
Abstract
Microbial genomics facilitates the analysis of microbial attributes, which can be applied in bioremediation of pollutants and oil recovery process. The biosurfactants derived from microbes can replace the chemical surfactants, which are ecologically detrimental. The aim of this work was to study the genetic organization responsible for biodegradation of aromatics and biosurfactant production in potential microbes isolated from polluted soil. Bacterial isolates were tested for biosurfactant production, wherein Bacillus sp. AKBS9 and Acinetobacter sp. AKBS16 exhibited highest biosurfactant production potential. Whole genome sequencing and annotations revealed the occurrence of sfp and NPRS gene in the Bacillibactin biosynthetic gene cluster in AKBS9 strain and emulsan biosynthetic gene cluster in AKBS16 strain for biosurfactant production. Various aromatic compound ring cleaving oxygenases scavenging organic molecules could be annotated for strain AKBS16 using RAST annotations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Surfactants are amphiphillic organic compounds that reduce surface and interfacial tensions by accumulating at the interface between two immiscible fluids like oil and water. The surface active property makes surfactants one of the most important and versatile class of product used in a variety of applications such as in household, industry, and agriculture [1]. Two classes of surfactants are available, which can be categorized as chemical surfactants and biosurfactants based on their synthesis process. The chemically synthesized surfactants are petroleum based and poorly biodegradable since they have a high degree of branching. Biosurfactants are surface active molecules secreted by microbes mostly found in aerobic environments. They have a significant role in bioremediation owing to their feasible application in in situ scenarios, low critical micelle amount, low environmental burden, enhanced biodegradability, and effective solubilization [2]. The first ever biosurfactant-producing bacteria were characterized in 1960s, which proved to be the ecological and biogeochemically important drivers of bioremediation. Current research focuses on biosurfactant production, since they increase the bioavailability of xenobiotics, making them more amenable to biodegradation [3, 4]. Biosurfactants exhibit excellent emulsifying properties that aid in effectively removing crude oil and also facilitate oil recovery process. Owing to broad range of applications, several biosurfactants have been commercialized, petroferm being the very first commercial industrial biosurfactant in the market was used for microbe-assisted oil recovery in 1987.
Diverse biosurfactants molecules are produced by different microorganisms, which are classified mainly by their chemical structure and microbial origin. Rhamnolipids from Pseudomonas aeruginosa, surfactin from Bacillus subtilis, emulsan from Acinetobacter calcoacetius, and sophorolipid from Candida bombicola are a few examples of microbe-derived surfactants [5].
Since the structure of DNA was discovered, continuous efforts have been made in understanding the genomic complexity and diversity using genetic material of organisms. The genomic and molecular approaches have been extensively used to understand environmental microbes and their metabolic functions [6, 7]. Molecular tools such as RAPD, 16S gene identification, PCRs, etc. have often been employed to identify and analyze microbial potentials and further derive industrial applications, thus help in improving the process economics [8,9,10]. Present day next generation sequencing techniques have further contributed to the understanding of microbial functions by serving as a cost-effective technique to understand complete genomes of organisms.
Present work aims at analyzing the genes/pathways responsible for biosurfactant production and biodegradation of aromatic molecules in two bacterial isolates obtained from contaminated soil. Petroleum-polluted soil samples were used to setup enrichments and bacterial isolation was performed. The isolated cultures were screened for their ability to produce biosurfactants by performing various confirmatory tests. The potent isolates, namely Bacillus sp. AKBS9 and Acinetobacter sp. AKBS16, were analyzed to study genes responsible for biosurfactant production and catabolic genes, respectively, by draft genome analysis.
Materials and Methods
Chemicals
Luria agar, Luria broth, and nutrient agar were purchased from HiMedia (India). Castor oil was purchased from Raj Chemicals (India). SDS, CTAB, and methylene blue were purchased from Sisco Research Lab (India). Mineral oil was purchased from Affymetrix, USB Chemicals (India) and Triton X-100 was purchased Sigma-Aldrich (USA). The human blood used in experimental procedures was purchased from VSPM’s hospital blood bank, Maharashtra, India.
Isolation of Biosurfactant Producing Microbes
We performed sampling at the petrol pump situated at our institute, CSIR-NEERI, Maharashtra, India. Five grams of soil was collected from distinct locations (sub-sampling) and was mixed to obtain a composite sample. Enrichments were set up with the sampled petroleum-contaminated soil using two different carbon sources, sugar molasses and canola oil in M9 media [7]. Incubation was carried out at 120 rpm at 30 °C for 4 weeks. At the interval of every week, the enrichment culture were transferred to fresh minimal media. After four weekly transfers, the enrichment was serially diluted and 10−4, 10−5, 10−6 and plated on minimal media agar plates containing 1 ml of crude oil per 100 ml media. Purified bacterial strains were obtained by repetitive streaking on M9 agar media plates with crude oil as a carbon source and further screened for its biosurfactant-producing ability. The isolates Bacillus sp. AKBS9 and Acinetobacter sp. AKBS16 were found positive for biosurfactant production based on hemolysis assay [11], CTAB agar plate method [12], and oil displacement assay [13] and hence were selected for genomic analysis to understand the genes responsible for biosurfactant production and their bioremediation potential.
Genomic Sequencing Assembly and Analysis
Genomic DNA was isolated using the CTAB and phenol-choloroform method. About 300 ng of isolated DNA was resolved on 0.8% agarose gel at 120 V for 60 min and the samples were checked by using Nanodrop for determining A260/280 ratios. The paired-end libraries were prepared using Illumina TruSeq Nano DNA Prep kit. The mean of the libraries fragment sizes ranged from 419 to 644 bp for both the draft genomes. The libraries were sequenced on NextSeq 500 using 2 × 150 bp chemistry. The sequenced raw data was processed to obtain high-quality reads using Trimmomatic v0.35 to remove adapter sequences, ambiguous reads, and low-quality reads (reads with more than 10% quality threshold (QV < 20 phred score). The overall steps involved in the analysis of AKBS9 and AKBS16 draft genomes have been depicted in Supplementary Fig. 1.
The de novo assembly and scaffolding steps were carried out for both the AKBS9 and AKBS16 genomes. The high-quality reads were assembled into scaffold using Velvet Optimiser (v.2.2.5), which works on the velvet algorithm for estimating insert lengths for paired-end libraries and optimizes assembly based on default optimization conditions. Genes were predicted from the assembled scaffolds using the Prodigal with the default parameters, i.e., Translational code: Standard bacteria, N’s to be regarded as masked RNA genes: no, allow edges to run off edges: yes. Further functional annotation of genes was performed using BLASTx program, which is a part of NCBI-Blast 2.3.0+ standalone tool, which searches the genes against the non-redundant protein database. Genome annotations were also derived using Rapid Annotation using Subsystem Technology (RAST) [14].
To visualize sequence similarity, we used Circoletto for both the draft genome datasets [15]. Here, the scaffolds were placed around a circle, in clockwise direction and the ideograms were placed in order to untaggle the ribbons maximally. The ribbons symbolized the local alignments created by BLAST, their width represents alignment length, and different colors code the alignment bitscore in four quartiles: worst score by blue for the first 25% of the maximum bitscore, green for the next 25%, orange for the third, and finally red for the top (i.e., best) bitscores of between 75 and 100% of the maximum bitscore. Black ribbons represented the best scoring local alignment for the corresponding query, whereas any twisted ribbon meant that the local alignment was inverted. The stacked histograms on top of the ideograms represented the frequency (simple count) and score (by aforementioned color) of the ribbons.
Gene ontology (GO)-based annotations of the genes were determined by using the Blast2GO platform. These results were used to study the functions of the predicted genes. The gene ontology mapping classifies the genes into three main domains: biological process, genes involved in molecular functions, and cellular components. The genes involved in biosurfactant production and responsible for aromatic compound degradation were mined using RAST v4.0 [14] and NCBI Prokaryotic Genomes Automatic Annotation Pipeline (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html) [16] The draft genome assemblies were analyzed for gene clusters involved in biosurfactant production using antiSMASH 3.0 at default parameters [17].
Nucleotide Sequence Submission
The whole genome shotgun sequence of Bacillus sp. AKBS9 and Acinetobacter sp. AKBS16 was deposited in GenBank under the accession numbers POYG00000000 and POYH00000000 version POYG00000000.1 and POYH00000000.1, respectively.
Quantification of Biosurfactant Production
Emulsification Assay
The cell-free supernatant broth was checked for biosurfactant production by testing its ability to emulsify with oil [12]. A milliliter of cell-free broth from overnight grown culture was added to 5 ml of 50 mM Tris buffer (pH 8.0) in a 50 ml of falcon tube. Five microliters of castor oil was added to cell-free broth and vortexed on a vortex mixer for 1 min. The emulsion mixture was allowed to remain upright for 20 min. The absorbance of the aqueous phase was measured by a spectrophotometer (Shimadzu, Japan) at a wavelength of 400 nm. Emulsification activity per milliliter was calculated by using following formula:
A negative control was maintained using buffer solution, crude oil, and a positive control containing Triton X-100 was used.
Oil Displacement Assay
Micelles formation is a unique quality of biosurfactant-producing bacteria which is often confirmed through oil displacement assays. This assay was performed following steps reported earlier [13]. Bacterial extracts were tested for their efficiency to displace oil from a water layer. Water-filled petri plates were subjected to 30 μl of sterile (filtered, 0.45 μm) mineral oil using a micropipette. Fifteen microliters of bacterial extract was spotted in the center of this oil surface. The clearing zone which appeared around the spotting site was measured. This clear zone diameter on the oil surface serves as a measure of surfactant activity.
Results and Discussion
Studies on molecular mechanisms responsible for biosurfactant production in microbes are of great significance as it assists in designing bioremediation and oil recovery process. A lack of sufficient genomic understanding has led us to an investigation of the biosurfactant production and bioremediation of aromatics by Bacillus sp. AKBS9 and Acinetobacter sp. AKBS16, respectively. The biosurfactants derived from microbes can replace the chemical surfactants, which are ecologically detrimental and because of their properties as eco-friendly, least toxicity, biodegradability, and high specificity [18]. Here, we describe the molecular mechanisms of both these microbes based on high-throughput omics and in silico analysis.
Biosurfactant Production
The oil displacement assay works on the principle that oil displaced is directly related to the amount of biosurfactant produced. The microbial extracts were found to displace 90% of the oil layer; moreover, both the strains exhibited β-hemolytic activity, indicated by a clear zone of 1.9 cm on blood agar plate, which confirms biosurfactant production of strain AM13. The emulsification assay depicted 90 and 69% for AKBS9 and AKBS16, respectively, emulsifying ability with respect to the positive control as seen in Supplementary Table 1. Based on these results, we selected the isolates AKBS9 and AKBS16 for genomic analysis.
Genome Features
Whole genome sequencing was performed for the AKBS9 and AKBS16 strains using NextSeq 500 with the 2*150 bp chemistry. The high quality of data obtained was 1.33 Gb with 4,458,219 reads for AKBS9 and 1.17 Gb with 3,973,627 for AKBS16 which were used for downstream analysis. High-quality reads were assembled using Velvet optimiser, as a result of which 80 scaffolds were generated with N50 value of 207,077 bp for AKBS9 and 111 scaffolds with N50 value of 75,000 bp for AKBS16. Detail genome sequence data statistics are given in Table 1. The gene ontology mapping could quantitatively categorize all genes into three groups, namely biological process, molecular function, and cellular components as seen in Supplementary Fig. 2a and b. About 10% genes belonged to each category like cell, cell part, binding, catalytic activity, and metabolic functions in both genomes. To investigate the long-range synteny across our strains and their respective closest reference genomes, we used the contiguator-assembled scaffold of our strain and the reference genome to perform alignment. The parameters used to generate Circos plot were set to show best hit per query and color ribbons by Blast scores to generate the plot. The Circos plot given in Supplementary Fig. 3a and b represents the strains of our study with respect to their closest reference genome available in NCBI database. The closest match to AKBA16 was Acinetobacter pittii PHEA-2 (g-proteobacteria), assembly number ASM19114V1 and that of AKBS9 was Bacillus cereus NCBI assembly ASM211746v1. The figure shows the synteny and clear representation of inter-genic relationships across the strains and their closest reference.
Gene Arrangement of the Features Involved in Biosurfactant Production in AKBS9 and AKBS16
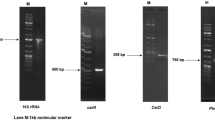
Bacillus genus is often reported for its ability to produce biosurfactant such as surfactin (lipopeptides). This attribute of Bacillus sp. is of prime importance as surfactin is the most potential type of lipoprotein exhibiting highest surface tension-reducing ability [5]. The sfp gene is responsible for activation of the seven peptidyl protein domains of surfactin synthase, i.e., SRF1,2,3. It functions by transferring the 4′-phosphopantetheinyl moiety of coenzyme A (CoA) to a serine residue. Hence, it is requisite for microbial cells of B. subtilis to produce lipopeptide antibiotics like surfactin and plipastatin B1. The genome analysis of AKBS9 revealed the presence of 750 bp long sfp gene on scaffold 64 as seen in Fig. 1a. Upstream to this gene, we could locate a nonribosomal peptide synthetase (NRPS) gene and the RAST annotations revealed its role as a Bacillibactin synthetase component F (EC 2.7.7.-). The NRPS gene cluster codes for diverse proteins having biological roles or pharmacological properties. They often function toward production of secondary metabolites, pigments, or toxins. The NRPS gene in AKBS9 was 7158-bp long and located near the sfp gene as seen in Fig. 1a. The gene arrangement of strain AKBS9 is seen in Fig. 1a, with respect to the closest matches available for sfp gene in RAST subsystem database, namely B. anthracis, Oceanobacillus iheyensis, and Streptomyces griseus. The analysis of scaffold 64 using antiSMASH revealed the presence of Bacillibactin biosynthetic gene cluster with 46% of genes showing similarity to similar cluster from Bacillus subtilis and Paenibacillus elgii to antiSMASH id BGC0000309 and BGC0000401, respectively (Fig. 1b). Another recent study reports the main role of a nonribosomal peptide synthetase (NRPS) cluster in production of surfactin in a strain AM13 through similar genomic analysis [19]. According to previous report, an environmental strain, Bacillus amyloliquefaciens FZB42, was sequenced and the genome analysis identified fen and srf operons responsible for the production of the antibiotic lipopeptide products like surfactin and fengycin in this strain [20].

a Gene organization of sfp gene in AKBS9. 1: 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase, 2:isochorismate synthase, 3: 2,3 dihydroxybenzoate-AMP ligase, 4: isochorismatase, 5: siderophore biosynthesis nrps module, 6:polymyxin synthetase PmxB, 7: polymixin synthetase PmxC, 8: 4-phosphopantetheinyl transferase, 9: uncharacterized protein in polymixin biosynthetic cluster, 10: DNA binding protein HBsu. b antiSMASH cluster analysis for NRPS gene. The figure represents the presence of biosurfactant (nonribosomal peptide synthetase) synthesizing genecluster in the draft genome of AKBS9. The scaffold 64 of AKBS9 draft genome is annotated by antiSMASH to its closest match Bacillus subtilis (BGC0000309) and Paenibacillus elgii (BGC0000401). The genes annotated were numerically represented as 1: 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase, 2:isochorismate synthase, 3: 2,3-dihydroxybenzoate-AMP ligase, 4: isochorismatase, 5: Nonribosomal peptide synthatase (NRPS). c antiSMASH cluster analysis for emulsan biosynthetic cluster in AKBS16. The genes numerically represented as 1: protein tyrosine kinase, 2: protein tyrosine phosphatise, 3: outer membrane protein, 4: USP N-acetylglucoasmine 2-epimerase, 5: NDP-N-acetyl-d-galactosaminouronic acid dehydrogenase, 6: galactoside acetyltransferase, 7: emulsan repeating unit flippase, 8: emulsan repeating unit polymerase, 9: glycosyl transferase, 10: unknown gene, 11: unknown gene, 12: gylcosyl transferase, 13: galactose phosphate transferase 14: acetyltransferase, 15:amino transferase, 16: dTDP-glucose-4,6 dehydratase, 17: UTP-glucose-1-phosphate-uridylyltransferase, 18: glucose dehydrogenase, 19: phosphoglucose isomerise, 20: UDP-glucose-4-epimerase
Acinetobacter sp. has been known to yield high molecular weight biosurfactants such as emulsan and alasan [21]. The strain AKBS16 was identified as Acinetobacter sp. and its draft genome analysis revealed the presence of emulsan biosynthetic gene cluster in scaffold 18 as seen in Fig. 1c. The gene cluster analysis revealed the homologies of emulsan biosynthetic gene cluster to its only available match, i.e., Acinetobacter lwoffii. The next homologies to capsular polysaccharide biosynthetic gene cluster of Mannheimia haemolytica (BGC0000736) were very poor. Genes such as emulsan-repeating unit flippase and emulsan-repeating unit polymerase could be annotated in the scaffold. There is inadequate information about the genes/pathways responsible for biosurfactant production in Acinteobacter genus. The emulsan biosynthetic cluster in our draft genome AKBS16 could be identified owing to its homologies to the wee gene operon coding for the biosynthesis of the bioemulsifier in the oil-degrading strain Acinetobacter lwoffii RAG-1 [22].
Genes Involved in Multiple Aromatic Compounds Degradation in Strain AKBS16
The in silico analysis of the AKBS16 genome revealed the genes coding for enzymes involved in the metabolic pathways for degradation of multiple aromatic compounds and their arrangement on the genome. As per SEED subsystem, about 146 genes coded for various degradation pathways for aromatic compounds, namely biphenyl, phenol, benzoate, chloroaromatics, gentisate, salicylate, hydroxybenzoate, etc. Oxygenase genes, responsible for the ring cleaving of aromatic compounds, were mined in the genome of AKBS16. A total of 56 different oxygenase coding gene sequences could be obtained in entire draft genome of which 17 belonged to the category of aromatic compound degradation (Table 2). We analyzed the phenol hydroxylase, benzoate 1,2-dioxygenase, catechol 1,2-dioxygenase, gentisate 1,2-dioxygenase, protocatechuate 3,4-dioxygenase, and phenylacetate-CoA oxygenase for their gene arrangement using RAST annotations (Fig. 2a–f).

a-f Genetic organization of aromatic compound cleaving oxygenases in AKBS16. a Genetic organization of benzoate 1,2-dioxygenase. 1:5′nucleotidase, 2: hypotheticalprotein, 3: isochorismatase, 4: 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase, 5: molybdenum transporter ATP binding protein, 6: molybdenum transporter permeaseprotein, 7: molybdenum transporter DNA binding protein, 8: benzoate 1,2-dioxygenasealpha subunit, 9: benzoate 1,2-dioxygenase beta subunit, 10: benzoate dioxygenaseferrodoxin reductase, 11: benABC transcriptional activator BenR, 12: protein involved in meta pathway of phenol degradation, 13: nitriloacetate monooxygenase component B, 14: putative oxygenase subunit, 15: short chin dehydrogenase, 16: dienelactone hydrolase. b Genetic organization of phenol oxygenase in AKBS16. 1: ferrodoxin, 2: Gfa like protein, 3: Cu(I) responsive transcriptional regulator, 4: heavy metal transcriptional ATPase, 5: copper chaperone, 6: positive regulator of phenol hydroxylase, 7:phenolhydroxylase assembly protein DmpK, 8: phenol hydroxylase, P1 oxygenase component DmpL, 9: phenol hydroxylase, P2 regulatory component DmpM, 10: pheol hydroxylaseP3 oxygenase component DmpN, 11: Phenol hydroxylase oxygenase component DmpO, 12: phenol hydroxylase, FAD and (2Fe-2S) containing reducatse DmpP, 13: protein involvedin meta pathway of phenol degradation, 14: aromatic hydrocarbonutilizing transcriptional regulator CatR, 15: benzoate 1,2 dioxygenase beta subunit. c Genetic organization of catechol 1,2-dioxygenase in AKBS16. 1: Nitrate/nitritetransporter, 2: l-arabonate dehydratase, 3: GntR family transcriptional regulator, 4: hypothetical protein, 5: aromatic hydrocarbon utilization transcriptional regulator CatR, 6: muconate cycloisomerase, 7: muconolactone isomerase, 8: catechol 1,2-dioxygenase, 9: 3-oxoadipate CoA-transferase subunit A, 10: 3-oxoadipate CoA-transferase subunit B. d Genetic organization of gentisate 1,2-dioxygenase in AKBS16. 1: arginine antiporter, 2: alkane sulfonate utilization regulator, 3: senescence marker protein, 4: d-glucaratepermease, 5: Transcriptional regulator of LysR family, 6: hypothetical protein, 7: hypothetical protein 8: LysR family transcriptional regulator, 9: gentisate 1,2dioxygenase, 10: maleylacetoacetase isomerase, 11: fumarylacetoacetase, 12: 4 hydroxybenzaote transporter, 13: benzoate specific porin, 14: MFS transporter protein. e Genetic organization of protocatechuate 3,4-dioxygeanse in AKBS16. 1: 4 hydroxybenzote transporter, 2: 4-carboxymuconolactone decarboxylase, 3: protocatechuate 3,4-dioxygenase alpha subunit, 3: protocatechuate 3,4-dioxygenase betasubunit, 4: 3-dehydroquinate dehydratase, 5: 3-dehydroshikimate dehydratase, 6: porinB precursor, 7: quinate dehydrogenase, 8: hypothetical protein, 9: transmembrane protein. f Genetic organization of phenylacetate Co-A oxygenase in AKBS16. 1: ketoglutarate semialdehyde dehydrogenase, 2: amino acid permease, 3: l-serine dehydratase, 4: phenylacetic acid degradation protein PaaN, ring opening aldehyde dehydrogenase, 5: phenylacetate Co-A oxygenase, PaaG subunit, 6: phenylacetate Co-A oxygenase, PaaHsubunit 7: phenylacetate Co-A oxygenase, PaaI subunit 8: phenylacetate Co-A oxygenase, PaaJ subunit 9: phenylacetate Co-A oxygenase, PaaK subunit, 10: phenylacetate degradation enoyl-CoA hydratase PaaA, 11: phenylacetate degradationenoyl-CoA hydratase PaaB, 12: 3-hydroxylacyl-CoA dehydrogenase PaaC, 13: Phenylacetate degradation protein PaaE ketothiolase
The benzoate 1,2 dioxygenase gene could be located in the draft genome assembly of AKBS16 on scaffold number 82. The benzoate degradation pathway was under the regulation of benR element in AKBS16 strain as seen in Fig. 2a. The gene arrangement of benzoate 1,2-dioxygenase in AKBS16 was very similar to that of the closest match in SEED subsystem, i.e., A. baumannii AB307-0294. The benzoate 1,2-dioxygenase gene sequence from AKBS16 genome exhibited 98% identity with the benzoate 1,2-dioxygenase gene of A. calcoaceticus.
The gene arrangement of phenol hydroxylase is depicted in Fig. 2b with respect to its closest matches available in RAST, i.e., Azotobacter vinelandii and Methylibium petroleiphil. The DmpKLMNOP subunit genes were located serially and under the effect of positive regulator of phenol hydroxylase DmpR, which was 1671 bp in size, all present on the scaffold 91 of the genome. AKBS16 genome possessed copper transporter and metabolism genes in the vicinity to the phenol hydroxylase which was distinct from its closest neighbors. Draft genome analysis of Pseudomonas sp. EGD-AKN5 revealed similar DMP operon for phenol degradation in addition to atrazine-degrading atz operon for atrazine degradation pathway [23].
The catechol 1,2-dioxygenase gene was located on the scaffold 33 of the AKBS16 draft genome. The metabolism of central aromatic intermediates is initiated by the action of catechol 1,2-dioxygenase, followed by subsequent reactions of gene products like 3 oxoadipate Co-A transferase subunits A and B, muconolactone isomerase, and muconate cycloisomerase, all present near catechol 1,2-dioxygenase as seen in Fig. 2c. The aromatic hydrocarbon utilization transcriptional regulator CatR family could be located upstream of the catechol 1,2-dioxygenase gene. Additionally, the distinct gene arrangement of catechol 1,2-dioxygenase from its available closest matches, namely Ralstonia eutropha JMP134, Burkholderia xenovorans LB400 was depicted in Fig. 2c.
The gentisate 1,2-dioxygenase also falls under the SEED subsystem of metabolism of central aromatic intermediates, and its gene arrangement can be seen in Fig. 2d. This oxygenase gene was located on scaffold 98 of the draft genome adjacent to the LysR family transcriptional activator and 4-hydroxybenzoate transporter genes. The action of gentisate 1,2-dioxygenase is followed by the action of maleylacetoacetate isomerase and fumarylacetoacetase; both these genes were also found on this scaffold.
Protocatechuate 3,4 dioxygenase gene coding for the protocatechuate branch of beta-ketoadipate pathway was found on scaffold 63 in the AKBS16 genome adjacent to the 4-carboxymuconolactone decarboxylase gene. All genes adjacent to the protocatechuate 3,4-dioxygenase were not annotated owing to missing homologies in RAST. The arrangement of genes was identical to that of Ralstonia solanacearum GMI1000, Roseobacter sp. MED193, and Bradyrhizobium japonicum USDA110 as seen in Fig. 2e.
All the pca genes coding for the phenylacetyl-CoA catabolic pathway were clustered in a single scaffold in the draft genome of AKBS16. The paaGHIJK genes were serially arranged followed by genes for subsequent steps of this pathway (paaN, paaA, paaB, paaC, paaE). The closest matches to this gene arrangement can be seen in Fig. 2f. An overview of the biochemical pathway for degradation of aromatics such as phenol, benzoate, salicylate, and catechol was presented in Fig. 3. The genes present in AKBS16 strain and annotated for these pathways were depicted with respect to the compounds of their subsequent intermediates eventually leading to the citrate cycle. The strain AKBS16 exhibited presence of genes for benzoate degradation via hydroxylation reactions. The central pathway for aromatic compound degradation en route via catechol 1,2 dioxygenase (ortho) pathway.

Recent genomic and phenotypic analysis of an Acinetobacter venetianus, known to produce emulsan, which helps interface between organism cell membranes, revealed its role in oil/n-alkanes biodegradation [24]. Studies have proved significant potential of Acinetobacter genus in bioremediation of organic molecules. Recent studies disclose that Acinetobacter sp. DW-1 isolated from water sources served as an attractive option for phenol biodegradation [25]. Our recent microbial diversity analysis with respect to sulfamethaxole degradation revealed the dominance of Acinetobacter genus as it ranked among the top 20 dominating genera in the sample [26]. In another recent metagenomic study for petroleum hydrocarbon-contaminated samples from distinct ecologies, Acinetobacter genus was capable of biodegradation and possessed genes for stress response [27].
Conclusion
The genome-wide analysis performed for the bacterial strains AKBS9 and AKBS16 revealed the genes/pathway for biosurfactant production in these organisms. The emulsan cluster could be mined in the draft genome of AKBS16 and its gene arrangement could be understood with reference to the closest available matches. Till date several organisms have been reported for biosurfactant production, but the genes/pathways for which have been frugally understood. Hence, we exclusively report the genomic analysis of catabolic oxygenases and biosurfactant production metabolism present in AKBS bacterial strains in this study.
References
Deleu, M., & Paquot, M. (2004). From renewable vegetables resources to microorganisms: New trends in surfactants. Comptes Rendus Chimie, 7(6-7), 641–646.
Owsianiak, M., Chrzanowski, Ł., Szulc, A., Staniewski, J., Olszanowski, A., Olejnik-Schmidt, A. K., & Heipieper, H. J. (2009). Biodegradation of diesel/biodiesel blends by a consortium of hydrocarbon degraders: Effect of the type of blend and the addition of biosurfactants. Bioresource Technology, 100(3), 1497–1500.
Bezza, F. A., & Chirwa, E. M. N. (2016). Biosurfactant-enhanced bioremediation of aged polycyclic aromatic hydrocarbons (PAHs) in creosote contaminated soil. Chemosphere, 144, 635–644.
Perez-Ameneiro, M., Vecino, X., Cruz, J. M., & Moldes, A. B. (2015). Wastewater treatment enhancement by applying a lipopeptide biosurfactant to a lignocellulosic biocomposite. Carbohydrate Polymers, 131, 186–196.
Desai, J. D., & Banat, I. M. (1997). Microbial production of surfactants and their commercial potential. Microbiology and Molecular Biology Reviews, 61(1), 47–64.
Kapley, A., De Baere, T., & Purohit, H. J. (2007). Eubacterial diversity of activated biomass from a common effluent treatment plant. Research in Microbiology, 158(6), 494–500.
Kapley, A., Prasad, S., & Purohit, H. J. (2007). Changes in microbial diversity in fed-batch reactor operation with wastewater containing nitroaromatic residues. Bioresource Technology, 98(13), 2479–2484.
Kapley, A., & Purohit, H. J. (2009). Diagnosis of treatment efficiency in industrial wastewater treatment plants: A case study at a refinery ETP. Environmental Science & Technology, 43(10), 3789–3795.
Thangaraj, K., Kapley, A., & Purohit, H. J. (2008). Characterization of diverse Acinetobacter isolates for utilization of multiple aromatic compounds. Bioresource Technology, 99(7), 2488–2494.
Kapley, A., Liu, R., Jadeja, N. B., Zhang, Y., Yang, M., & Purohit, H. J. (2015). Shifts in microbial community and its correlation with degradative efficiency in a wastewater treatment plant. Applied Biochemistry and Biotechnology, 176(8), 2131–2143.
Nwaguma, I. V., Chikere, C. B., & Okpokwasili, G. C. (2016). Isolation, characterization, and application of biosurfactant by Klebsiella pneumoniae strain IVN51 isolated from hydrocarbon-polluted soil in Ogoniland, Nigeria. Bioresources and Bioprocessing, 3(1), 40.
Shoeb, E., Ahmed, N., Akhter, J., Badar, U., Siddiqui, K., Ansari, F., Waqar, M., Imtiaz, S., Akhtar, N., Shaikh, Q. U. A., & BAIG, R. (2015). Screening and characterization of biosurfactant-producing bacteria isolated from the Arabian Sea coast of Karachi. Turkish Journal of Biology, 39, 210–216.
Morikawa, M., Daido, H., Takao, T., Murata, S., Shimonishi, Y., & Imanaka, T. (1993). A new lipopeptide biosurfactant produced by Arthrobacter sp. strain MIS38. Journal of Bacteriology, 175(20), 6459–6466.
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Parrello, B., Shukla, M., & Vonstein, V. (2013). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Research, 42, 206–214.
Darzentas, N. (2010). Circoletto: Visualizing sequence similarity with Circos. Bioinformatics, 26(20), 2620–2621.
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., Lomsadze, A., Pruitt, K. D., Borodovsky, M., & Ostell, J. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Research, 44(14), 6614–6624.
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., Lee, S. Y., Fischbach, M. A., Müller, R., Wohlleben, W., & Breitling, R. (2015). antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Research, 43, 237–243.
Santos, D. K. F., Rufino, R. D., Luna, J. M., Santos, V. A., & Sarubbo, L. A. (2016). Biosurfactants: Multifunctional biomolecules of the 21st century. International Journal of Molecular Sciences, 17(3), 401.
Shaligram, S., Kumbhare, S. V., Dhotre, D. P., Muddeshwar, M. G., Kapley, A., Joseph, N., Purohit, H. P., Shouche, Y. S., & Pawar, S. P. (2016). Genomic and functional features of the biosurfactant producing Bacillus sp. AM13. Functional & Integrative Genomics, 16(5), 557–566.
Koumoutsi, A., Chen, X. H., Henne, A., Liesegang, H., Hitzeroth, G., Franke, P., Vater, J., & Borriss, R. (2004). Structural and functional characterization of gene clusters directing nonribosomal synthesis of bioactive cyclic lipopeptides in Bacillus amyloliquefaciens strain FZB42. Journal of Bacteriology, 186(4), 1084–1096.
Bach, H., Berdichevsky, Y., & Gutnick, D. (2003). An exocellular protein from the oil-degrading microbe Acinetobacter venetianus RAG-1 enhances the emulsifying activity of the polymeric bioemulsifier emulsan. Applied and Environmental Microbiology, 69(5), 2608–2615.
Nakar, D., & Gutnick, D. L. (2001). Analysis of the wee gene cluster responsible for the biosynthesis of the polymeric bioemulsifier from the oil-degrading strain Acinetobacter lwoffii RAG-1. Microbiology, 147(7), 1937–1946.
Bhardwaj, P., Sharma, A., Sagarkar, S., & Kapley, A. (2015). Mapping atrazine and phenol degradation genes in Pseudomonas sp. EGD-AKN5. Biochemical Engineering Journal, 102, 125–134.
Fondi, M., Maida, I., Perrin, E., Orlandini, V., La Torre, L., Bosi, E., Negroni, A., Zanaroli, G., Fava, F., Decorosi, F., & Giovannetti, L. (2016). Genomic and phenotypic characterization of the species Acinetobacter venetianus. Scientific Reports, 6(1), 21985.
Gu, Q., Wu, Q., Zhang, J., Guo, W., Wu, H., & Sun, M. (2017). Acinetobacter sp. DW-1 immobilized on polyhedron hollow polypropylene balls and analysis of transcriptome and proteome of the bacterium during phenol biodegradation process. Scientific Reports, 7, 4863.
Rudrashetti, A. P., Jadeja, N. B., Gandhi, D., Juwarkar, A. A., Sharma, A., Kapley, A., & Pandey, R. A. (2017). Microbial population shift caused by sulfamethoxazole in engineered-Soil Aquifer Treatment (e-SAT) system. World Journal of Microbiology and Biotechnology, 33(6), 121.
Mukherjee, A., Chettri, B., Langpoklakpam, J. S., Basak, P., Prasad, A., Mukherjee, A. K., Bhattacharyya, M., Singh, A. K., & Chattopadhyay, D. (2017). Bio informatic approaches including predictive metagenomic profiling reveal characteristics of bacterial response to petroleum hydrocarbon contamination in diverse environments. Scientific Reports, 7(1), 1108.
Acknowledgements
The authors acknowledge the Council of Scientific and Industrial Research, India, CSIR-network project ESC-0108-MESER, for supporting this research. Niti B Jadeja (SRF) is grateful to the CSIR. We are also grateful to Director, CSIR-NEERI, Nagpur, for the support. The manuscript has been checked for plagiarism using iThenticate Software under assigned KRC No.: CSIR-NEERI/KRC/2018/JAN/DRC/1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Table 1
Results of confirmatory tests for Biosurfactant production in strains AKBS9 and AKBS16. (DOCX 12 kb)
Supplementary Figure 1
In-silico steps involved in the analysis of AKBS9 and AKBS16 draft genomes. (PNG 171 kb)
Supplementary Figure 2
Graphical representation of gene ontologies for draft genome assemblies a) AKBS9 and b) AKBS16. (PNG 820 kb)
(PNG 1019 kb)
Supplementary Figure 3
Circos plot of AKBS9 and closest reference match Bacillus cereusb) Circos plot of AKBS16 with closest reference match Acinetobacter pittii. (PNG 4396 kb)
(PNG 4851 kb)
Rights and permissions
About this article

Cite this article
Jadeja, N.B., Moharir, P. & Kapley, A. Genome Sequencing and Analysis of Strains Bacillus sp. AKBS9 and Acinetobacter sp. AKBS16 for Biosurfactant Production and Bioremediation. Appl Biochem Biotechnol 187, 518–530 (2019). https://doi.org/10.1007/s12010-018-2828-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-018-2828-x