 2 inflammation in this asthma subgroup. Conversely, more recent studies do indicate concurrence between levels of blood and airway eosinophils in asthmatics [48]. The relationship between peripheral blood eosinophilia and the risk of recurrent exacerbations has been replicated in adult and pediatric cohorts [49, 50]. Various AEC cutoff values between 150 and 400 cells/μl have been used to predict response to anti-IL-5 inhibitors. In general, a higher blood eosinophil cutoff is associated with a larger reduction in exacerbation rates and greater improvements in lung function [50].
Beyond the diagnosis of T2-high asthma, monitoring of blood eosinophil counts does not reflect response to biologics. The suppression of peripheral eosinophilia by anti-IL-5 therapy does not always correlate with clinical response or normalization of sputum eosinophilia [51]. Despite these issues, the peripheral eosinophil count is commonly obtained due to ease of performance and clinical accessibility. However, it is important to be aware of the caveats in its use and the potential limitations as a biomarker for phenotyping refractory asthma.
Serum Total IgE
In accordance with its central role in the development of allergic asthma, serum IgE is a good biomarker for atopy status. Serum IgE levels correlate positively with asthma severity in adults and children [52, 53]. The probability of wheeze and reduced lung function also increases parallel with serum IgE values [53]. Serum total IgE is used to predict responsiveness to anti-IgE therapy but is not useful for monitoring response. However, bronchial mucosal IgE+ mast cells might be a useful biomarker [54].
Allergen Sensitization Panel
The NIH Asthma Outcomes Task Force recommends the assessment of aeroallergen sensitization as a core biomarker for classification of asthma [55]. There is a direct relationship between the degree of allergen sensitization, as reflected by serum-specific IgE, and the likelihood of expression of asthma symptoms [52, 53]. This association with allergen-specific IgE titers is especially marked in children, and increased specific IgE in children likely reflects a T2-high profile. When combined with childhood symptoms of atopy and asthma, positive IgE testing helps to confirm early-onset allergic asthma. However, high IgE in T2-high asthma is not always indicative of atopy. Also, the role of IgE as a biomarker of disease activity or control is unclear and should be evaluated in future studies of novel biologic agents in severe asthma.
Fractional Excretion of Nitric Oxide (FeNO)
FeNO measurement can be a valuable tool and has the advantage of standardized and expedient measurement in exhaled breath. Nitric oxide (NO) is produced by airway epithelial cells, eosinophils, and macrophages, through the conversion of the amino acid l-arginine to l-citrulline by the enzyme nitric oxide synthase (NOS). T2-high airway inflammation drives the transcription of inducible NOS (iNOS), thus increasing NO production. FeNO predominantly signifies IL-4 and IL-13 activity, with FeNO concentrations greater than 50 ppb (> 35 ppb in children) alluding to eosinophilic airway inflammation and steroid responsiveness [56]. Thus, the role of FeNO may be additive as a biomarker in relation to asthma morbidity. In conjunction with peripheral eosinophilia, an elevated FeNO is a risk factor for airway hyperreactivity and uncontrolled asthma [48]. However, despite being a reasonable indicator of T2-driven asthma, there are no specific guidelines on how FeNO can guide therapy with biologics.
Periostin
Periostin is an extracellular matrix protein whose expression is consequent to IL-4 and IL-13 activity in AECs and lung fibroblasts. It stimulates eosinophil degranulation as well as the generation of superoxide anions and production of TGF-β and cysLTs from eosinophils. Periostin expression in bronchial tissue is not associated with asthma severity but has been shown to be a biomarker of persistent eosinophilic airway inflammation despite corticosteroid use [57]. The potential use of serum periostin levels is the assessment of greater response to anti-T2-based therapies [58].
Dipeptidyl Peptidase 4 (DPP-4)
CD26 is a membrane-anchored enzyme with DPP-4 activity that is found on CD4+ T cells and detectable in high concentrations in lung connective tissue. DPP-4 is the circulating form of CD26 and is found in blood and biologic fluids, such as BAL. It is still unclear whether DPP-4 activity plays a role in either up- or down-regulating asthmatic inflammation. Nonetheless, increased DPP-4 mRNA expression following IL-13 stimulation of bronchial epithelial cells has been demonstrated in asthma patients [59]. Interestingly, serum DPP-4 can predict responses to anti–IL-13 therapy [59]. Hence, DPP-4 may be an important biomarker in anti-IL-13 therapies.
Urinary LTE4
A high level of urinary LTE4 is a highly sensitive discriminator of AERD from aspirin-tolerant asthma. Receiver operator characteristic analysis of 24-h urinary LTE4 in one analysis established a cutoff value of 166 pg/mg Cr for prediction of aspirin sensitivity with 89% specificity [60]. Due to its high negative predictive value, it could potentially be used as a clinical test to assess the risk of AERD in asthma patients with concomitant nasal polyps.
Non-T2-High (T2-Low) Endotype
T2-biased airway inflammation is observed in only half of patients with asthma [61] and only in 37% of patients with severe asthma from airway epithelial transcriptome analysis [62]. The imbalance of T1/T2 cytokines has long recognized T2-high asthma onset; however, T2-low disease is relatively under-studied. While the underpinnings of T2-low asthma are yet an enigma, it is typified by the absence of markers of T2-high disease, such as eosinophilia. It is generally characterized by neutrophilic (sputum neutrophils > 40–60%) or paucigranulocytic (i.e., normal sputum levels of both eosinophils and neutrophils) inflammation and a lack of response to corticosteroid therapy. T2-low asthma has been linked with the activation of Th1 and/or Th17 cells, and recent studies have found that the imbalance of Th17/Treg cells may play an important role in steroid-resistant, severe, and neutrophilic asthma.
The mechanisms underlying recruitment and maintenance of neutrophilic airway inflammation are yet unknown. Severe neutrophilic asthma has been associated with chronic infection with atypical bacteria [63], obesity, smoking, and poorly understood underlying smooth muscle abnormalities. The pathogenesis has also been demonstrated to involve activation of the NLRP3 inflammasome and elevated IL-1β [64]. However, the role of the neutrophil itself is in debate because the presence of neutrophils may merely represent an off-target effect of high corticosteroid doses [65]. It is also possible that some T2-low asthma is labeled as such only because steroid therapy has masked the T2 signature below the threshold for detection. Thus, T2-low asthma must take into consideration the effects of concomitant therapies.
Th1-High Inflammatory Signature
Raundhal et al. recently reported a Th1 skewed signature marked by the production of IFN-γ in approximately 50% of patients with severe asthma [66]. Elevated IFN-γ was associated with high airway resistance, increased inflammatory infiltrates, and corticosteroid refractoriness. The authors suggested that IFN-γ-induced downregulation of secretory leukocyte protease inhibitor (SLPI) in AECs is responsible for increased AHR in severe asthma. The same group also suggested that corticosteroids may not only be inefficacious in these patients but may actually exacerbate the underlying inflammatory state through increased Th1 recruitment [67].
Th17-High Inflammatory Signature
Th17-high asthma is generally characterized by a steroid-dependent, refractory phenotype. Th17 cytokines are key players in T2-low disease with increased levels of IL-17A and IL-17F found in the bronchial walls of severe asthmatics and associated with neutrophilic infiltration, AHR, and steroid resistance. IL-17A/IL-22 contributes to asthma pathology by increasing smooth muscle cell proliferation and IL-17A also drives collagen deposition [68]. Even more recently, an IL-17F/frequent exacerbator endotype has been described [69].
IL-17 activity manifests as neutrophilic inflammation and IL-8 drives neutrophil recruitment. Bronchial TRPV1 expression is higher among severe asthmatic patients compared to healthy controls, and stimulation of the channel leads to production of IL-8 (and potential neutrophilic inflammation) in bronchial epithelial cells [70] suggesting mechanisms of increased neutrophil recruitment in these severe asthmatics.
Classification of Non-T2 or T2-Low Asthma Phenotypes
T2-low phenotypes have been classified according to clinical characteristics that include obesity, smoking, and age.
Obesity Associated
Obesity is an important risk factor for asthma morbidity. The prototypical patient with obesity-associated asthma is the non-atopic, middle-aged woman with severe symptoms despite a moderately preserved lung function. While the obese-asthma syndrome is complex and multifaceted, the bulk of evidence points toward non-eosinophilic inflammatory mechanisms at the molecular level [71]. Interestingly, obesity biases CD4 cells toward Th1 differentiation, which is associated with steroid refractory asthma [72]. Additional innate immune responses involving Th17 pathways and ILCs have also been implicated. Uniquely, it is the type 3 ILCs (ILC3) that express both IL-17 and IL-22 that have been associated with obesity-related asthma [71].
Another important cytokine, IL-6, has also been recently shown to cause systemic inflammation in a subgroup of asthma patients with obesity and severe disease [33]. The notable observation in this study was increased plasma IL-6 levels in the subset of obese patients with more severe asthma and not in all obese asthmatics. Consequently, both IL-17, IL-22, and IL-6 rather than the T2 cytokines may be clinically relevant in obese patients with severe asthma.
Smoking Associated
The mechanisms underlying smoking-associated asthma is unclear, but it has been considered a T2-low neutrophilic, steroid-resistant phenotype [73]. Putative mechanisms include oxidative stress that mediates epigenetic modifications causing neutrophil and macrophage activation [73]. However, smoking also increases the risk of sensitization to allergens and increases total IgE demonstrating the link between asthma and COPD. The recently described term “asthma-COPD overlap syndrome (ACOS)” demarcates those patients with a significant smoking history and consequent airflow obstruction but also have overlapping features of asthma (bronchodilator reversibility, eosinophilia, atopy). The current joint task force of the Global Initiative for Asthma (GINA) and Global Initiative for Chronic Obstructive Lung Disease (GOLD) published a consensus document outlining diagnostic criteria for ACOS [50]. The committee recommends the presence of all three of the following major criteria and at least one minor criterion for diagnosis. The major criteria include persistent airflow limitation in individuals > 40 years of age with at least 10 pack-years of tobacco smoking, and onset of asthma at < 40 years of age. The minor criteria include a history of atopy, significant bronchodilator reversibility, and peripheral eosinophilia. Although all COPD patients have not responded to the new biologic agents [74], the ACOS subset may actually benefit.
Very Late Onset
The age cutoff for the diagnosis of very late-onset asthma is not consistent but defined as > 50 years in some studies [75] and > 65 years in others [76]. The aging lung is associated with decreased lung function due to loss of elastic recoil and mechanical disadvantages. In addition to these consequences of normal aging, immunosenescence likely has important consequences in elderly asthmatics [77]. While mechanisms have not been fully elucidated, emerging data suggest that older asthmatics have increased sputum neutrophilia secondary to Th1 and Th17 inflammation [78, 79].
T2-Low Biomarkers
While progress has been made in profiling T2-high-driven asthma, there is a high unmet need in the endotype-driven approach for the T2-low asthma. Thus far, biomarkers for T2-low asthma have not been tested extensively. Neutrophilic inflammation cannot be detected based on currently available biomarkers. A recent study by Maes et al. [80] has suggested that certain micro-RNAs in sputum might be able to accurately identify neutrophilic airway inflammation, but most clinicians are unable to obtain satisfactory sputum samples from these patients.
Proposed biomarkers include blood/sputum neutrophilia; however, this is associated with inherent limitations. It remains unknown whether these neutrophils are clinically relevant or merely a byproduct of the local inflammatory response. Also, the finding of neutrophilic inflammation can be secondary to a variety of unrelated causes such as concomitant high-dose corticosteroid therapy, exposure to environmental pollution or cigarette smoke, or intercurrent bacterial infection. Another potential marker is metalloproteinase 9 (MMP9) which is implicated in inflammation and remodeling of asthmatic airways [81].
Several cytokines are associated with neutrophilic airway inflammation. IL-6 is a pleiotropic cytokine produced by various cell types in response to a wide range of inflammatory stimuli. It is considered an indicator of metabolic dysfunction as well as asthma severity and has been identified as a potential candidate biomarker in a study with obese asthmatic patients [82]. There is a strong association between IL-6 concentrations and disease severity both before and after correction for BMI but may also increase in the setting of viral infections, which is a major cause of asthma exacerbations.
T-Cell Plasticity and Heterogeneity in Asthma
Adding to this already complex situation, it has been demonstrated that the majority of T-helper cell subpopulations in asthma do not represent finally differentiated static conditions. Rather, they show a degree of plasticity depending on environmental influences and may transdifferentiate into other effector CD4 cell types. For instance, there is evidence for concomitant Th1 and Th2 responses in chronic asthma [83], and crosstalk between Th2 and Th1 cytokines contributes to airway wall remodeling in this situation. In a cluster analysis of AEC gene expression from asthma patients, the two clusters with the most severe Th2 expression also showed upregulation of IFN-related genes. This suggests that type-2 inflammation may occur concomitant with additional innate and adaptive immune pathways. This can dampen the effect of steroids and may underlie incomplete response to type 2–directed therapy [84]. The Th1/IFN-γ axis is thought to modulate Th2 inflammation through a balance between its suppressive effect on Th2 activation and its promoting effect on Th2 cell recruitment into the airways. However, it is yet not fully clear how a Th1 response is able to potentiate Th2 inflammation.
Similarly, an overlap of Th2 and Th17 pathways has also been evidenced in severe asthma [85]. The presence of dual positive Th2/Th17 cells in BAL fluid implies a more severe phenotype as compared to those with Th2 or Th17 cells alone. A possible mechanism why both IL-17 + Th2 cells are more highly pathogenic is that these cytokines act in synergy to enhance the recruitment of leukocytic airway infiltration while stimulating mucus secretion by AECs [85].
Akin to T-helper cells, heterogeneity and plasticity in relation to environmental signals in asthma have also recently been described in ILCs [86]. Together, both plasticity of T cell and ILCs add to the complexity of inflammation in severe asthma.
These findings raise the consideration that as opposed to highly polarized and unambiguous endotypes, severe asthma might represent a spectrum of overlapping inflammatory states. The relative expression of T2-high and T2-low gene signatures may be transient depending on ongoing circumstances. The most prominent expression of any of these molecular profiles at a given time can serve as a therapeutic guide. This may account for incomplete response to currently available T2-directed therapies, since additional mechanisms driving pathology may dilute the drug effect. It is also important to note that these patients with mixed granulocytic inflammation generally exhibit the highest disease burden in terms of symptoms, lung function, and asthma control [87].
Future Directions in Asthma Phenotyping
The application of cluster analysis in asthma has gained increasing attention as a departure from traditional hypothesis-based approaches to phenotyping asthma. This analysis is accomplished through “-omics” methods, which refers to a large dataset derived from a single sample to gain insight into previously unrecognized molecular host–environment interactions and mechanisms of disease. Minimally invasive analytic tools have been reported for asthma, using blood, sputum, or bronchial brushings. Several datasets apply clustering algorithms that measure the behavior of several clinical parameters (e.g., demographics, lung function, BMI, ACQ, atopy, and blood eosinophils) to large cohorts to identify asthma phenotypes. These include the Severe Asthma Research Program (SARP) [3], the Unbiased Biomarkers for the Prediction of Respiratory Disease Outcome (U-BIOPRED) [4], and Airways Disease Endotyping for Personalized Therapeutics (ADEPT) [5]. The heuristic approach of these novel “-omics” methods are designed to avoid the imposition of preexisting hypotheses, allowing latent phenotypes to arise from the data. This approach thus eliminates investigator bias and fosters the generation of novel paradigms.
RNA Transcriptomics (Study of Gene Expression)
Bronchial Tissue Transcriptomics
The seminal paper by Woodruff et al. first applied gene expression profiling to endobronchial brushings of adults with mild to moderate asthma [61]. This study segregated Th2-high and Th2-low inflammatory endotypes based on enhanced expression of three epithelial genes: CLCA1, periostin, and serpinB2, in Th2-high subjects. Only these patients had eosinophilic airway obstruction and responded well to steroids.
However, eosinophilic inflammation may also be overwhelming to the point of steroid unresponsiveness. U-BIOPRED applied this approach to severe asthmatics [4] and identified two eosinophilic subgroups with limited response to steroids—one with elevated mucosal eosinophilia, elevated FeNO, and oral steroid use. The other eosinophilic subgroup had high airway eosinophils and higher BMI. The U-BIOPRED group also developed a computational model for predicting responsiveness to steroids.
Most of the omics studies are cross-sectional, thus corresponding to a single time point and do not represent the evolution of gene expression. The ADEPT study [5] is a phenotyping study that validated adult clusters longitudinally using clinical and biomarker characteristics. The phenotypes included (1) mild, type 2, early-onset disease and preserved lung function; (2) moderately controlled Th2-high asthma with mild reversible airflow obstruction; (3) moderately controlled Th2-low asthma with fixed obstruction, and (4) severe asthma with uncontrolled reversible airflow obstruction and mixed inflammation. This classification was further validated in the U-BIOPRED subset.
Blood Transcriptomics
The U-BIOPRED group used blood transcriptomics to identify nearly 1700 genes differentially expressed in adult asthmatics compared with controls, with the biggest effect size in severe asthmatics [88]. However, they were unable to reconcile these transcriptomic changes with any specific clinical cluster, underlining the complexity and our incomplete understanding of asthma.
Sputum Transcriptomics
The adult U-BIOPRED cohort has used sputum transcriptomics to recognize clusters in a population that included subjects with mild to severe asthma [62, 89]. The investigators attempted to elucidate differential gene expression in Th2-high and Th2-low endotypes [62]. Analysis of sputum omics data in severe asthmatics revealed three molecular phenotypes. One cluster was characterized by canonical Th2 inflammation and severe airway obstruction. An inflammasome-associated gene signature was found in the second phenotype that was associated with accumulation of sputum neutrophils. This is in concordance with previous associations of elevated gene expression of NLRP3, caspase-1, and IL-1beta in sputum macrophages among neutrophilic asthma patients [64]. In the final cluster, the authors reported enhancement of genes of metabolic pathways, ubiquitination, and mitochondrial function, and was clinically characterized by paucigranulocytic inflammation and preserved lung function. This unbiased approach provides an overall idea of the various pathways associated with these three phenotypes of asthma.
Another application of sputum transcriptomics has been the quantification of T2 inflammation in asthmatic airways based on the measurement of the Th2 cytokines, IL-4, IL-5, and IL-13, in induced sputum cells [90]. The authors proposed the “type 2 gene mean” (T2GM) as a combined multigene metric of IL-4, IL-5, and IL-13 expression in sputum cells. This concept of a type 2 gene expression profile was recently corroborated in a SARP sub-cohort [33].
Metabolomics
Metabolomics refers to the measurement of mediators or metabolic products in tissues or exhaled air and is the omics field that is closest to phenotype expression and potentially reflects genome–environmental interactions in asthma. Several circulating metabolites in asthma differ from those in healthy individuals [91]. Recently, a systematic review concluded that exhaled breath volatile organic compounds (VOCs), mainly alkanes, are promising biomarkers for asthma diagnosis [92]. Exhaled breath analysis (breathomics) is of particular interest in view of being non-invasive, and thus facilitates repeat testing to evaluate the ever-changing and fluid milieu of asthmatic airways.
Evidence from metabolomics-based studies has substantiated the existence of a distinct obesity-asthma phenotype at the molecular level [93]. Metabolomics has also helped to establish candidate biomarkers for asthma severity and steroid resistance [94]. However, additional studies are needed to standardize the approach to metabolomics in asthma.
The Problems with Omics
Omics is yet a nascent field and there are inherent concerns with cluster analysis. The disparate findings from all these various omics approaches may reflect the heterogeneity of the populations recruited in terms of asthma severity, in addition to the experimental settings. However, the observations made by published cohorts to date do serve to support prior observations describing T2-high and T2-low disease. With several T2-directed biologics on the horizon, omics may facilitate future sub-phenotyping for more precise treatment selection. Cluster analysis is currently of limited clinical relevance in this regard. Otherwise, beyond the establishment of potential connections, it is difficult to separate association from causation. Furthermore, the long-term stability of these phenotypes is just not known. However, the integration of clinical characteristics and the microenvironment does represent an important step forward in delineating the relationship between asthma endotypes and phenotypes even though more studies are needed.
Conclusions
As more and more innate and adaptive immune cell types and cytokines are identified as important drivers of asthma, it is evident that asthma endotype definitions are still fluid and continue to evolve. At this point, due to the availability of therapies targeted toward T2 cytokines and the identification of relatively simple biomarkers associated with T2 inflammation, the approach is to divide patients into those with T2-high and T2-low asthma. There continue to be critical unanswered questions in severe asthma, mainly since our understanding of the inflammatory microenvironment in the lower airway and the contributions to clinical expression of disease remains incomplete. Recent advances have provided further insight into molecular mechanisms underlying steroid resistance, tissue remodeling, and disease exacerbations. The -omics approach has been helpful in understanding the molecular mechanisms of T2-low asthma. The potential chemosensory and remodeling signatures recently described in asthmatic airways may point to new endotypes relevant in T2-low patients. The accurate translation of discoveries from these studies will require careful clinical characterization for the design of clinical trials and development of new biologic therapies.
References
Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL et al (1999) Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med 160(3):1001–1008
Miranda C, Busacker A, Balzar S, Trudeau J, Wenzel SE (2004) Distinguishing severe asthma phenotypes: role of age at onset and eosinophilic inflammation. J Allergy Clin Immunol 113(1):101–108
Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D'Agostino R Jr, Castro M, Curran-Everett D, Fitzpatrick AM, Gaston B, Jarjour NN, Sorkness R, Calhoun WJ, Chung KF, Comhair SA, Dweik RA, Israel E, Peters SP, Busse WW, Erzurum SC, Bleecker ER, National Heart, Lung, and Blood Institute's Severe Asthma Research Program (2010) Identification of asthma phenotypes using cluster analysis in the severe asthma research program. Am J Respir Crit Care Med 181(4):315–323
Shaw DE, Sousa AR, Fowler SJ, Fleming LJ, Roberts G, Corfield J, Pandis I, Bansal AT, Bel EH, Auffray C, Compton CH, Bisgaard H, Bucchioni E, Caruso M, Chanez P, Dahlén B, Dahlen SE, Dyson K, Frey U, Geiser T, Gerhardsson de Verdier M, Gibeon D, Guo YK, Hashimoto S, Hedlin G, Jeyasingham E, Hekking PP, Higenbottam T, Horváth I, Knox AJ, Krug N, Erpenbeck VJ, Larsson LX, Lazarinis N, Matthews JG, Middelveld R, Montuschi P, Musial J, Myles D, Pahus L, Sandström T, Seibold W, Singer F, Strandberg K, Vestbo J, Vissing N, von Garnier C, Adcock IM, Wagers S, Rowe A, Howarth P, Wagener AH, Djukanovic R, Sterk PJ, Chung KF, U-BIOPRED Study Group (2015) Clinical and inflammatory characteristics of the European U-BIOPRED adult severe asthma cohort. Eur Respir J 46(5):1308–1321
Loza MJ, Djukanovic R, Chung KF, Horowitz D, Ma K, Branigan P et al (2016) Validated and longitudinally stable asthma phenotypes based on cluster analysis of the ADEPT study. Respir Res 17(1):165
Heijink IH, Kies PM, Kauffman HF, Postma DS, van Oosterhout AJ, Vellenga E (2007) Down-regulation of E-cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor-dependent Th2 cell-promoting activity. J Immunol 178(12):7678–7685
Sweerus K, Lachowicz-Scroggins M, Gordon E, LaFemina M, Huang X, Parikh M, Kanegai C, Fahy JV, Frank JA (2017) Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J Allergy Clin Immunol 139(1):72–81.e1
Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, von Mutius E, Farrall M, Lathrop M, Cookson WOCM, GABRIEL Consortium (2010) A large-scale, consortium-based genomewide association study of asthma. N Engl J Med 363(13):1211–1221
Guo Z, Wu J, Zhao J, Liu F, Chen Y, Bi L, Liu S, Dong L (2014) IL-33 promotes airway remodeling and is a marker of asthma disease severity. J Asthma 51(8):863–869
Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, Robinson D, Zhang G, Zhao J, Lee TH, Corrigan C (2005) Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol 174(12):8183–8190
Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, Lemiere C, Martin JG, Hamid Q (2009) Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol 183(8):5094–5103
Cheng D, Xue Z, Yi L, Shi H, Zhang K, Huo X, Bonser LR, Zhao J, Xu Y, Erle DJ, Zhen G (2014) Epithelial interleukin-25 is a key mediator in Th2-high, corticosteroid-responsive asthma. Am J Respir Crit Care Med 190(6):639–648
Al-Sajee D, Sehmi R, Hawke TJ, El-Gammal A, Howie KJ, Watson RM et al (2018) The expression of IL-33 and TSLP and their receptors in asthmatic airways following inhaled allergen challenge. Am J Respir Crit Care Med
Yang Q, Ge MQ, Kokalari B, Redai IG, Wang X, Kemeny DM, Bhandoola A, Haczku A (2016) Group 2 innate lymphoid cells mediate ozone-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol 137(2):571–578
Chen R, Smith SG, Salter B, El-Gammal A, Oliveria JP, Obminski C et al (2017) Allergen-induced increases in sputum levels of group 2 innate lymphoid cells in subjects with asthma. Am J Respir Crit Care Med 196(6):700–712
Hirose K, Iwata A, Tamachi T, Nakajima H (2017) Allergic airway inflammation: key players beyond the Th2 cell pathway. Immunol Rev 278(1):145–161
Guo L, Huang Y, Chen X, Hu-Li J, Urban JF Jr, Paul WE (2015) Innate immunological function of TH2 cells in vivo. Nat Immunol 16(10):1051–1059
Paul WE (2010) What determines Th2 differentiation, in vitro and in vivo? Immunol Cell Biol 88(3):236–239
Stone KD, Prussin C, Metcalfe DD (2010) IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol 125(2):S73–S80
Persson C (2013) Lysis of primed eosinophils in severe asthma. J Allergy Clin Immunol 132(6):1459–1460
Durrani SR, Viswanathan RK, Busse WW (2011) What effect does asthma treatment have on airway remodeling? Current perspectives. J Allergy Clin Immunol 128(3):439–448 quiz 49-50
Chung KF (2000) Airway smooth muscle cells: contributing to and regulating airway mucosal inflammation? Eur Respir J 15(5):961–968
Cayrol C, Girard JP (2018) Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev 281(1):154–168
Fanning LB, Boyce JA (2013) Lipid mediators and allergic diseases. Ann Allergy Asthma Immunol 111(3):155–162
Kim BS, Wang K, Siracusa MC, Saenz SA, Brestoff JR, Monticelli LA, Noti M, Tait Wojno ED, Fung TC, Kubo M, Artis D (2014) Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol 193(7):3717–3725
Kim BS, Wojno ED, Artis D (2013) Innate lymphoid cells and allergic inflammation. Curr Opin Immunol 25(6):738–744
Kepley CL, McFeeley PJ, Oliver JM, Lipscomb MF (2001) Immunohistochemical detection of human basophils in postmortem cases of fatal asthma. Am J Respir Crit Care Med 164(6):1053–1058
Macfarlane AJ, Kon OM, Smith SJ, Zeibecoglou K, Khan LN, Barata LT, McEuen AR, Buckley MG, Walls AF, Meng Q, Humbert M, Barnes NC, Robinson DS, Ying S, Kay AB (2000) Basophils, eosinophils, and mast cells in atopic and nonatopic asthma and in late-phase allergic reactions in the lung and skin. J Allergy Clin Immunol 105(1 Pt 1):99–107
Samitas K, Delimpoura V, Zervas E, Gaga M (2015) Anti-IgE treatment, airway inflammation and remodelling in severe allergic asthma: current knowledge and future perspectives. Eur Respir Rev 24(138):594–601
Brown JM, Wilson TM, Metcalfe DD (2008) The mast cell and allergic diseases: role in pathogenesis and implications for therapy. Clin Exp Allergy 38(1):4–18
Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW et al (2011) Mast cell phenotype, location, and activation in severe asthma. Data from the severe asthma research program. Am J Respir Crit Care Med 183(3):299–309
Fajt ML, Gelhaus SL, Freeman B, Uvalle CE, Trudeau JB, Holguin F, Wenzel SE (2013) Prostaglandin D(2) pathway upregulation: relation to asthma severity, control, and TH2 inflammation. J Allergy Clin Immunol 131(6):1504–1512
Peters MC, Kerr S, Dunican EM, Woodruff PG, Fajt ML, Levy BD, Israel E, Phillips BR, Mauger DT, Comhair SA, Erzurum SC, Johansson MW, Jarjour NN, Coverstone AM, Castro M, Hastie AT, Bleecker ER, Wenzel SE, Fahy JV (2018) Refractory airway type 2 inflammation in a large subgroup of asthmatic patients treated with inhaled corticosteroids. J Allergy Clin Immunol
Tomassen P, Vandeplas G, Van Zele T, Cardell LO, Arebro J, Olze H et al (2016) Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol 137(5):1449–56.e4
Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, Bleecker ER, National Heart, Lung, and Blood Institute Severe Asthma Research Program (2010) Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol 125(5):1028–36.e13
Liu T, Kanaoka Y, Barrett NA, Feng C, Garofalo D, Lai J, Buchheit K, Bhattacharya N, Laidlaw TM, Katz HR, Boyce JA (2015) Aspirin-exacerbated respiratory disease involves a cysteinyl leukotriene-driven IL-33-mediated mast cell activation pathway. J Immunol 195(8):3537–3545
Buchheit KM, Cahill KN, Katz HR, Murphy KC, Feng C, Lee-Sarwar K, Lai J, Bhattacharyya N, Israel E, Boyce JA, Laidlaw TM (2016) Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol 137(5):1566–76.e5
Lluis A, Schedel M, Liu J, Illi S, Depner M, von Mutius E, Kabesch M, Schaub B (2011) Asthma-associated polymorphisms in 17q21 influence cord blood ORMDL3 and GSDMA gene expression and IL-17 secretion. J Allergy Clin Immunol 127(6):1587–94.e6
Castillo JR, Peters SP, Busse WW (2017) Asthma exacerbations: pathogenesis, prevention, and treatment. J Allergy Clin Immunol Pract 5(4):918–927
Edwards MR, Strong K, Cameron A, Walton RP, Jackson DJ, Johnston SL (2017) Viral infections in allergy and immunology: how allergic inflammation influences viral infections and illness. J Allergy Clin Immunol 140(4):909–920
Guarnieri M, Balmes JR (2014) Outdoor air pollution and asthma. Lancet 383(9928):1581–1592
Undem BJ, Taylor-Clark T (2014) Mechanisms underlying the neuronal-based symptoms of allergy. J Allergy Clin Immunol 133(6):1521–1534
Colsoul B, Nilius B, Vennekens R (2009) On the putative role of transient receptor potential cation channels in asthma. Clin Exp Allergy 39(10):1456–1466
Bertin S, Aoki-Nonaka Y, de Jong PR, Nohara LL, Xu H, Stanwood SR, Srikanth S, Lee J, To K, Abramson L, Yu T, Han T, Touma R, Li X, González-Navajas JM, Herdman S, Corr M, Fu G, Dong H, Gwack Y, Franco A, Jefferies WA, Raz E (2014) The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4(+) T cells. Nat Immunol 15(11):1055–1063
Hahn C, Islamian AP, Renz H, Nockher WA (2006) Airway epithelial cells produce neurotrophins and promote the survival of eosinophils during allergic airway inflammation. J Allergy Clin Immunol 117(4):787–794
Biomarkers Definitions Working G (2001) Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 69(3):89–95
Hastie AT, Moore WC, Li H, Rector BM, Ortega VE, Pascual RM, Peters SP, Meyers DA, Bleecker ER, National Heart, Lung, and Blood Institute's Severe Asthma Research Program (2013) Biomarker surrogates do not accurately predict sputum eosinophil and neutrophil percentages in asthmatic subjects. J Allergy Clin Immunol 132(1):72–80
Wagener AH, de Nijs SB, Lutter R, Sousa AR, Weersink EJ, Bel EH et al (2015) External validation of blood eosinophils, FE(NO) and serum periostin as surrogates for sputum eosinophils in asthma. Thorax 70(2):115–120
Fitzpatrick AM, Jackson DJ, Mauger DT, Boehmer SJ, Phipatanakul W, Sheehan WJ, Moy JN, Paul IM, Bacharier LB, Cabana MD, Covar R, Holguin F, Lemanske RF Jr, Martinez FD, Pongracic JA, Beigelman A, Baxi SN, Benson M, Blake K, Chmiel JF, Daines CL, Daines MO, Gaffin JM, Gentile DA, Gower WA, Israel E, Kumar HV, Lang JE, Lazarus SC, Lima JJ, Ly N, Marbin J, Morgan W, Myers RE, Olin JT, Peters SP, Raissy HH, Robison RG, Ross K, Sorkness CA, Thyne SM, Szefler SJ, NIH/NHLBI AsthmaNet (2016) Individualized therapy for persistent asthma in young children. J Allergy Clin Immunol 138(6):1608–18.e12
Nair P, O'Byrne PM (2016) Measuring eosinophils to make treatment decisions in asthma. Chest 150(3):485–487
Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E et al (2009) Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med 360(10):985–993
Burrows B, Martinez FD, Cline MG, Lebowitz MD (1995) The relationship between parental and children's serum IgE and asthma. Am J Respir Crit Care Med 152(5 Pt 1):1497–1500
Gerald JK, Gerald LB, Vasquez MM, Morgan WJ, Boehmer SJ, Lemanske RF Jr, Mauger DT, Strunk RC, Szefler SJ, Zeiger RS, Bacharier LB, Bade E, Covar RA, Guilbert TW, Heidarian-Raissy H, Kelly HW, Malka-Rais J, Sorkness CA, Taussig LM, Chinchilli VM, Martinez FD (2015) Markers of differential response to inhaled corticosteroid treatment among children with mild persistent asthma. J Allergy Clin Immunol Pract 3(4):540–6.e3
Pillai P, Chan YC, Wu SY, Ohm-Laursen L, Thomas C, Durham SR, Menzies-Gow A, Rajakulasingam RK, Ying S, Gould HJ, Corrigan CJ (2016) Omalizumab reduces bronchial mucosal IgE and improves lung function in non-atopic asthma. Eur Respir J 48(6):1593–1601
Szefler SJ, Wenzel S, Brown R, Erzurum SC, Fahy JV, Hamilton RG, Hunt JF, Kita H, Liu AH, Panettieri RA Jr, Schleimer RP, Minnicozzi M (2012) Asthma outcomes: biomarkers. J Allergy Clin Immunol 129(3 Suppl):S9–S23
Lim HF, Nair P (2018) Airway inflammation and inflammatory biomarkers. Semin Respir Crit Care Med 39(1):56–63
Jia G, Erickson RW, Choy DF, Mosesova S, Wu LC, Solberg OD, Shikotra A, Carter R, Audusseau S, Hamid Q, Bradding P, Fahy JV, Woodruff PG, Harris JM, Arron JR, Bronchoscopic Exploratory Research Study of Biomarkers in Corticosteroid-refractory Asthma (BOBCAT) Study Group (2012) Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol 130(3):647–54.e10
Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR et al (2011) Lebrikizumab treatment in adults with asthma. N Engl J Med 365(12):1088–1098
Shiobara T, Chibana K, Watanabe T, Arai R, Horigane Y, Nakamura Y, Hayashi Y, Shimizu Y, Takemasa A, Ishii Y (2016) Dipeptidyl peptidase-4 is highly expressed in bronchial epithelial cells of untreated asthma and it increases cell proliferation along with fibronectin production in airway constitutive cells. Respir Res 17:28
Divekar R, Hagan J, Rank M, Park M, Volcheck G, O'Brien E, Meeusen J, Kita H, Butterfield J (2016) Diagnostic utility of urinary LTE4 in asthma, allergic rhinitis, chronic rhinosinusitis, nasal polyps, and aspirin sensitivity. J Allergy Clin Immunol Pract 4(4):665–670
Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Arron JR, Koth LL, Fahy JV (2009) T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med 180(5):388–395
Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, Hoda U, Rossios C, Sousa A, Wilson SJ, Howarth P, Dahlen B, Dahlen SE, Chanez P, Shaw D, Krug N, Sandstrӧm T, de Meulder B, Lefaudeux D, Fowler S, Fleming L, Corfield J, Auffray C, Sterk PJ, Djukanovic R, Guo Y, Adcock IM, Chung KF, U-BIOPRED Project Team ‡ (2017) A transcriptome-driven analysis of epithelial brushings and bronchial biopsies to define asthma phenotypes in U-BIOPRED. Am J Respir Crit Care Med 195(4):443–455
Carr TF, Kraft M (2016) Chronic infection and severe asthma. Immunol Allergy Clin N Am 36(3):483–502
Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG (2014) Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J 43(4):1067–1076
Marwick JA, Dorward DA, Lucas CD, Jones KO, Sheldrake TA, Fox S, Ward C, Murray J, Brittan M, Hirani N, Duffin R, Dransfield I, Haslett C, Rossi AG (2013) Oxygen levels determine the ability of glucocorticoids to influence neutrophil survival in inflammatory environments. J Leukoc Biol 94(6):1285–1292
Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, Huff R, Pilewski J, Holguin F, Kolls J, Wenzel S, Ray P, Ray A (2015) High IFN-gamma and low SLPI mark severe asthma in mice and humans. J Clin Invest 125(8):3037–3050
Gauthier M, Chakraborty K, Oriss TB, Raundhal M, Das S, Chen J, et al. Severe asthma in humans and mouse model suggests a CXCL10 signature underlies corticosteroid-resistant Th1 bias. JCI Insight. 2017;2(13)
Al-Ramli W, Prefontaine D, Chouiali F, Martin JG, Olivenstein R, Lemiere C et al (2009) T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol 123(5):1185–1187
Ricciardolo FLM, Sorbello V, Folino A, Gallo F, Massaglia GM, Favata G et al (2017) Identification of IL-17F/frequent exacerbator endotype in asthma. J Allergy Clin Immunol 140(2):395–406
McGarvey LP, Butler CA, Stokesberry S, Polley L, McQuaid S, Abdullah H et al (2014) Increased expression of bronchial epithelial transient receptor potential vanilloid 1 channels in patients with severe asthma. J Allergy Clin Immunol 133(3):704–12.e4
Peters U, Dixon AE, Forno E (2018) Obesity and asthma. J Allergy Clin Immunol 141(4):1169–1179
Rastogi D, Fraser S, Oh J, Huber AM, Schulman Y, Bhagtani RH, Khan ZS, Tesfa L, Hall CB, Macian F (2015) Inflammation, metabolic dysregulation, and pulmonary function among obese urban adolescents with asthma. Am J Respir Crit Care Med 191(2):149–160
Takahashi K, Pavlidis S, Ng Kee Kwong F, Hoda U, Rossios C, Sun K, et al. Sputum proteomics and airway cell transcripts of current and ex-smokers with severe asthma in U-BIOPRED: an exploratory analysis. Eur Respir J. 2018;51(5)
Tripple JW, McCracken JL, Calhoun WJ (2017) Biologic therapy in chronic obstructive pulmonary disease. Immunol Allergy Clin N Am 37(2):345–355
Pite H, Pereira AM, Morais-Almeida M, Nunes C, Bousquet J, Fonseca JA (2014) Prevalence of asthma and its association with rhinitis in the elderly. Respir Med 108(8):1117–1126
Gibson PG, McDonald VM, Marks GB (2010) Asthma in older adults. Lancet 376(9743):803–813
Dunn RM, Busse PJ, Wechsler ME (2018) Asthma in the elderly and late-onset adult asthma. Allergy 73(2):284–294
Nyenhuis SM, Schwantes EA, Evans MD, Mathur SK (2010) Airway neutrophil inflammatory phenotype in older subjects with asthma. J Allergy Clin Immunol 125(5):1163–1165
Schmitt V, Rink L, Uciechowski P (2013) The Th17/Treg balance is disturbed during aging. Exp Gerontol 48(12):1379–1386
Maes T, Cobos FA, Schleich F, Sorbello V, Henket M, De Preter K et al (2016) Asthma inflammatory phenotypes show differential microRNA expression in sputum. J Allergy Clin Immunol 137(5):1433–1446
Grzela K, Litwiniuk M, Zagorska W, Grzela T (2016) Airway remodeling in chronic obstructive pulmonary disease and asthma: the role of matrix metalloproteinase-9. Arch Immunol Ther Exp (Warsz) 64(1):47–55
Peters MC, McGrath KW, Hawkins GA, Hastie AT, Levy BD, Israel E, Phillips BR, Mauger DT, Comhair SA, Erzurum SC, Johansson MW, Jarjour NN, Coverstone AM, Castro M, Holguin F, Wenzel SE, Woodruff PG, Bleecker ER, Fahy JV, National Heart, Lung, and Blood Institute Severe Asthma Research Program (2016) Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med 4(7):574–584
Krug N, Madden J, Redington AE, Lackie P, Djukanovic R, Schauer U, Holgate ST, Frew AJ, Howarth PH (1996) T-cell cytokine profile evaluated at the single cell level in BAL and blood in allergic asthma. Am J Respir Cell Mol Biol 14(4):319–326
Modena BD, Tedrow JR, Milosevic J, Bleecker ER, Meyers DA, Wu W, Bar-Joseph Z, Erzurum SC, Gaston BM, Busse WW, Jarjour NN, Kaminski N, Wenzel SE (2014) Gene expression in relation to exhaled nitric oxide identifies novel asthma phenotypes with unique biomolecular pathways. Am J Respir Crit Care Med 190(12):1363–1372
Wang YH, Voo KS, Liu B, Chen CY, Uygungil B, Spoede W, Bernstein JA, Huston DP, Liu YJ (2010) A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med 207(11):2479–2491
Li BWS, Stadhouders R, de Bruijn MJW, Lukkes M, Beerens D, Brem MD et al (2017) Group 2 innate lymphoid cells exhibit a dynamic phenotype in allergic airway inflammation. Front Immunol 8:1684
Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD (2015) Asthma. Nat Rev Dis Primers 1:15025
Bigler J, Boedigheimer M, Schofield JPR, Skipp PJ, Corfield J, Rowe A, Sousa AR, Timour M, Twehues L, Hu X, Roberts G, Welcher AA, Yu W, Lefaudeux D, Meulder B, Auffray C, Chung KF, Adcock IM, Sterk PJ, Djukanović R, U-BIOPRED Study Group ‖ (2017) A severe asthma disease signature from gene expression profiling of peripheral blood from U-BIOPRED cohorts. Am J Respir Crit Care Med 195(10):1311–1320
Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F et al (2017) U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol 139(6):1797–1807
Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV (2014) Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J Allergy Clin Immunol 133(2):388–394
Reinke SN, Gallart-Ayala H, Gomez C, Checa A, Fauland A, Naz S, et al. Metabolomics analysis identifies different metabotypes of asthma severity. Eur Respir J. 2017;49(3)
Rufo JC, Madureira J, Fernandes EO, Moreira A (2016) Volatile organic compounds in asthma diagnosis: a systematic review and meta-analysis. Allergy 71(2):175–188
Maniscalco M, Paris D, Melck DJ, D'Amato M, Zedda A, Sofia M, Stellato C, Motta A (2017) Coexistence of obesity and asthma determines a distinct respiratory metabolic phenotype. J Allergy Clin Immunol 139(5):1536–47.e5
Park YH, Fitzpatrick AM, Medriano CA, Jones DP (2017) High-resolution metabolomics to identify urine biomarkers in corticosteroid-resistant asthmatic children. J Allergy Clin Immunol 139(5):1518–24.e4
Acknowledgments
We would like to thank Sandhya Khurana and Jen Kwong for their excellent comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
F.E.-H.L. is the founder of MicroBplex, Inc. M.E.K. and G.B.L. have no conflicts of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Kuruvilla, M.E., Lee, F.EH. & Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clinic Rev Allerg Immunol 56, 219–233 (2019). https://doi.org/10.1007/s12016-018-8712-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-018-8712-1