
Abstract
Osteoporosis (OP) is a disease associated with bone loss and microstructure degradation. Recent studies have shown that iron accumulation may be a risk factor for OP. Bone marrow mesenchymal stem cells (MSCs) are multipotent cells and precursors to osteoblasts. MSCs play an important role in OP. Therefore, we evaluated the correlation between MSCs and OP in an environment of iron accumulation. Serum P1NP was decreased in iron accumulation mice. Micro-CT revealed that iron accumulation decreased bone mineral density and spatial structural parameters. Iron accumulation inhibited MSC quantity in bone marrow. However, the iron chelator deferoxamine (DFO) rescued the suppression. Iron accumulation also changed the MSC cell cycle. Iron elevated MSC cell ROS level and NOX4 protein expression. MSC apoptosis was increased, and more caspase3 was cleaved after iron intervention. Our data suggests that iron accumulation inhibits MSC quantity and induces MSC apoptosis. Bone loss from iron accumulation may correlate with the inhibition of MSCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoporosis is a disease characterized by trabecular structure degradation and brittle fractures [1].In 2006, Weinberg proposed iron loading as a risk factor for osteoporosis [2]. Clinical and animal research has demonstrated a close relationship between iron accumulation and bone loss. The association between iron accumulation and osteoporosis in patients with hereditary hemochromatosis has been observed in clinical, and the degree of bone loss was associated with the severity of iron overload [3]. Iron accumulation may cause osteoporosis in patients with thalassemia major, and iron chelators may abrogate the effects by reducing osteoclast activity [4]. The incidence of postmenopausal osteoporosis (type 1 osteoporosis), over the age of 50 years, is approximately 50% [5]. Iron accumulation occurs in postmenopausal women [6], and an intervention for iron accumulation may improve postmenopausal osteoporosis. Clinical studies have shown that iron accumulation may be a risk factor for postmenopausal osteoporosis [7,8,9,10]. However, the underlying mechanism of iron accumulation induced osteoporosis is still unknown. Tsay et al. demonstrated that iron overload in mice results in increased bone resorption and oxidative stress, leading to changes in bone microarchitecture and material properties and thus bone loss [11]. N-acetyl-L-cysteine, an antioxidant, prevented the effects of iron overload. The inhibition of osteoblast activity, mineralization, and specific gene expression may be attributed to the ferroxidase activity of ferritin [12].
Bone marrow mesenchymal stem cells (MSCs), also known as marrow stromal cells, are a type of mesodermal pluripotent stem cell [13] that can differentiate into osteoblasts, chondrocytes, fat cells, muscle cells, endothelial cells, and myocardial cells [14]. MSCs are progenitor cells of osteoblasts. Traditionally, researchers consider osteoporosis to be associated with aging MSCs in tissues. The decline in MSC population decreases bone formation, bone mass, and bone strength, destroying the bone’s microenvironment. Lower differentiation rates of MSCs results in low quality bone remodeling and reduces the replacement of aged or damaged bone tissue. In vitro, umbilical cord blood MSCs increased cell apoptosis and ROS levels and reduced colony formation after intervention with ferric ammonium citrate (FAC). The study found that iron accumulation produced ROS that damaged MSC proliferation and inhibited the ability of MSCs to support hematopoiesis [15]. Further study found that iron intervention in MSCs increased apoptosis and was accompanied by the upregulation of p-p38MAPK, p38MAPK, and p53 protein expression levels, revealing that the apoptosis mechanism may be related to stimulation of the p38MAPK-p53 pathway [16]. Iron accumulation suppressed MSCs; however, the iron chelator deferoxamine (DFO) inhibited the suppression [17]. Hepcidin, a hormone of iron metabolism in the body, has effects on the osteogenic differentiation of MSCs. Hepcidin stimulated the intracellular BMP2/Smad and MAPK/p38 pathway of MSCs to promote osteogenic differentiation [18]. MSCs play an important role in OP. Therefore, we evaluated the correlation between MSCs and OP in an environment of iron accumulation.
Materials and Methods
In Vivo Experiments
Animal Experiments
All animal experiments were approved by the Animal Care Committee of Soochow University. The mice were obtained by the Soochow University Experiment Animal Center and housed in the specific pathogen-free barrier system with normal diet and water in the animal facility of Soochow University. Eight-week-old C57 male mice (weight 25 ± 1 g) were randomly divided into a control group (Ctrl), an iron intervention group (FAC), and a DFO group (FAC + DFO). Each group included six mice. The FAC group was intraperitoneally injected with 0.1 g/kg of ferric ammonium citrate (FAC) for 8 weeks [11]. DFO was dissolved in normal saline to a concentration of 1%. The (FAC + DFO) group was intraperitoneally injected with 0.1 g/kg of FAC for 8 weeks, and 0.2 mg/kg of DFO was administered intraperitoneally from the fourth to eighth week [19]. The control group mice were injected with 0.1 g/kg of normal saline.
Serum Ferritin and Bone Turnover Detection
Blood was taken by orbital bleed and then centrifuged at 1000×g for 10 min to acquire the serum. Serum ferritin (ABCam) and serum P1NP (USCNlife) were detected by ELISA.
Histology
Tibiae were collected and fixed over night at 4 °C, decalcified in 14% EDTA for 1 week and paraffin embedded. Serial sections (5 μm) were deparaffinized and rehydrated to water for hematoxylin and eosin (H&E) staining.
Micro-CT Scan
Micro-CT (SkyScan 1176; Kontich, Belgium) scans were conducted for distal femoral three-dimensional reconstruction and parameter analysis. Femora were dissected after soft tissue was removed. Samples of Femora were stored in 4% paraformaldehyde. The details were as described previously [20, 21]. Parameter analysis included bone mineral density (BMD), bone volume/tissue volume (BV/TV), trabecular thickness (Tb. Th), and trabecular number (Tb. N).
MSC Proportion Detection
Mouse femora and tibiae were stripped, and bone marrow cells were washed with a PBS suspension. CD45 PerCP/Ter119 PE/Sca-1 APC/PDGFR FITC marked MSCs were the proportion of MSCs detected by flow cytometry (Aria III, BD Biosciences). Antibody staining and flow cytometric analysis were conducted as described previously [22].
Quantitative RT-PCR Analysis
Tibiae were dissected after soft tissue was removed. Total RNA was prepared from the femora by liquid nitrogen and mixed in 1 ml Trizol. Total RNA was reverse-transcribed into cDNA using a reverse transcription kit (Invitrogen) following the manufacturer’s instructions. The cDNA was mixed as described previously [23]. The following primer were used: runt-related transcription factor 2, Runx2; bone γ-carboxyglutamate protein, BGLAP. Each experiment was performed in duplicate and the results were standardized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer-probe sets used are presented in Table 1.
In Vitro Experiment
Cell Culture
Bone marrow MSCs were derived from C57 mice femoral bone marrow. The bone marrow was washed with PBS and centrifuged at 500×g for 5 min. The lower precipitation contained 10% FBS, 100 μg/mL penicillin, and 100 μg/mL α-MEM in 5% CO2 at 37 °C. The cells were divided into the control group and experimental group. The experimental group was treated with 100 μmol/L FAC for 24 h. The control group was treated with PBS for 24 h. The FAC + DFO group was treated with FAC for 24 h, and then treated with 10 μmol/L DFO for another 24 h [24].
Cell Cycle
Cells were washed in cold PBS with 70% alcohol twice at 4 °C overnight. Cells were incubated in 0.5 mL of 50 μg/mL propidium iodide (PI) solution containing 20 μg/mL RNase A in the dark. The cell suspension was for antibody staining, and the cells were detected by flow cytometry. Details were described previously [25].
Cell ROS
According to the manufacturer’s instruction steps, ROS was measured by incubating cells with the CellROX (Molecular the Probes) dye (10 μM) at 37 °C for 15 min. Flow cytometry was used to detect mitochondrial peroxide. Details were described previously [26].
Cell Apoptosis
MSCs were collected and washed with PBS. The cells were stained with PI and the Annexin-V kit (US Everbright Inc.), and cell apoptosis was measured by flow cytometry. Details were described previously [27].
Western Blot
MSCs were collected and washed with PBS. Protein levels were measured using the BCA protein assay. The proteins (30 μg/well) were separated by 12% SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidenedifiuoride (PVDF) membrane. The membrane was incubated in 5% skim milk for 1 h at room temperature with the primary antibody and at 4 °C overnight. The following antibodies were used: anti-NOX4 antibody (Santa-Cruz Biotechnology) and anti-caspase3(Cell Signaling Technology). The antibodies were applied to probe the membranes. After washing five times in TBST and incubating with HRP-conjugated secondary antibody (anti-GAPDH, Sigma-Aldrich Corp) diluted 1:2000 in TBST for 1 h, the reagent was developed using an ECL kit (Biological Industries). The results were analysis by ImageJ.
Statistical Analysis
The experimental data are presented as the mean ± s.d. Each group included six samples. The differences between each group were evaluated using one-way ANOVA followed by Bonferroni post-tests. The statistical data were analyzed by SPSS19.0 software. P < 0.05 was considered statistically significant.
Results
Iron Accumulation Leads to Bone Loss in the Animal Model
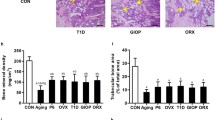
The iron accumulation model was verified by serum ferritin levels. The results of the ELISA analysis showed that the ferritin level of the FAC group was significantly higher than that of the control group (Fig. 1a). Serum P1NP is an indicator of osteogenic function. The results revealed that FAC inhibited serum P1NP levels compared with the control group (Fig. 1b). Animals in the FAC group exhibited normal body weight (Fig. 1c). H&E stain of bone tissue revealed that the trabeculae were destroyed in the FAC group (Fig. 1d). H&E staining revealed iron deposition in liver tissue (Fig. 1e). Runx2 and Bglap are osteogenesis genes. Quantitative real-time polymerase chain reaction (RT-PCR) indicated that Runx2 and Bglap expression were decreased (Fig. 1f). Micro-CT three-dimensional reconstruction showed that the structure of trabeculae was destroyed, bone trabeculae became spars, and continuity declined after 8 weeks of the FAC intervention (Fig. 2a). Micro-CT indicated that the bone volume fraction, number of trabeculae and trabecular thickness were decreased in the FAC group; however, trabecular spacing was increased (Fig. 2b–g). *P < 0.05.

Iron accumulation animal model. a Serum ferritin (FER) Elisa result in control (Ctrl) group, ferric ammonium citrate (FAC) group and DFO (FAC + DFO) group. Iron accumulation induced high level of FER. b Serum osteogenesis index P1NP Elisa result. Iron accumulation inhibited serum P1NP. c Body weight in both group. Both groups have normal body weight. d H&E stain in bone tissue. FAC group trabecular was destroyed. e H&E stain in liver tissue. Iron deposition was seen in FAC group. f Quantitative real-time polymerase chain reaction (RT-PCR) analysis of expression of osteogenesis gene Runx2 and Bglap. Iron accumulation inhibited Runx2 and Bglap mRNA expression. Each group included six mice. *P < 0.05

Iron accumulation causes low bone mass. DFO is iron chelator. DFO rescues bone loss induced by iron accumulation. a Representative miro-CT three-dimensional reconstruction of femur. The upper is cortical bone and the bottom is trabecula. Iron accumulation destroyed trabecula and thinned cortical bonel. b–g Micro-CT parameter analysis of femur. Micro-CT parameter of FAC group was inhibited. BMD, bone mineral density; BV/TV, bone volume per total tissue volume; Tb.Th, trabecular thickness; Tb.Sp, trabecular separation; Tb.N, trabecular number. Each group included six mice.*P < 0.05
FAC Inhibited MSCs In Vivo and In Vitro
In the animal model, MSCs were marked by CD45 PerCP/Ter119 PE/Sca-1 APC/PDGFR FITC in bone marrow cells. The MSC proportion in bone marrow cells was reduced in the FAC group (Fig. 3a, b). In vitro, MSCs were cultured and treated with FAC. We also tested the cell cycle. The cell cycle results revealed that FAC changed the MSC G0/G1 cell cycle ratio relative to the control group (Fig. 3c, d). *P < 0.05.

a, b MSC proportion of bone marrow cells was detected by flow cytometry. FAC inhibited MSC proportion. DFO rescued decreased MSC proportion induced by FAC.*P < 0.05. c MSC cell cycle was detected. M1 represents G0/G1. M2 represents S. M3 represents G2/M. FAC changes G0/G1. Each group included six samples.*P < 0.05
DFO Rescued the Suppression of MSCs Induced by FAC
Iron accumulation leads to bone loss and suppressed MSCs. The iron chelator, DFO, inhibited the suppression. Serum ELISA results revealed that DFO decreased iron accumulation and increased P1NP (Fig. 1a, b). The DFO and FAC group both exhibited normal body weight (Fig. 1c). Micro-CT revealed that DFO inhibited iron accumulation induced bone loss (Fig.2a, b–g). DFO also attenuated suppression of MSC numbers (Fig. 3a, b). *P < 0.05.
FAC Generated More ROS in MSCs
Iron accumulation may activate cellular ROS levels. To test whether FAC activates ROS in MSCs, we measured ROS levels in MSCs. The flow cytometry results revealed that FAC increased ROS levels in MSCs. The iron chelator, DFO, decreased ROS levels (Fig. 4a, b). NOX4 protein, which is crucial to ROS activity, was assessed. The western blot results showed that FAC increased the level of NOX4 protein, which corresponds to increased ROS. DFO decreased NOX4 protein expression (Fig. 4c). *P < 0.05.

a, b MSC ROS level was measured by flow cytometry. *P < 0.05. Iron accumulation elevated ROS level in MSCs. Iron chelator, DFO, decreased ROS level to normal. c NOX4 is a basic activator of ROS. NOX4 protein expression was detected in MSCs in iron accumulation environment. Iron accumulation increased NOX4 protein expression. DFO attenuated this effect. Each group included six samples. *P < 0.05
FAC Induced MSCs Apoptosis
The proportion of MSCs among bone marrow cells was reduced by the FAC intervention. MSC apoptosis was tested in vitro. FAC induced MSC apoptosis. Treatment with the DFO decreased cell apoptosis (Fig. 5a, b). Subsequently, we evaluated the levels of caspase3 protein, which is involved in the classical apoptotic pathway after being cleaved. The results indicated that FAC promoted cleavage of caspase3. DFO inhibited this trend (Fig. 5c). *P < 0.05.

a, b MSC apoptosis is tested by flow cytometry. Iron accumulation induced MSC cell apoptosis. DFO rescued the trend. *P < 0.05. c Caspase3 is involved in classical apoptotic pathways after cleavage. Cleaved caspase3 protein expression was detected. Iron accumulation increased cleaved caspase3 in MSCs. Each group included six samples. *P < 0.05
Discussion
Our results showed that iron intervention resulted in higher serum ferritin levels, which illustrated that we successfully established an iron accumulation animal model. Micro-CT three-dimensional reconstruction of mouse distal femur revealed structural changes of trabecular thinning and bone trabecular space reduction in response to the iron intervention, suggesting that iron accumulation is associated with decreased bone mass. Our research determined that iron accumulation destroyed trabecular and cortical bone and confirmed the finding that iron overload mice exhibited cortical thinning of bone. Hepcidin is the key regulator of iron homeostasis. Hepcidin1 gene knockout mice exhibit iron accumulation. Lower cortical bone thickness was observed in Hepcidin1 gene knockout mice [28].Tsay reported that iron accumulation had a negative effect on bone volume in this animal model, which may be related to the accumulation of iron leading to elevated ROS [11]. Changes included trabecular and cortical bone thinning, which were associated with increased resorption. In addition, iron accumulation facilitated osteoclast differentiation and bone resorption through the production of ROS [29].
Bone mass loss may be associated with inhibited function in osteogenesis. MSCs are capable of differentiating into precursor osteoblasts cells and eventually differentiate into mature osteoblasts [30]. Reductions in serum P1NP, Runx2 and BGLAP suggested that osteoblasts were reduced, which results from a decreased number of precursor osteoblasts and upstream MSCs. The proportion of CD45-Ter119-Sca-1+PDGFR+ marked MSC bone marrow cells [22] were reduced in the iron accumulation model. Previous studies revealed that iron accumulation was antagonistic towards MSCs; Lu found that iron accumulation produced ROS, which inhibited MSC proliferation [15]. The results indicated that iron accumulation inhibited MSCs, which are the precursor cells of osteoblasts, leading to a better understanding of osteoporosis risk.
Previous studies have suggested that iron accumulation may augment ROS levels, leading to diseases such as osteoporosis [31]. ROS production is stimulated by changes in cell metabolism and the environment such as altered mitochondrial respiration, damaged DNA, and the availability of lipids and proteins. ROS production is catalyzed by the Fenton reaction, and our previous research showed that iron accumulation inhibited osteoblast differentiation and mineralization by producing ROS [32]. ROS affects osteogenic transcriptional proteins, including Runx2, Wnt-βcatenin, and ERK [33,34,35]. In addition, iron influences osteoclast differentiation and bone absorption by producing ROS [11]. The experimental FAC intervention in vitro revealed that iron accumulation increased MSC ROS levels, suggesting that iron uses ROS to suppress MSCs.
The NOX family has seven subtypes and uses the pyridine nucleotide NADPH as an electron donor and molecular oxygen as an electron acceptor; this family causes a catalytic decline in subprime peroxide and generates ROS [36]. NOX subtypes may affect MSC differentiation. ROS and bone disease are induced by NOX, and NOX4 is a basic activator of ROS. NOX4 special nucleotide polymorphisms result in a higher bone turnover index, and decreased BMD was observed in NOX4 knockout mice; NOX4 expression is thought to reduce BMD [37]. High expression of NOX4 can promote the differentiation of MSCs into adipocytes [38,39,40]. Our experimental western blot results indicated that iron intervention increased NOX4 expression.
This study found that MSC apoptosis was induced by increasing iron accumulation, resulting in a decline in cell number. Apoptosis is regulated, programmed cell death that includes a variety of biological and morphological changes and pathways. Proteolytic enzymes play an important role in signaling pathways in apoptosis; the caspase (cysteinyl aspartate-specific proteases) family plays a key role in the apoptosis process [41]. Caspase proteolysis augments and integrates apoptotic pathways [42]. Caspase3 is an important protein in classic apoptosis that is activated by the intrinsic (mitochondrial) and extrinsic (death ligand) apoptotic pathways [43, 44]. The caspase3 proenzyme itself is not active; the inactive proenzyme of caspase3 needs to be activated by protease p17 or p12. Caspase3 is involved in classical apoptotic pathways after cleavage [45, 46]. To detect the possible mechanism underlying MSC apoptosis, our experiment examined caspase3 expression. Our result revealed that cleaved caspase3 expression increased, suggesting that apoptosis in MSCs was induced by iron accumulation. The cell apoptosis mechanism may be related to caspase3 activation. In addition, caspase3 is also found in other cells with FAC-induced apoptosis.
In summary, the functional decline in osteogenesis from iron accumulation is correlated with MSC inhibition. This mechanism may be related to iron induced NOX4 expression and ROS activation. Our research leads to a better understanding of osteoporosis risk and serves as a potential target for clinical therapeutics.
References
Moriwaki S, Suzuki K, Muramatsu M, Nomura A, Inoue F, Into T, Yoshiko Y, Niida S (2014) Delphinidin, one of the major anthocyanidins, prevents bone loss through the inhibition of excessive osteoclastogenesis in osteoporosis model mice. PLoS One 9:e97177. https://doi.org/10.1371/journal.pone.0097177
Weinberg ED (2006) Iron loading: a risk factor for osteoporosis. Biometals 19:633–635. https://doi.org/10.1007/s10534-006-9000-8
Valenti L, Varenna M, Fracanzani AL, Rossi V, Fargion S, Sinigaglia L (2009) Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos Int 20:549–555. https://doi.org/10.1007/s00198-008-0701-4
Rossi F, Perrotta S, Bellini G, Luongo L, Tortora C, Siniscalco D, Francese M, Torella M, Nobili B, Di Marzo V, Maione S (2014) Iron overload causes osteoporosis in thalassemia major patients through interaction with transient receptor potential vanilloid type 1 (TRPV1) channels. Haematologica 99:1876–1884. https://doi.org/10.3324/haematol.2014.104463
Kanis JA, McCloskey EV, Johansson H, Cooper C, Rizzoli R, Reginster JY (2013) European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int 24:23–57. https://doi.org/10.1007/s00198-012-2074-y
Kim BJ, Ahn SH, Bae SJ, Kim EH, Lee SH, Kim HK, Choe JW, Koh JM, Kim GS (2012) Iron overload accelerates bone loss in healthy postmenopausal women and middle-aged men: a 3-year retrospective longitudinal study. J Bone Miner Res 27:2279–2290. https://doi.org/10.1002/jbmr.1692
Jian J, Pelle E, Huang X (2009) Iron and menopause: does increased iron affect the health of postmenopausal women? Antioxid Redox Signal 11:2939–2943. https://doi.org/10.1089/ars.2009.2576
Huang X, Xu Y, Partridge NC (2013) Dancing with sex hormones, could iron contribute to the gender difference in osteoporosis? Bone 55:458–460. https://doi.org/10.1016/j.bone.2013.03.008
Li GF, Pan YZ, Sirois P, Li K, Xu YJ (2012) Iron homeostasis in osteoporosis and its clinical implications. Osteoporos Int 23:2403–2408. https://doi.org/10.1007/s00198-012-1982-1
Weinberg ED (2008) Role of iron in osteoporosis. Pediatr Endocrinol Rev 6(Suppl 1):81–85. https://doi.org/10.1007/s00198-012-1982-1
Tsay J, Yang Z, Ross FP, Cunningham-Rundles S, Lin H, Coleman R, Mayer-Kuckuk P, Doty SB, Grady RW, Giardina PJ, Boskey AL, Vogiatzi MG (2010) Bone loss caused by iron overload in a murine model: importance of oxidative stress. Blood 116:2582–2589. https://doi.org/10.1182/blood-2009-12-260083
Zarjou A, Jeney V, Arosio P, Poli M, Zavaczki E, Balla G, Balla J (2010) Ferritin ferroxidase activity: a potent inhibitor of osteogenesis. J Bone Miner Res 25:164–172. https://doi.org/10.1359/jbmr.091002
Jackson WM, Nesti LJ, Tuan RS (2012) Concise review: clinical translation of wound healing therapies based on mesenchymal stem cells. Stem Cells Transl Med 1:44–50. https://doi.org/10.5966/sctm.2011-0024
Chamberlain G, Fox J, Ashton B, Middleton J (2007) Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells 25:2739–2749. https://doi.org/10.1634/stemcells.2007-0197
Lu WY, Zhao MF, Chai X, Meng JX, Zhao N, Rajbhandary S, Xu XN, Ma L, Li YM (2013) Reactive oxygen species mediate the injury and deficient hematopoietic supportive capacity of umbilical cord derived mesenchymal stem cells induced by iron overload. Zhonghua Yi Xue Za Zhi 93:930–934
Lu WY, Zhao MF, Sajin R, Zhao N, Xie F, Xiao X, Mu J, Li YM (2013) Effect and mechanism of iron-catalyzed oxidative stress on mesenchymal stem cells. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 35:6–12. https://doi.org/10.3881/j.issn.1000-503X.2013.01.002
Lu W, Zhao M, Rajbhandary S, Xie F, Chai X, Mu J, Meng J, Liu Y, Jiang Y, Xu X, Meng A (2013) Free iron catalyzes oxidative damage to hematopoietic cells/mesenchymal stem cells in vitro and suppresses hematopoiesis in iron overload patients. Eur J Haematol 91:249–261. https://doi.org/10.1111/ejh.12159
Lu H, Lian L, Shi D, Zhao H, Dai Y (2015) Hepcidin promotes osteogenic differentiation through the bone morphogenetic protein 2/small mothers against decapentaplegic and mitogen-activated protein kinase/P38 signaling pathways in mesenchymal stem cells. Mol Med Rep 11:143–150. https://doi.org/10.3892/mmr.2014.2769
Link G, Ponka P, Konijn AM, Breuer W, Cabantchik ZI, Hershko C (2003) Effects of combined chelation treatment with pyridoxal isonicotinoyl hydrazone analogs and deferoxamine in hypertransfused rats and in iron-loaded rat heart cells. Blood 101:4172–4179. https://doi.org/10.1182/blood-2002-08-2382
Wang L, Zhou F, Zhang P, Wang H, Qu Z, Jia P, Yao Z, Shen G, Li G, Zhao G, Li J, Mao Y, Xie Z, Xu W, Xu Y, Xu Y (2017) Human type H vessels are a sensitive biomarker of bone mass. Cell Death Dis 8:e2760. https://doi.org/10.1038/cddis.2017.36
Wang L, Jia P, Shan Y, Hao Y, Wang X, Jiang Y, Yuan Y, Du Q, Zhang H, Yang F, Zhang W, Sheng M, Xu Y (2017) Synergistic protection of bone vasculature and bone mass by desferrioxamine in osteoporotic mice. Mol Med Rep 16:6642–6649. https://doi.org/10.3892/mmr.2017.7451
Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, Suzuki S, Miyauchi-Hara C, Nagoshi N, Sunabori T, Shimmura S, Miyawaki A, Nakagawa T, Suda T, Okano H, Matsuzaki Y (2009) Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med 206:2483–2496. https://doi.org/10.1084/jem.20091046
Xiao W, Beibei F, Guangsi S, Yu J, Wen Z, Xi H, Youjia X (2015) Iron overload increases osteoclastogenesis and aggravates the effects of ovariectomy on bone mass. J Endocrinol 226:121–134. https://doi.org/10.1530/JOE-14-0657
Zhao GY, Zhao LP, He YF, Li GF, Gao C, Li K, Xu YJ (2012) A comparison of the biological activities of human osteoblast hFOB1.19 between iron excess and iron deficiency. Biol Trace Elem Res 150:487–495. https://doi.org/10.1007/s12011-012-9511-9
Cao Y, Zhang A, Cai J, Yuan N, Lin W, Liu S, Xu F, Song L, Li X, Fang Y, Wang Z, Wang Z, Wang J, Zhang H, Zhao W, Hu S, Zhang S, Wang J (2015) Autophagy regulates the cell cycle of murine HSPCs in a nutrient-dependent manner. Exp Hematol 43:229–242. https://doi.org/10.1016/j.exphem.2014.11.002
Cao Y, Fang Y, Cai J, Li X, Xu F, Yuan N, Zhang S, Wang J (2016) ROS functions as an upstream trigger for autophagy to drive hematopoietic stem cell differentiation. Hematology 21:613–618. https://doi.org/10.1080/10245332.2016.1165446
Yuan N, Song L, Zhang S, Lin W, Cao Y, Xu F, Fang Y, Wang Z, Zhang H, Li X, Wang Z, Cai J, Wang J, Zhang Y, Mao X, Zhao W, Hu S, Chen S, Wang J (2015) Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica 100:345–356. https://doi.org/10.3324/haematol.2014.113324
Shen GS, Yang Q, Jian JL, Zhao GY, Liu LL, Wang X, Zhang W, Huang X, Xu YJ (2014) Hepcidin1 knockout mice display defects in bone microarchitecture and changes of bone formation markers. Calcif Tissue Int 94:632–639. https://doi.org/10.1007/s00223-014-9845-8
Jia P, Xu YJ, Zhang ZL, Li K, Li B, Zhang W, Yang H (2012) Ferric ion could facilitate osteoclast differentiation and bone resorption through the production of reactive oxygen species. J Orthop Res 30:1843–1852. https://doi.org/10.1002/jor.22133
Neve A, Corrado A, Cantatore FP (2011) Osteoblast physiology in normal and pathological conditions. Cell Tissue Res 343:289–302. https://doi.org/10.1007/s00441-010-1086-1
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247. https://doi.org/10.1038/35041687
He YF, Ma Y, Gao C, Zhao GY, Zhang LL, Li GF, Pan YZ, Li K, Xu YJ (2013) Iron overload inhibits osteoblast biological activity through oxidative stress. Biol Trace Elem Res 152:292–296. https://doi.org/10.1007/s12011-013-9605-z
Hinoi E, Fujimori S, Wang L, Hojo H, Uno K, Yoneda Y (2006) Nrf2 negatively regulates osteoblast differentiation via interfering with Runx2-dependent transcriptional activation. J Biol Chem 281:18015–18024. https://doi.org/10.1074/jbc.M600603200
Bai XC, Lu D, Bai J, Zheng H, Ke ZY, Li XM, Luo SQ (2004) Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem Biophys Res Commun 314:197–207
Almeida M, Han L, Martin-Millan M, O'Brien CA, Manolagas SC (2007) Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem 282:27298–27305. https://doi.org/10.1074/jbc.M702811200
Atashi F, Modarressi A, Pepper MS (2015) The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review. Stem Cells Dev 24:1150–1163. https://doi.org/10.1089/scd.2014.0484
Goettsch C, Babelova A, Trummer O, Erben RG, Rauner M, Rammelt S, Weissmann N, Weinberger V, Benkhoff S, Kampschulte M, Obermayer-Piesch B, Hofbauer LC, Brandes RP, SchroderK (2013) NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J Clin Invest 123:4731–4738. https://doi.org/10.1172/JCI67603
Schroder K, Wandzioch K, Helmcke I, Brandes RP (2009) Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol 29:239–245. https://doi.org/10.1161/ATVBAHA.108.174219
Kanda Y, Hinata T, Kang SW, Watanabe Y (2011) Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sci 89:250–258. https://doi.org/10.1016/j.lfs.2011.06.007
Mouche S, Mkaddem SB, Wang W, Katic M, Tseng YH, Carnesecchi S, Steger K, Foti M, Meier CA, Muzzin P, Kahn CR, Ogier-Denis E, Szanto I (2007) Reduced expression of the NADPH oxidase NOX4 is a hallmark of adipocyte differentiation. Biochim Biophys Acta 1773:1015–1027. https://doi.org/10.1016/j.bbamcr.2007.03.003
Della-Fera MA, Li C, Baile CA (2003) Resistance to IP leptin-induced adipose apoptosis caused by high-fat diet in mice. Biochem Biophys Res Commun 303:1053–1057
Rahmani M, Davis EM, Bauer C, Dent P, Grant S (2005) Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem 280:35217–35227. https://doi.org/10.1074/jbc.M506551200
Salvesen GS (2002) Caspases: opening the boxes and interpreting the arrows. Cell Death Differ 9:3–5. https://doi.org/10.1038/sj.cdd.4400963
Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, Los M (2009) Apoptosis and cancer: mutations within caspase genes. J Med Genet 46:497–510. https://doi.org/10.1136/jmg.2009.066944
Walters J, Pop C, Scott FL, Drag M, Swartz P, Mattos C, Salvesen GS, Clark AC (2009) A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem J 424:335–345. https://doi.org/10.1042/BJ20090825
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376:37–43. https://doi.org/10.1038/376037a0
Funding
This work was supported by grants from the Natural Science Foundation of China (81572179 and 81773439), the Clinical Medicine Technology Project of Jiangsu Province (BL2014044), the Minsheng Science and Technology Project of Suzhou City (SS201634), Clinical Medical Center Project of Suzhou City (SZZX201504), Advantage Discipline Groups of the Second Affiliated Hospital of Soochow University (XKQ2015001), the Second Affiliate Hospital of Soochow University Pre-research Foundation (SDFEYQN1608), Jiangsu Province’s Young Medical Talents Program (QNRC2016880), and Soochow University Youth Teacher Science Fund Project (SDY2011A28).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yuan, Y., Xu, F., Cao, Y. et al. Iron Accumulation Leads to Bone Loss by Inducing Mesenchymal Stem Cell Apoptosis Through the Activation of Caspase3. Biol Trace Elem Res 187, 434–441 (2019). https://doi.org/10.1007/s12011-018-1388-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-018-1388-9