
Abstract
Changes in redox state are described in the early stages of ethanol-induced cardiac toxicity. Here, we evaluated whether nebivolol would abrogate ethanol-induced redox imbalance in the heart. Male Wistar rats were treated with a solution of ethanol (20% v/v) for 3 weeks. Treatment with nebivolol (10 mg/kg/day; p.o. gavage) prevented the increase of both superoxide (O2•-) and thiobarbituric acid reactive substances (TBARS) in the left ventricle of rats chronically treated with ethanol. Neither ethanol nor nebivolol affected the expression of Nox4, p47phox, or Rac-1. Nebivolol prevented ethanol-induced increase of Nox2 expression in the left ventricle. Superoxide dismutase (SOD) activity as well as the concentration of reduced glutathione (GSH) was not altered by ethanol or nebivolol. Augmented catalase activity was detected in the left ventricle of both ethanol- and nebivolol-treated rats. Treatment with nebivolol, but not ethanol increased eNOS expression in the left ventricle. No changes in the activity of matrix metalloproteinase (MMP)2 or in the expressions of MMP2, MMP9, and tissue inhibitor metalloproteinase (TIMP)1 were detected after treatment with ethanol or nebivolol. However, ethanol increased the expression of TIMP2, and this response was prevented by nebivolol. Our results provided novel insights into the mechanisms underlying the early stages of the cardiac injury induced by ethanol consumption. We demonstrated that Nox2/NADPH oxidase-derived ROS play a role in ethanol-induced lipoperoxidation and that this response was prevented by nebivolol. In addition, we provided evidence that MMPs are not activated in the early stages of ethanol-induced cardiac toxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alcoholic cardiomyopathy is a term used to describe a specific heart muscle disease that is induced by long-term consumption of high doses of ethanol [1]. The period of exposure to ethanol is directly related to the development of alcoholic cardiomyopathy, suggesting that the cardiac effects of ethanol are time related. In this sense, longer periods of ethanol consumption are more often associated with heart failure symptoms such as diastolic and systolic dysfunction [2, 3]. Although excessive consumption of ethanol is regarded to be detrimental to cardiovascular health, several studies provided evidence that low-to-moderate doses of ethanol display cardioprotective effects, further confirming the observation that the effects of ethanol are dose related [4, 5].
Oxidative stress is described as one of the primary mechanisms whereby ethanol initiates the array of its adverse cardiac changes [3]. Ethanol induces redox imbalance in cardiomyocytes by increasing the generation of superoxide (O2•−). This response is mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is a multi-subunit enzyme composed by membrane and cytosolic components [6, 7]. There are seven catalytic subunits of NADPH oxidase (Nox 1-5 and Duox 1-2) [8]. In the myocardium, up-regulation of Nox2 has been associated with overproduction of ROS and cardiac dysfunction [9]. Importantly, ethanol consumption was found to increase the expression of Nox2 in the heart, suggesting that this is one mechanism by which ethanol increases ROS generation in the myocardium [10, 11]. Lipid peroxidation is one important consequence of ethanol-induced ROS generation in cardiomyocytes [7, 12]. Elevated levels of ROS may also reduce the bioavailability of nitric oxide (NO) [13]. In addition, ethanol consumption alters the enzymatic and nonenzymatic antioxidant systems in the myocardium [14, 15]. Altogether, these observations show that in cardiomyocytes alterations of the redox state arise from distinctive actions of ethanol [3].
Collagen accumulation and fibrosis are common structural changes found in the late stages of the cardiac damage induced by ethanol [16, 17]. Matrix metalloproteinases (MMPs) are tissue‐remodeling enzymes that are critically involved in extracellular matrix recycling. Both MMP2 and MMP9 play an essential role in cardiac remodeling and left ventricle dysfunction associated with cardiovascular disorders [18, 19]. The activity of MMPs can be augmented by intracellular ROS. This response results in increased extracellular matrix remodeling, which will ultimately contribute to cardiac hypertrophy [19]. However, the expression/activity of MMPs during the initial stages of the cardiac injury induced by ethanol has not been studied.
Nebivolol is a selective β1-receptor antagonist, which is widely used in the management of cardiovascular diseases such as heart failure, hypertension and ischemic heart disease. This drug displays additional effects including blockade of α1-receptors, NO production and antioxidant activity [20]. The latter is mainly mediated by direct O2•− scavenging and inhibition of NADPH oxidase activity/expression [21]. Additionally, nebivolol decreases the activity of MMPs in the heart of hypertensive rats leading to a reduction in collagen deposition and hypertrophy [19]. Of note, treatment with antioxidants is described to exert cardioprotective effects by controlling ethanol-induced redox imbalance [16, 22]. Considering that nebivolol displays cardioprotective effects due to its antioxidant properties, we hypothesized that nebivolol would prevent ethanol-induced changes in oxidative state. Here, we sought to determine whether nebivolol would prevent redox changes during the early stages of the cardiac toxicity induced by ethanol. The harmful effects of ethanol in the heart are related to the duration of drinking and the dose of ethanol. In this context, long periods of consumption of high doses of ethanol are more often associated with cardiac toxicity [3]. Since our goal was to evaluate the cardiac changes during the early stages of ethanol consumption, animals were treated for a short period with a high dose of ethanol.
Materials and Methods
Animals and Grouping
The detailed protocol for treatment of male Wistar rats with ethanol or nebivolol has been previously described [23, 24]. Briefly, rats (40- to 50-day old) were randomly distributed in the following groups (n = 18 for each group): control, ethanol, nebivolol, and ethanol + nebivolol. Rats from the nebivolol group had free access to water and were treated with nebivolol (10 mg/kg/day, p.o., gavage). Rats from the control group had free access to water and received vehicle orally (carboxymethylcellulose 0.25%, gavage). Animals from the ethanol groups had free access to solutions of ethanol of 5% and 10% (v/v) during the first and second weeks of treatment, respectively. From the third to the fifth weeks of treatment, rats had free access to a solution of ethanol of 20% (v/v). Animals from the ethanol group received an oral solution of carboxymethylcellulose 0.25%, while rats from the ethanol + nebivolol group were treated with nebivolol (10 mg/kg/day, p.o., gavage). At the end of the treatment, half of animals of each group (n = 9) was used for biochemical analysis and half (n = 9) for protein isolation. The left ventricle of each animal was isolated and fragmented. For protein determination, the whole left ventricle was used. The dose and route of administration of nebivolol were chosen based on previous reports describing that nebivolol decreases heart rate and exerts antioxidant effects in the heart [19, 25, 26]. The animals were weighed, and the consumption of standard laboratory chow and liquid (water or ethanol solution) was measured weekly. Urethane (1.25 g/kg, i.p., Sigma-Aldrich, St. Louis, IL, USA) was used to anaesthetize the rats which were then killed by aortic exsanguination. The left ventricle was isolated and maintained at – 80 °C. Blood samples were collected from vena cava using heparinized syringes and then centrifuged at 6500×g for 15 min (at 4 °C). Serum was separated and stored at – 80 °C for creatine kinase (CK)-MB determination. All protocols were approved by the Animal Ethics Committee of the University of São Paulo—Campus of Ribeirão Preto (#14.1.357.53.2). Animal experiments comply with the ARRIVE guidelines and were carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
Determination of Serum CK-MB Activity
The activity of CK-MB (U/l) was evaluated to estimate cardiac damage. The activity of CK-MB was determined colorimetrically using a commercially available kit (#118, Labtest Diagnostica, Lagoa Santa, MG, Brazil).
Determination of O2 •−, H2O2, Nitrate/Nitrite (NOx) and Thiobarbituric Acid Reactive Substances (TBARS) Levels in the Left Ventricle
A glass-to-glass homogenizer was used to homogenize the left ventricle. The samples (50 µl) were then transferred to 96-wells white plates. The luminescence that results from the reaction of O2•- with lucigenin was used to estimate the production of O2•− as previously described [27]. Luminescence [relative light units (RLU)/mg protein] was determined using an Orion II luminometer (Berthold Detection Systems, Pforzheim, Germany). The concentration of extracellular H2O2 (nmol H2O2/mg protein) in samples of the left ventricle was determined using the fluorogenic substrate Amplex®Red (#A22188, Invitrogen, Carlsbad, CA, USA) as previously described [20]. A colorimetric assay kit (#780001, Cayman Chemical, Ann Arbor, MI, USA) was used to determine the concentration of NOx (nmol/mg protein) in the left ventricle. A commercially available kit (#10009055, Cayman Chemical, Ann Arbor, MI, USA) was used to measure the concentration of TBARS (nmol/mg protein) colorimetrically at 540 nm.
Determination of Reduced Glutathione (GSH) Levels and Activities of Superoxide Dismutase (SOD) and Catalase in the Left Ventricle
The concentration of GSH (µg/mg protein) was determined as previously described [28]. In brief, the left ventricle was homogenized in 300 μl of phosphate buffered saline (PBS, pH 7.4) and centrifuged (10,000×g, 15 min, 4 °C). Then, supernatants were incubated with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) which oxidizes GSH forming 5'-thio-2-nitrobenzoic (TNB), a yellow product that is detected at 415 nm.
In order to determine SOD activity, the left ventricle was homogenized in 300 μl of PBS (pH 7.4) and centrifuged (10,000×g, 12 min, 4 °C). The supernatants were used for the determination of total SOD activity (% inhibition rate/mg protein) using a commercially available kit (#19160, Sigma-Aldrich, St. Louis, IL, USA). The activity of catalase (nmol/min/mg protein) was determined based on the consumption of H2O2 using a spectrophotometer (at 240 nm) as previously described by Gonzaga et al. [28].
Western Immunoblotting
Samples were prepared as previously described by Gonzaga et al. [28]. Proteins (60 µg) were separated by electrophoresis (90 min at 150 V) on 10% polyacrylamide gels and then transferred (90 min at 100V) onto nitrocellulose membranes. In order to block nonspecific binding, the membranes were exposed to skimmed milk (7%) and then washed out three times in Tris-buffered saline containing Tween 20 (TBS-T). The membranes were incubated overnight with one of the following primary antibodies: anti-gp91phox (1:250, sc-5827, Santa Cruz Biotechnology, Dallas, Texas, USA), anti-Nox4 (1:250, sc-30141, Santa Cruz Biotechnology), anti-p47phox (1:500, sc-14015, Santa Cruz Biotechnology), anti-Rac-1 (1:500, sc-217, Santa Cruz Biotechnology), anti-eNOS (1:500, N3893, Sigma-Aldrich, St. Louis, IL, USA), anti-MMP2 (1:500, ab110186, Abcam), anti-MMP9 (1:500, ab38898, Abcam), anti-TIMP1 (1:500, sc-21734, Santa Cruz Biotechnology), or anti-TIMP2 (1:500, sc-21735, Santa Cruz Biotechnology). After incubation with primary antibodies, membranes were washed out three times in TBS-T and incubated with secondary antibodies at room temperature for 90 min. Signals were revealed using a chemiluminescent reagent and visualized using a ChemiDoc™ XRS+ (Bio-Rad, Hercules, CA, USA). Proteins were quantified by densitometry, and β-actin (1:1000, sc-4778, Santa Cruz Biotechnology) was used as an internal control.
Determination of MMP2 Activity in the Left Ventricle by Gelatin Zymography
The activity of MMP2 isoforms (64, 72 and 75 kDa) was determined by gelatin zymography as previously described [29]. In brief, proteins (30 µg) were separated by electrophoresis on 12% polyacrylamide gel electrophoresis (SDS-PAGE) co-polymerized with gelatin (1%). After electrophoresis, gels were incubated (1 h at room temperature) in a Triton X-100 solution (2%). Then, the gels were incubated at 37 °C for 18 h in Tris-HCl buffer (Tris 50 mM and ZnCl2 1 μM, pH 7.4) and CaCl2 (10 mM). Gels were stained with 0.05% Coomassie Brilliant Blue (CBB) G-250 and then destained using a solution of methanol (30%) and acetic acid (10%). Gelatinolytic activity was detected as an unstained band against the background of gelatin stained by CBB. The ChemiDoc™ XRS+ (Bio-Rad, Hercules, CA, USA) was used to quantify band densitometry. The results were given as arbitrary units.
Statistical Analysis
Data shown as means ± standard error of the mean (S.E.M.) were analyzed using two-way analysis of variance (ANOVA) followed by Bonferroni’s comparison test. Statistical tests with p < 0.05 were considered as significant. All statistical analyses were performed using the program GraphPad® Prism 5.01 (GraphPad Software Inc., San Diego, CA, USA).
Results
Body Mass, Chow Consumption, and Liquid Intake
Before treatment, rats showed mean body masses of 370 ± 10 g (control group), 374 ± 7 g (ethanol group), 371 ± 7 g (nebivolol group), and 374 ± 8 g (ethanol + nebivolol group). Rats from the ethanol groups showed reduced weight gain when compared to rats from control and nebivolol groups (Table 1). Both chow and liquid intake were reduced in rats from the ethanol groups when compared to rats from control and nebivolol groups (Table 1).
Effects of Chronic Ethanol Consumption and Nebivolol on CK-MB Activity, ROS Generation, and Lipoperoxidation in the Rat Left Ventricle
Ethanol consumption did not alter CK-MB activity (Fig. 1a). Treatment with ethanol increased lucigenin-derived chemiluminescence in the left ventricle, and this response was prevented by nebivolol (Fig. 1b). No changes in the concentration of H2O2 were detected after treatment with either ethanol or nebivolol (Fig. 1c). Treatment with nebivolol prevented ethanol-induced increase in cardiac TBARS concentration (Fig. 1d).

Effects of chronic ethanol consumption and nebivolol on serum CK-MB levels, ROS generation, and lipoperoxidation in the rat left ventricle. Serum levels of CK-MB were determined colorimetrically (a). The generation of O2•− was evaluated by the lucigenin-derived chemiluminescence assay (b). The concentration of H2O2 was determined fluorometrically (C). The levels of TBARS were determined colorimetrically (D). Results are given as means ± S.E.M. of 7 to 9 animals. *Compared to control, nebivolol and ethanol+nebivolol groups (p < 0.05, two-way ANOVA followed by Bonferroni’s comparison test)
Effects of Chronic Ethanol Consumption and Nebivolol in the Expression of Nox2, Nox4, Rac-1, and p47phox in the Rat Left Ventricle
Nebivolol prevented the increase of Nox2 expression detected in the left ventricle of ethanol-treated rats (Fig. 2a). Neither ethanol nor nebivolol affected the expression of Nox4 (Fig. 2b), Rac-1 (Fig. 2c) or p47phox (Fig. 2d) in the left ventricle.

Effects of chronic ethanol consumption and nebivolol in Nox2, Nox4, Rac-1, and p47phox expression in the rat left ventricle. Top panels: representative immunoblots for Nox isoforms, Rac-1, and p47phox. Bottom panels: corresponding bar graphs show densitometric data for protein expression of Nox2 (a), Nox4 (b), Rac-1 (c), and p47phox (d). Results are given as means ± S.E.M. of 5 to 6 animals. *Compared to control, nebivolol, and ethanol + nebivolol groups (p < 0.05, two-way ANOVA followed by Bonferroni’s comparison test)
Effects of Chronic Ethanol Consumption and Nebivolol in NOx Levels and eNOS Expression in the Rat Left Ventricle
Neither ethanol nor nebivolol affected the concentrations of NOx in the left ventricle (Fig. 3c). No changes in the expression of eNOS were found in the left ventricle of ethanol-treated rats. However, increased expression of eNOS was found in the left ventricle of nebivolol-treated rats (Fig. 3b).

Effects of chronic ethanol consumption and nebivolol treatment in NOx levels and eNOS expression in the rat left ventricle. The concentration of NOx was evaluated colorimetrically (a). Top panels: representative immunoblots for eNOS. Bottom panels: corresponding bar graph shows densitometric data for protein expression of eNOS (b). Results are given as means ± S.E.M. of 7 to nine animals. *Compared to control and ethanol groups (p < 0.05, two-way ANOVA followed by Bonferroni’s comparison test)
Effects of Chronic Ethanol Consumption and Nebivolol in the Activities of SOD and Catalase and in the Concentration of GSH in the Rat Left Ventricle
No changes in total SOD activity were detected in the left ventricle of rats treated with either ethanol or nebivolol (Fig. 4a). Treatment with ethanol or nebivolol increased catalase activity in the left ventricle (Fig. 4b). No changes in the concentration of GSH were found in the left ventricle of rats treated with ethanol or nebivolol (Fig. 4c).

Effects of chronic ethanol consumption and nebivolol treatment in the activities of SOD and catalase and in the concentration of GSH in the rat left ventricle. Total SOD activity was determined by colorimetric assay (a). Catalase activity was determined spectrophotometrically by measuring H2O2 decomposition (b). The concentration of GSH was determined colorimetrically (c). Results are given as means ± S.E.M. of eight to nine animals. *Compared to control group; **Compared to control, ethanol, and nebivolol groups (p < 0.05, two-way ANOVA followed by Bonferroni’s comparison test)
Effects of Chronic Ethanol Consumption and Nebivolol in the Expression of MMP2, MMP9, TIMP1, and TIMP2 in the Rat Left Ventricle
No changes in the expression of MMP2 (Fig. 5a), MMP9 (Fig. 5b), or TIMP1 (Fig. 5c) were detected in the left ventricle of rats treated with ethanol or nebivolol. Conversely, increased expression of TIMP2 (Fig. 5d) was found in the left ventricle of ethanol-treated rats.

Effects of chronic ethanol consumption and nebivolol treatment in MMP2, MMP9, TIMP1, and TIMP2 expression in the rat left ventricle. Top panels: representative immunoblots for MMP2, MMP9, TIMP1, and TIMP2. Bottom panels: corresponding bar graphs show densitometric data for protein expression of MMP2 (a), MMP9 (b), TIMP1 (c), and TIMP2 (d). Results are given as means ± S.E.M. of five to six animals. *Compared to control, nebivolol, and ethanol+nebivolol groups (p < 0.05, two-way ANOVA followed by Bonferroni’s comparison test)
Effects of Chronic Ethanol Consumption and Nebivolol in the Activity of MMP2 isoforms in the Rat Left Ventricle
No differences were found in the activities of MMP2 isoforms (Fig. 6a–d). Similarly, no changes of total MMP2 activity were detected after treatment with ethanol or nebivolol (Fig. 6e).

Effects of chronic ethanol consumption and nebivolol treatment in MMP2 isoforms activities in the rat left ventricle. Representative SDS-PAGE gelatin zymogram of left ventricle extracts (a). Molecular weights of MMP2 bands (75, 72, and 64 kDa) were identified after electrophoresis on 12% SDS-PAGE. Bar graphs show the values for the activity of MMP2 isoforms at 75 kDa (b), 72 kDa (c), and 64 kDa (d) and total (75 + 72 + 64 kDa) MMP2 activity (e). Results are given as means ± S.E.M. of four to six animals. BFS bovine fetal serum
Discussion
The existence of a potential cause-and-effect relationship between oxidative stress and ethanol-induced cardiac damage has been described. Redox imbalance is pointed out as an early and initiating mechanism by which ethanol induces heart muscle injury [3]. Corroborating previous studies, we found that exposure to ethanol for a short period did not induce heart muscle injury but increased ROS generation within the myocardium [16, 17, 30].
NADPH oxidase is the major source of ROS in cardiomyocytes [31, 32]. In the heart, up-regulation of Nox2 (a catalytic subunit of NADPH oxidase) is associated with overproduction of ROS [9]. We detected an increase in the expression of Nox2, but not Nox4 in the left ventricle of ethanol-treated rats. This finding corroborated previous studies showing up-regulation of Nox2 in the heart of ethanol-treated rats [10, 11]. Based on such finding, we may suggest that Nox2/NADPH oxidase mediates ethanol-induced O2•− generation in the left ventricle. Treatment with nebivolol prevented the up-regulation of Nox2 as well as the increase in O2•− generation, suggesting that nebivolol may exert cardioprotective effects by attenuating Nox2/NADPH oxidase-derived ROS. However, our results do not exclude the possibility that the antioxidant effect of nebivolol was due to its direct ROS-scavenging action. In addition, clinical and experimental studies have shown that the beneficial effects of nebivolol are associated to its capacity of decreasing cardiac work [33, 34]. Using the same dose and route of administration used in the present investigation, Górska et al. [26] described that nebivolol decrease heart rate in both normotensive and hypertensive rats. Thus, it is possible that nebivolol-induced decrease in cardiac work may also be contributing to its beneficial effects in our model.
The seven members of the Nox family (Nox1-5 and Duox1-2) are distinctly activated. While Nox4 is constitutively active, Nox2 activation requires cytoplasmic subunits including Rac-1 and p47phox [8]. Our present findings showed that neither ethanol nor nebivolol altered the expression of Rac-1 or p47phox, suggesting that up-regulation of regulatory subunits of NADPH oxidase did not play a role in ethanol-induced ROS generation. In some tissues, nebivolol impairs the assembly of NADPH oxidase by decreasing the expression of its regulatory subunits [23, 35]. However, the antioxidant activity of nebivolol here described is not mediated by down-regulation of Rac-1 and p47phox.
The redox system is essential in maintaining cellular homeostasis. Loss of redox balance favors lipid peroxidation, a metabolic process that is initiated by the reaction of ROS (e.g., O2•−) with a lipid substrate [36]. The consequences of the oxidative degradation of lipids include decreased lipid fluidity, altered membrane permeability, and changes in ion transport. These are initial steps found in the cardiac toxicity induced by ethanol [12, 22]. Here, we found increased concentrations of TBARS in the left ventricle of ethanol-treated rats, suggesting an increase in lipid peroxidation. NADPH oxidase-derived O2•− may be responsible for the increase in lipid peroxidation induced by ethanol. This idea is strengthen by the fact that nebivolol prevented the increase in the concentration of both O2•− and TBARS.
Oxidative stress may reduce NO bioavailability by affecting eNOS expression. Reaction of O2•− with NO is also a mechanism whereby oxidative stress decreases NO levels [37]. In the heart, NO influences both inotropic and chronotropic responses [38]. Our findings demonstrated that ethanol consumption did not alter NO levels or the expression of eNOS in the left ventricle. These observations are in accordance with previous findings of Silva et al. [30]. Corroborating previous studies where nebivolol was shown to increase eNOS expression in distinctive tissues [34, 39, 40], we found up-regulation of eNOS in the left ventricle of nebivolol-treated rats. Using the same protocol for nebivolol treatment, we have previously described that this drug increased the concentration of reduced glutathione (GSH) in plasma and decreased the expression of PKCδ and Rac-1, which are regulatory subunits of the enzyme NADPH oxidase [23, 24]. Altogether these findings show that the protocol for treatment with nebivolol was effective.
Accumulation of oxidizing molecules may lead to disruptions of redox homeostasis. For this reason, enzymatic and nonenzymatic antioxidant systems balance the intracellular levels of oxidizing molecules [41]. A number of studies have provided evidence that ethanol changes cell antioxidant defenses in a tissue-specific manner [23, 24, 42]. Antioxidant defense mechanisms may be trigged soon after ethanol drinking to protect the myocardium against the oxidative damage induced by ethanol. For example, increased activity of catalase was found in cardiomyocytes in early stages of ethanol-induced cardiac toxicity [14, 15]. Corroborating these previous studies, we found increased catalase activity in the left ventricle of ethanol-treated rats. Treatment with nebivolol also increased catalase activity as previously shown by El-Sheikh et al. [43] and Gao et al. [44]. The combination of ethanol and nebivolol showed synergistic effects in augmenting catalase activity. A possible explanation for such result is that these drugs act through distinctive mechanisms to increase catalase activity. Catalase is an enzyme that decomposes H2O2 into water and oxygen. However, despite the increase in catalase activity, we detected no changes in the concentration of H2O2 in the left ventricle of ethanol-treated rats.
The three isoforms of superoxide dismutase (SOD1-3) are expressed in different subcellular compartments where they catalyze the dismutation of O2•− to molecular oxygen and H2O2. Thus, these enzymes are considered the first line of defense against the cellular damage induced by ROS [45]. The effects of ethanol in SOD activity are time related, and for this reason, increases, decreases, and no changes in SOD activity were reported in the myocardium after treatment with ethanol [7, 46]. Here, we found no changes in total SOD activity in the left ventricle of ethanol-treated rats. Similarly, ethanol consumption did not alter the levels of GSH, a nonenzymatic antioxidant peptide [47]. Both SOD and catalase buffer free radicals using H2O2 as an intermediate. Thus, given that oxidant levels are increased by ethanol, one might have expected an increased activity of both catalase and SOD. The reason for a catalase-specific response to ethanol is unclear. As mentioned earlier, the contribution of each enzyme for detoxification may be related to the period of exposure to ethanol. Ribière et al. [46] described increased SOD activity in the heart of rats treated with ethanol for 4 weeks. Since SOD acts as the first line of defense against oxidative stress, it is possible that SOD was activated prior to catalase in our model.
MMPs are tissue‐remodeling enzymes that recycles cardiac extracellular matrix. Increased activity and/or expression of these enzymes results in fibrosis and cardiac remodeling. Both MMP2 and MMP9 play an essential role in cardiac remodeling and left ventricle dysfunction associated with cardiovascular disorders [18, 19]. Ethanol consumption was found to increase the activity/expression of MMPs in distinctive tissues [48, 49]. Here, we found no changes in the expression or activity of MMPs, suggesting that these enzymes do not play a role in the initial steps of the cardiac injury induced by ethanol. TIMPs are endogenous inhibitors of MMPs, and decreased expression of these proteins may lead to an increased activity of MMPs [18]. Ethanol consumption did not alter TIMP1 expression in the left ventricle, while increased expression of TIMP2 was found after treatment with ethanol. Reasons for such response are unclear, but they may be part of a regulatory mechanism to prevent an overactivity of MMP2.
Using this model of ethanol feeding, we have previously found blood ethanol levels in the range of 35–40 mM [24, 35]. Thus, the effects of ethanol on the cardiac oxidative stress as well as the protective action of nebivolol here described are physiologically relevant since this concentration of ethanol is within that found in the bloodstream of humans after ethanol consumption [50].
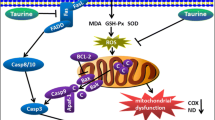
In summary, our results provided novel insights into the mechanisms underlying the early stages of the cardiac injury induced by ethanol consumption. We demonstrated that Nox2/NADPH oxidase-derived ROS play a role in ethanol-induced lipoperoxidation and that this response was prevented by nebivolol. In addition, we provided evidence that MMPs are not activated in the early stages of ethanol-induced cardiac toxicity. A schematic representation of the possible events that follows ethanol consumption is shown in Fig. 7.

Schematic illustration of the possible mechanisms whereby nebivolol acts in the initial stages of ethanol-induced toxicity in the mice heart. Nox2 catalytic subunit of the enzyme NADPH oxidase, O2•- superoxide, eNOS endothelial nitric oxide synthase, TIMP2 tissue inhibitor metalloproteinase
References
Mirijello, A., Tarli, C., Vassallo, G. A., Sestito, L., Antonelli, M., d’Angelo, C., et al. (2017). Alcoholic cardiomyopathy: What is known and what is not known. European Journal of Internal Medicine, 43, 1–5.
Lazarevic, A. M., Nakatani, S., Neskovic, A. N., Marinkovic, J., Yasumura, Y., Stojicic, D., et al. (2000). Early changes in left ventricular function in chronic asymptomatic alcoholics: relation to the duration of heavy drinking. Journal of the American College of Cardiology, 35, 1599–1606.
Piano, M. R., & Phillips, S. A. (2014). Alcoholic cardiomyopathy: pathophysiologic insights. Cardiovascular Toxicology, 14, 291–308.
Guiraud, A., de Lorgeril, M., Boucher, F., Berthonneche, C., Rakotovao, A., & de Leiris, J. (2004). Cardioprotective effect of chronic low dose ethanol drinking: insights into the concept of ethanol preconditioning. Journal of Molecular and Cellular Cardiology, 36(4), 561–566.
Miyamae, M., Kaneda, K., Domae, N., & Figueredo, V. M. (2010). Cardioprotection by regular ethanol consumption: Potential mechanisms and clinical application. Current Drug Abuse Reviews, 3(1), 39–48.
Zhang, B., Turdi, S., Li, Q., Lopez, F. L., Eason, A. R., Anversa, P., & Ren, J. (2010). Cardiac overexpression of insulin-like growth factor 1 attenuates chronic alcohol intake-induced myocardial contractile dysfunction but not hypertrophy: Roles of Akt, mTOR, GSK3beta, and PTEN. Free Radical Biology and Medicine, 49, 1238–1253.
Nakashima, M. A., Silva, C. B. P., Gonzaga, N. A., Simplicio, J. A., Omoto, A. C. M., Tirapelli, L. F., et al. (2019). Chronic ethanol consumption increases reactive oxygen species generation and the synthesis of pro-inflammatory proteins in the heart through TNFR1-dependent mechanisms. Cytokine, 121, 154734.
Urner, S., Ho, F., Jha, J.C., Ziegler, D., Jandeleit-Dahm, K. (2020). NADPH Oxidase Inhibition: Preclinical and Clinical Studies in Diabetic Complications. Antioxid Redox Signal in press.
Murdoch, C. E., Chaubey, S., Zeng, L., Yu, B., Ivetic, A., Walker, S. J., et al. (2014). Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. Journal of the American College of Cardiology, 63, 2734–2741.
Brandt, M., Garlapati, V., Oelze, M., Sotiriou, E., Knorr, M., Kröller-Schön, S., et al. (2016). NOX2 amplifies acetaldehyde-mediated cardiomyocyte mitochondrial dysfunction in alcoholic cardiomyopathy. Scientific Reports, 6, 325–354.
Mustroph, J., Wagemann, O., Lebek, S., Tarnowski, D., Ackermann, J., Drzymalski, M., et al. (2018). SR Ca2+-leak and disordered excitation-contraction coupling as the basis for arrhythmogenic and negative inotropic effects of acute ethanol exposure. Journal of Molecular and Cellular Cardiology, 116, 81–90.
Vendemiale, G., Grattagliano, I., Altomare, E., Serviddio, G., Portincasa, P., Prigigallo, F., & Palasciano, G. (2001). Mitochondrial oxidative damage and myocardial fibrosis in rats chronically intoxicated with moderate doses of ethanol. Toxicology Letters, 123, 209–216.
Khanna, D., Kan, H., Fang, Q., Xie, Z., Underwood, B. L., Jain, A. C., et al. (2007). Inducible nitric oxide synthase attenuates adrenergic signaling in alcohol fed rats. Journal of Cardiovascular Pharmacology, 50, 692–696.
Kino, M. J. (1981). Chronic effects of ethanol under partial inhibition of catalase activity in the rat heart: light and electron microscopic observations. Journal of Molecular and Cellular Cardiology, 13, 5–21.
Vikhert, A.M., Tsiplenkova, V.G., Cherpachenko, N.M. (1986). Alcoholic cardiomyopathy and sudden cardiac death. Journal of the American College of Cardiology, 8(1 Suppl A), 3A-11A.
Tan, Y., Li, X., Prabhu, S. D., Brittian, K. R., Chen, Q., Yin, X., et al. (2012). Angiotensin II plays a critical role in alcohol-induced cardiac nitrative damage, cell death, remodeling, and cardiomyopathy in a protein kinase C/nicotinamide adenine dinucleotide phosphate oxidase-dependent manner. Journal of the American College of Cardiology, 59, 1477–1586.
Zhang, R. H., Gao, J. Y., Guo, H. T., Scott, G. I., Eason, A. R., Wang, X. M., & Ren, J. (2013). Inhibition of CYP2E1 attenuates chronic alcohol intake-induced myocardial contractile dysfunction and apoptosis. Biochimica et Biophysica Acta, 1832, 128–141.
Berry, E., Bosonea, A. M., Wang, X., & Fernandez-Patron, C. (2013). Insights into the activity, differential expression, mutual regulation, and functions of matrix metalloproteinases and a disintegrin and metalloproteinases in hypertension and cardiac disease. Journal of Vascular Research, 50, 52–68.
Rizzi, E., Guimarães, D. A., Ceron, C. S., Prado, C. M., Pinheiro, L. C., Martins-Oliveira, A., et al. (2014). β1-Adrenergic blockers exert antioxidant effects, reduce matrix metalloproteinase activity, and improve renovascular hypertension-induced cardiac hypertrophy. Free Radical Biology and Medicine, 73, 308–317.
Carda, A. P., Marchi, K. C., Rizzi, E., Mecawi, A. S., Antunes-Rodrigues, J., Padovan, C. M., & Tirapelli, C. R. (2015). Acute restraint stress induces endothelial dysfunction: role of vasoconstrictor prostanoids and oxidative stress. Stress, 18, 233–243.
Vanhoutte, P. M., & Gao, Y. (2013). Beta blockers, nitric oxide, and cardiovascular disease. Current Opinion of Pharmacology, 13, 265–273.
Jing, L., Jin, C. M., Li, S. S., Zhang, F. M., Yuan, L., Li, W. M., et al. (2012). Chronic alcohol intake-induced oxidative stress and apoptosis: role of CYP2E1 and calpain-1 in alcoholic cardiomyopathy. Molecular and Cellular Biochemistry, 359, 283–292.
Do Vale, G. T., Gonzaga, N. A., Simplicio, J. A., & Tirapelli, C. R. (2017). Nebivolol prevents ethanol-induced reactive oxygen species generation and lipoperoxidation in the rat kidney by regulating NADPH oxidase activation and expression. European Journal of Pharmacology, 799, 33–40.
Do Vale, G. T., Simplicio, J. A., Gonzaga, N. A., Yokota, R., Ribeiro, A. A., Casarini, D. E., et al. (2018). Nebivolol prevents vascular oxidative stress and hypertension in rats chronically treated with ethanol. Atherosclerosis, 274, 67–76.
Ma, L., Gul, R., Habibi, J., Yang, M., Pulakat, L., Whaley-Connell, A., et al. (2012). Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the transgenic (mRen2) rat. American Journal of Physiology: Heart and Circulatory Physiology, 302, H2341–H2351.
Górska, D., Dudarewicz, M., Czarnecka, E., & Andrzejczak, D. (2010). Does nebivolol influence serum concentrations of proinflammatory cytokines in hypertensive (SHR) and normotensive (WKY) rats? Pharmacology Reports, 62(1), 86–94.
Yogi, A., Callera, G. E., Mecawi, A. S., Batalhão, M. E., Carnio, E. C., Antunes-Rodrigues, J., et al. (2012). Acute ethanol intake induces superoxide anion generation and mitogen-activated protein kinase phosphorylation in rat aorta: A role for angiotensin type 1 receptor. Toxicology and Applied Pharmacology, 264, 470–478.
Gonzaga, N. A., Callera, G. E., Yogi, A., Mecawi, A. S., Antunes-Rodrigues, J., Queiroz, R. H., et al. (2014). Acute ethanol intake induces mitogen-activated protein kinase activation, platelet-derived growth factor receptor phosphorylation, and oxidative stress in resistance arteries. Journal of Physiology Biochemistry, 70, 509–523.
Ceron, C. S., Rizzi, E., Guimaraes, D. A., Martins-Oliveira, A., Cau, S. B., Ramos, J., et al. (2012). Time course involvement of matrix metalloproteinases in the vascular alterations of renovascular hypertension. Matrix Biology, 31, 261–270.
Silva, C. B. P., Gómez, J. P. S., & do Vale, G.T., Simplicio, J.A., Gonzaga, N.A., Tirapelli, C.R. . (2020). Interleukin-10 limits the initial steps of the cardiorenal damage induced by ethanol consumption. Life Science, 242, 117239.
Froogh, G., Kandhi, S., Duvvi, R., Le, Y., Weng, Z., Alruwaili, N., et al. (2020). The contribution of chymase-dependent formation of ANG II to cardiac dysfunction in metabolic syndrome of young rats: roles of fructose and EETs. American Journal of Physiology: Heart and Circulatory Physiology, 318, H985–H993.
Okabe, K., Matsushima, S., Ikeda, S., Ikeda, M., Ishikita, A., Tadokoro, T., et al. (2020). DPP (Dipeptidyl Peptidase)-4 inhibitor attenuates ang II (Angiotensin II)–induced cardiac hypertrophy via GLP (Glucagon-Like Peptide)-1–dependent suppression of nox (nicotinamide adenine dinucleotide phosphate oxidase) 4-HDAC (histone deacetylase) 4 pathway. Hypertension, 75, 991–1001.
Karabacak, M., Doğan, A., Tayyar, S., Ozaydin, M., & Erdogan, D. (2015). Carvedilol and nebivolol improve left ventricular systolic functions in patients with non-ischemic heart failure. Anatolian Journal of Cardiology, 15(4), 271–276.
Zhang, Z., Ding, L., Jin, Z., Gao, G., Li, H., Zhang, L., et al. (2014). Nebivolol protects against myocardial infarction injury via stimulation of beta 3-adrenergic receptors and nitric oxide signaling. PLoS One, 9, e98179.
Oelze, M., Daiber, A., Brandes, R. P., Hortmann, M., Wenzel, P., Hink, U., et al. (2006). Nebivolol Inhibits Superoxide Formation by NADPH Oxidase and Endothelial Dysfunction in Angiotensin II-treated Rats. Hypertension, 48, 677–684.
Yin, H., Xu, L., & Porter, N. A. (2011). Free radical lipid peroxidation: mechanisms and analysis. Chemical Reveiws, 111, 5944–5972.
Förstermann, U., Xia, N., & Li, H. (2017). Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circulation Research, 120, 713–735.
Rastaldo, R., Pagliaro, P., Cappello, S., Penna, C., Mancardi, D., Westerhof, N., & Losano, G. (2007). Nitric oxide and cardiac function. Life Science, 81, 779–793.
Zhou, X., Ma, L., Habibi, J., Whaley-Connell, A., Hayden, M. R., Tilmon, R. D., et al. (2010). Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the Zucker obese rat. Hypertension, 55, 880–888.
Wang, Y., Zhang, F., Liu, Y., Yin, S., Pang, X., Li, Z., & Wei, Z. (2017). Nebivolol alleviates aortic remodeling through eNOS upregulation and inhibition of oxidative stress in L-NAME-induced hypertensive rats. Clinical and Experimental Hypertension, 39, 628–639.
Snezhkina, A. V., Kudryavtseva, A. V., Kardymon, O. L., Savvateeva, M. V., Melnikova, N. V., Krasnov, G. S., & Dmitriev, A. A. (2019). ROS generation and antioxidant defense systems in normal and malignant cells. Oxidative Medicine and Cellular Longevity, 2019, 6175804.
Simplicio, J. A., do Vale, G. T., Gonzaga, N. A., Leite, L. N., Hipólito, U. V., Pereira, C. A., et al. (2017). Reactive oxygen species derived from NAD(P)H oxidase play a role on ethanol-induced hypertension and endothelial dysfunction in rat resistance arteries. Journal of Physiology and Biochemisty, 73, 5–16.
El-Sheikh, A. A. K., Morsy, M. A., & Abdel-Latif, R. G. (2019). Modulation of eNOS/iNOS by nebivolol protects against cyclosporine A-mediated nephrotoxicity through targeting inflammatory and apoptotic pathways. Environmental Toxicology and Pharmacology, 69, 26–35.
Gao, J., Xie, Q., Wei, T., Huang, C., Zhou, W., & Shen, W. (2019). Nebivolol improves obesity-induced vascular remodeling by suppressing NLRP3 activation. Journal of Cardiovascular Pharmacology, 73, 326–333.
Younus, H. (2018). Therapeutic potentials of superoxide dismutase. International Journal of Health Science (Qassim), 12, 88–93.
Ribière, C., Hininger, I., Rouach, H., & Nordmann, R. (1992). Effects of chronic ethanol administration on free radical defence in rat myocardium. Biochemical Pharmacology, 44, 1495–1500.
Forman, H. J., Zhang, H., & Rinna, A. (2009). Glutathione: Overview of its protective roles, measurement, and biosynthesis. Moleulcar Aspects of Medicine, 30, 1–12.
Muniz, J. J., Leite, L. N., Lacchini, R., Tanus-Santos, J. E., & Tirapelli, C. R. (2018). Dysregulated mitogen-activated protein kinase and matrix metalloproteinase in ethanol-induced cavernosal dysfunction. Canadian Journal of Physiology and Pharmacology, 96, 266–274.
Cernigliaro, C., D’Anneo, A., Carlisi, D., Giuliano, M., Gammazza, M. A., Barone, R., et al. (2019). Ethanol-mediated stress promotes autophagic survival and aggressiveness of colon cancer cells via activation of Nrf2/HO-1 pathway. Cancers (Basel), 11, 505–526.
Urso, T., Gavaler, J. S., & Van Thiel, D. H. (1981). Blood ethanol levels in sober alcohol users seen in an emergency room. Life Science, 28, 1053–1056.
Funding
This work was funded by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) [Grant Number 2013/15824-9] and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) [Grant Number 304027/2017-0]. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES) [Finance Code 001].
Author information
Authors and Affiliations
Contributions
GTDV: Data curation; Formal analysis; Investigation; Methodology; Project administration; Roles/Writing - original draft. CBPDS: Data curation; Formal analysis; Investigation; Methodology. AHDS: Data curation; Formal analysis; Investigation; Methodology. Natália de AG: Data curation; Formal analysis; Investigation; Methodology. KMDA: Data curation; Formal analysis. JMP: Data curation; Formal analysis; Investigation; Methodology. MMDC: Funding acquisition; Investigation; Resources; Supervision; Writing - review & editing. CRT: Conceptualization; Formal analysis; Funding acquisition; Investigation; Project administration; Resources; Supervision; Roles/Writing - original draft; Writing - review & editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
All protocols were approved by the Animal Ethics Committee of the University of São Paulo—Campus of Ribeirão Preto (#14.1.357.53.2). Animal experiments comply with the ARRIVE guidelines and were carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
Additional information
Handling Editor: Vincent FM Segers.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
do Vale, G.T., da Silva, C.B.P., Sousa, A.H. et al. Nebivolol Prevents Up-Regulation of Nox2/NADPH Oxidase and Lipoperoxidation in the Early Stages of Ethanol-Induced Cardiac Toxicity. Cardiovasc Toxicol 21, 224–235 (2021). https://doi.org/10.1007/s12012-020-09614-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-020-09614-1